
Abstract
Objectives
Vaccination is the most effective preventive strategy for influenza disease. As the virus undergoes high antigenic drift, it requires a constant reformulation to obtain high protection.
Results
Immunogenicity of a purified chimeric protein containing conserved regions of influenza A/H1N1 viruses including the Hemagglutinin stalk domain, Nucleoprotein, and Matrix protein produced in a prokaryotic system was assessed in vitro and in vivo, alone or in combination with adjuvants by evaluating antibody responses, cytokine production, lymphocyte proliferative assay, and mortality rate after challenge. The animals that received the chimeric protein had specific antibody responses, elicited memory CD4 cells, cytokines of Th1 and Th2 cells and showed 75% protection against influenza virus lethal challenge. The animals injected with the chimeric protein supplemented with Alum showed improved immune responses, but they had 67% protection. In other words, although Alum adjuvant enriched the chimera specific immune responses potently, it could not enhance its protectivity.
Conclusion
Regarding the immunogenicity and protectivity of the chimeric protein construct against influenza, findings of the study suggested that the chimeric protein could be considered as a promising influenza vaccine candidate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza disease as a worldwide public health threat imposes a high social and economic burden (Lee et al. 2018). It can adversely affect 5–20% of the general population in the world resulting in severe disease and even death especially in the high-risk groups (Grohskopf et al. 2017). Human influenza viruses are respiratory pathogens readily transmitted through respiratory droplets and by contact (Deng et al. 2015).
Influenza outbreaks are caused by those subtypes of influenza A viruses, which have been already spread among human populations while influenza pandemic strains are generated through reassortment between human and avian viruses (Steel and Lowen 2014). Currently, vaccination, as the most effective strategy used to prevent influenza disease. Licensed influenza vaccines mostly provoke immunity against surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA), which are exposed to a high degree of genetic variations. So, vaccine compositions require to be updated approximately every year (Deng et al. 2015). According to the data published from the Centre for Disease Prevention and Control (CDC), the effectiveness of commercial influenza vaccines is up to 60% among the overall population, only if circulating influenza viruses are well-matched to the influenza vaccine (CDC 2018).
There are some influenza vaccines targeting conserved epitopes of viral glycoproteins. They do not need an annual update; however, they may have a significant proper effect on human health worldwide in a probable pandemic flu. In this regard, researches have attempted to focus on conserved or less variable epitopes of the proteins (Deng et al. 2015).
The extracellular domain of Matrix 2 peptide (M2e) is a conserved antigen of influenza A viruses and has been regarded as an encouraging epitope for developing a global influenza vaccine (Lee et al. 2015).
Hemagglutinin, as the main target for induction of strain-specific and protective influenza virus antibodies, comprises of HA1 and HA2 in which HA2 is less variable and causes cross-protection reaction among various subtypes of the virus (Impagliazzo et al. 2015; Maamary et al. 2017).
Nucleoprotein (NP), as another conserved protein of the virus, plays an important role in the virus infection process and induces cross-reactive immune responses, especially known as a strong T-cell inducer (Grant et al. 2013).
Conserved proteins of influenza viruses have also been studied as cocktail vaccines (Yassine et al. 2015). Accordingly, the present study was conducted to evaluate the immunogenicity of a chimeric protein containing conserved regions of HA (HA2), M2e, and NP derived from influenza A/H1N1 viruses in a mouse model. Our previous studies showed that the above-mentioned conserved proteins were not individually competent to confer high protection in virus-challenged animals. Therefore, in this study, purified chimeric protein (3M2e-HA2-NP) was prepared in a prokaryotic system and its protective immunity was evaluated in the presence of two different adjuvants.
Materials and methods
Materials
Mouse-adapted human influenza A virus (H1N1, PR/8/34), Madin-Darby Canine Kidney (MDCK) cell line, E. coli strains of Top10f′ and BL21 (DE3) as well as female BALB/c mice aged 6–8 weeks old were supplied by the Pasteur Institute of Iran, Tehran, Iran. Roswell Park Memorial Institute (RPMI)-1640 cell culture medium, penicillin/streptomycin (Pen/Strep), and Fetal Bovine Serum (FBS) were purchased from Gibco Company (USA). Isopropyl β-d-1-Thiogalactopyranoside (IPTG), Tetramethylbenzidine (TMB), (3-(4,5-dimethyl tetrazolyl-2) 2,5 diphenyl) tetrazolium bromide (MTT), Horseradish Peroxidase (HRP) conjugated anti-mouse Immunoglobulin G (IgG), HRP-conjugated rabbit anti-goat IgG, mouse IgG subclasses kit (ISO2), and also 96-well plates were obtained from Sigma-Aldrich Company (USA). Acrylamide and SDS were obtained from Merck Company (Germany). Protino™ Ni-TED-IDA 1000 kit and DNA extraction kit were supplied by Macherey Nagel™ Company (Germany) and Bioneer Company (South Korea), respectively. All materials were of analytical grade. Alum adjuvant was prepared from Alhydrogel®2% (Brenntag Biosector, Denmark) (CAS Number: 21645-51-2). MF59 emulsion adjuvant was also prepared according to the recommendation proposed in the study by Calabro et al. (2013), and its components (Tween 80, Span85, and Squalene) were purchased from Gibco Company (USA).
Construction of recombinant vector
Recombinant pET/3M2e-HA2-NP vector was constructed beforehand in the Department of Influenza and other Respiratory Viruses, Pasteur Institute of Iran (Tehran, Iran) (Hatami et al. 2017). The construct was made up from conserved regions of the 3 major vaccine candidate proteins, that its B-cell epitopes had been predicted using epitope prediction tool available at https://tools.iedb.org/ellipro/ (Ponomarenko et al. 2008).
Complementary sequence genome of 3 tandem repeat of M2 extracellular domain (3M2e) (accession number: ACF41880), HA2 subunit (accession number: HQ419001), and NP (accession number: LC120392) was fused to each other as shown in Fig. 1. Briefly, the HA2 sequence (including amino acids 339 to 566) was amplified by RT-PCR using the Influenza A virus (A/Tehran/18/2010(H1N1)) genome and was cloned in the pET28a vector into the target site of BamHI and HindIII (Farahmand et al. 2017). Then, 3M2e synthetic fragment (Biomatik, Denmark) was cloned upstream of the HA2 in the BamHI enzymatic site (Jalili et al. 2016). In the final step, NP fragment (including amino acids 1 to 100) was amplified by RT-PCR using the Influenza A virus (A/Puerto Rico/8/1934(H1N1)) genome and was subcloned downstream of the HA2 gene at the HindIII and XhoI sites. In structural design, the chimeric protein was embedded between two histidine tag sequences (Hatami et al. 2017). Accuracy of the chimeric construct was evaluated by enzymatic digestion and sequencing.

Schematic diagram of the recombinant pET/3M2e-HA2-NP vector construct
Preparation and purification of chimeric protein
The confirmed recombinant vector was expressed in E. coli BL21 (DE3) strain and the protein was verified using SDS-PAGE and western blotting (Laemmli 1970; Zamani et al. 2017). Briefly, competent bacteria were transformed by the recombinant vector and were cultured in LB agar plates supplemented with kanamycin (50 µg/ml) and tetracycline (10 µg/ml) antibiotics. Next, the plates were incubated for 16 h at 37 °C. Then, isolated colonies were sub-cultured in 10 ml of LB broth medium supplemented with antibiotics for 2–3 h at 37 °C in shaker incubator (at 185 rpm), that was induced with IPTG at final concentration of 0.5 mM and was incubated in shaker incubator once again (at 37 °C, 195 rpm). Protein expression was evaluated at various time intervals by SDS-PAGE. Scale-up production of the recombinant protein was performed in LB broth medium inoculated with a single colony containing a recombinant vector. Then, bacterial suspensions were centrifuged (at 4 °C, 10,000 rpm, for 10 min), and the pellets were stored at − 20 °C.
The Lysis-Equilibration-Wash (LEW) buffer, urea (8 M), and sonication (25–30 pulses, 80–90% pulse rate, 30 s on and off intervals) were used for protein extraction. Urea and LEW buffer containing 50 mM NaH2PO4 and 300 mM NaCl were added to the pellet at a ratio of 1:5 W/W. The suspension was homogenized with pipetting, and was incubated (at 4 °C, for 30 min) and was sonicated (25–30 pulses, 80–90% pulse rate, 30 s on and off intervals), and then it was centrifuged (at 10,000 rpm, 4 °C, for 5 min). The supernatant and the pellet were individually evaluated by the SDS-PAGE technique. The process of protein extraction was repeated until the whole pellet was dissolved. The recombinant protein was purified using Protino™ Ni-TED affinity column according to the manufacturer’s instruction. Briefly, the lysate was applied to the column, and the column was washed by 5 mM imidazole solution (pH 8).
Next, the protein was eluted with 300 mM imidazole elution buffer containing 8 M urea, and to remove the urea, the recombinant protein was dialyzed against Phosphate Buffered Saline (PBS) (pH 7.2) at 4 °C overnight.
Protein concentration was calculated using the Bradford method. Lipopolysaccharide content of the recombinant chimeric protein was measured by LAL clot assay, and the results showed no significant endotoxin activity. The LAL clot detection limit was < 1 EU/ml (LONZA N289-06).
Immunization procedure of animals
Immunogenicity of chimeric protein (3M2e-HA2-NP) was evaluated in BALB/c mice. All animals were housed in a well-lighted (12 h light/dark cycle), air-conditioned room (26 ± 1 °C) with 50 ± 10% of humidity. They had free access to standard diet and water. All animal experiments were approved and performed based on the guidelines introduced by the Ethics Committee of the Pasteur Institute of Iran (IR.PII.REC.1395.82). The animals were randomly divided into 3 case groups (15 mice/each group), and the animals individually received chimeric protein either alone or supplemented with adjuvants of Alum or MF59. The mice that received PBS, Alum, or MF5 were considered vehicle control groups. The compounds were administered intradermally in a total volume of 100 µl containing 15 µg of the chimeric protein for three times at 15-day interval (Table 1).
Measurement of specific anti-3M2e-HA2-NP antibodies
Blood samples were collected from 5 mice of each group 2 weeks after each immunization, and the blood sera were used to measure specific IgG, IgG1, and IgG2a antibodies using the ELISA technique. Briefly, 96-well ELISA plates were coated overnight at 4 °C using 100 μl of 10−4 mg/ml of the M2e synthetic peptide (GenScript: RP20206), 10−2 mg/ml of the NP synthetic peptide (Biomatik: RPC20340), or 10−3 mg/ml of the HA2 synthetic peptide (Biomatik: RPC25219) individually. Sera of the mice were diluted in PBS (1:1000). The concentration of coated antigen and the serum dilution was obtained by a checkerboard titration assay. Phosphate buffer saline with a pH of 7.4 (10 mM) containing 0.05% of Tween 20 (PBS-T) was used for washing steps and PBS-T containing 5% of bovine serum albumin was used as blocking buffer to prevent non-specific binding of antigens and antibodies to a microtiter plate (Shokouhi et al. 2018).
To evaluate specific IgG, HRP-conjugated goat anti-mouse IgG (A8924) was used as a secondary antibody and optical density was measured at 450 nm of wavelength. To measure IgG subclasses, goat anti-mouse IgG1 (M5532) and IgG2a (M5657) subtype antibodies and HRP-conjugated rabbit anti-goat IgG (A5420) were used as secondary and tertiary antibodies, respectively.
In vitro protection assay of immune sera
To assess reactivity of the sera, the virus neutralization test was performed. Madin-Darby Canine Kidney (MDCK) cell line was cultured in DMEM medium supplemented with 10% of FBS, 100 U/ml of penicillin, and 100 µg/ml of streptomycin. The sera were inactivated at 56 °C for 30 min, and two-fold serial dilutions were carried out using PBS starting from a ratio of 1:2 in a 96-well plate (50 µl/well), and was mixed with equal volume of influenza A/H1N1/PR8 virus (100 TCID50), and then was incubated at 37 °C for one hour. Then, 3 × 104 MDCK cells were added to each well and were incubated at 37 °C and 5% CO2 for 24 h. Cytopathic effect was examined and hemagglutination assays were performed to detect the presence of viral replication. To this end, 50 µl of each well was added to an equal volume of 0.5% chicken red blood cells in a 96-well U-bottom plate, and the plate was incubated for 1 h at room temperature. The absence of hemagglutination was considered as neutralization (Truelove et al. 2016).
Lymphocyte proliferation assay
Lymphocyte Proliferation Assay (LPA) was performed using MTT. Briefly, 3 mice from each group were sacrificed and their splenocytes were isolated 10 days after the last immunization, using a 40-μm cell strainer. Red blood cells were lysed using the NH4Cl lysis solution and the splenocytes were washed using RPMI-1640 medium (2×). Then, splenocytes were cultured in RPMI-1640 medium supplemented with 10% of FBS, 1% of 4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid (HEPES), 1% of pen/strep, and 2 mM l-glutamine to reach a final concentration of 2 × 105 cells/ml. Then, the splenocytes were cultured in 96-well plates and were incubated at 37ºC and 5% CO2 for 4 h. Then, the cells were stimulated with 1 µg/ml of 3M2e-HA2-NP chimeric protein and recombinant NP (RPC20340) or left without stimulation as a control group. After 48 h of incubation, lymphoproliferation assay was performed using MTT assay, and the results were expressed as Stimulation index (SI) using the following formula:
where, Cs and Cu are OD values of stimulated cells with recombinant protein and OD values of unstimulated cells, respectively. All the tests were performed in triplicate for each mouse.
Cytokine ELISA
The splenocytes obtained from lymphocyte proliferation assay (in above Section) were used to measure Interleukin-4 (IL-4) and Interferon-gamma (IFN-γ) cytokines as well. To do this, the cells were cultured and were stimulated as mentioned above. Some wells left without stimulation were considered as control. Supernatants of the culture media were collected after 72 h and secreted IFN-γ and IL-4 were measured by commercially available sandwich-based ELISA kits (R&D systems, DuoSet ELISA, USA) based on the manufacturer’s instructions. Then, optical densities were read at 450 nm, and cytokine level was measured for each sample based on standard curves. For each mouse, all the tests were performed in triplicate.
Viral challenge
12 mice from each group were challenged intranasally (i.n.) with one LD90 titer of A/H1N1/PR8 virus 15 days after the last immunization, and were kept under the class II laminar flow safety cabinets. Mortality rate and body weight loss were monitored twice daily during 15 days. Obtained results were plotted as survival rate versus days of post-challenge. Given the ethical permission provided by the Institutional Animal Care and Use Committee (IACUC), the mice with a body weight loss over 25% of total body weight were allowed to be euthanized (Standard A.C.U. 2016).
2.10. Statistical Analysis
The data obtained from cytokine assay and measurements of specific antibodies were analyzed with Excel and Graphpad Prism 6.0 software. The Kaplan–Meier curve was used for the analysis of survival rate. Statistical significance was determined by Chi-Square tests. P-values of < 0.05 were considered statistically significant. All values were expressed as means ± SD.
Results
Analysis of construction and structure using the software
Bioinformatic analysis of epitopes on the chimeric protein showed that the epitopes of all 3 fragments determined in a three-dimensional structure are presented to the immune system (Supplementary Table 1, and Supplementary Fig. 1).
The recombinant vector expressing chimeric protein was constructed using the conserved region of influenza A virus M2, NP, and HA proteins as mentioned in the Materials and Methods Section. Cloning and subcloning steps were evaluated by enzymatic digestion and the final construct was confirmed by sequencing (data not shown).
Preparation of chimeric protein
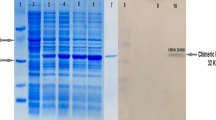
The chimeric protein (3M2e-HA2-NP) was prepared at large scale in BL21 cells and was purified using Protino™ Ni-TED-IDA kit, and then was dialyzed against PBS to remove urea and imidazole. Results of SDS-PAGE analysis confirmed the expression and production of the chimeric protein with a molecular weight of 51 KD, as expected (Fig. 2). The final concentration of the chimeric protein was estimated at up to 1 mg/ml.

Analysis of expression and purification of the recombinant protein in BL21 (DE3) E.coli by SDS-PAGE electrophoresis and western blotting: Lane (1) marker protein; lane (2) before induction; lanes (3, 4, 5, 6, 7) 1, 2, 3, 4 h and O/N after induction by IPTG, respectively; lane (8) purified chimeric protein; lane (9) marker protein (rainbow); lanes (10, 11) immunostaining of recombinant protein before and after induction, respectively
Measurement of specific antibodies
The potency of the recombinant protein in production of specific antibodies was evaluated in the mouse model. The protein was administered individually and in combination with 2 different adjuvants, namely Alum and MF59. The mice were bled and specific antibody responses against M2e, HA2, and NP recombinant proteins were evaluated using ELISA test 2 weeks after the first, second, and third immunizations. Blood samples obtained prior to immunization were considered as the negative control. Antibody titer increased significantly after the third injection compared to the first and second immunizations (data not shown). Results of the last immunization showed that all elicited chimer-receiving groups developed antibody responses compared to the control groups (Fig. 3). Although both adjuvants ameliorated antibody responses against M2e and NP antigens, none of them could improve specific anti-HA2 antibodies.

Measurement of IgG antibodies in sera of vaccinated mice using ELISA: Values for individual serum were measured at 450 nm of OD and were expressed as mean ± SD obtained from the 3 independent experiments. The differences between all treatment and control groups were statistically significant according to ANOVA results (P < 0.0001). Other differences were indicated in the figure. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001
Results of IgG sub-typing showed that the chimeric protein induced both IgG1 and IgG2a antibodies. Anti-M2e and anti-NP antibodies mostly belonged to the IgG2a subclass, while anti-HA2 antibodies belonged to the IgG1 subclass (Table 2).
The viral neutralizing test was used to show if antibodies detected by ELISA could inhibit virus propagation in vitro through binding to specific receptor binding sites and blocking viral infection. Neutralization of the virus was defined as loss of infection through a virus reaction to a specific antibody. Results of VNT showed that the sera of mice immunized with the chimeric protein could not neutralize the virus (data not shown).
Lymphocyte proliferation assay
To evaluate specific cellular immunity against the chimeric protein, the splenocytes isolated from mice in different groups were recalled with the chimeric protein. Results of the lymphocyte proliferation assay showed that the chimeric protein could stimulate memory CD4 cells to arm the immune system against viral infection, and Alum, as adjuvant improved lymphocyte responses significantly (P < 0.0001). Unexpectedly, mice immunized with the chimeric protein supplemented with MF59 had weaker responses even compared to the mice received the chimeric protein alone (P < 0.0001) (Fig. 4a).

Results obtained regarding lymphocyte proliferation after in vitro stimulation with: a chimeric protein; b chimeric protein and NP comparisingly. Splenocyte proliferation levels of 3 mice per each immunized group were evaluated by the MTT method 10 days after the last immunization. The values were expressed as mean ± SD obtained from the 3 independent experiments. The differences between all treatment and control groups were statistically significant according to ANOVA results (P < 0.0001). *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001
In addition to the chimeric protein, splenic lymphocyte cells were also stimulated with the recombinant nucleoprotein to determine its contribution to total response. Results showed the nucleoprotein as the main target of cell-mediated immunity (Fig. 4b).
Cytokine ELISA
To determine the potency of the chimeric protein for induction of various immune cells, secretion of IFN-γ (as a Th1 cytokine) and IL-4 (as a Th2 cytokine) was evaluated in spleen cells of immunized mice. Splenocytes of the mice were removed 10 days after the last immunization and were stimulated with a specific antigen (the chimeric protein). The concentration of IFN-γ and IL-4 secreted in the supernatant of stimulated spleen cells was measured (Fig. 5). Results showed that secretion of IFN-γ was significantly higher in mice that received the chimeric protein plus Alum than those received the chimeric protein plus MF59, or the chimeric protein alone (P < 0.001). In addition, results indicated that the concentration of IL-4 was higher in the chimera group than the other groups, although the differences observed between chimera and chimera-Alum-receiving groups were not statistically significant (P < 0.001).

Results obtained regarding cytokine assay (IFN-γ and IL-4). Splenocytes from 3 immunized mice of each group were obtained 10 days after the last immunization and were stimulated with chimeric protein. Cytokine concentration in splenocytes culture supernatants was measured using ELISA. The values were expressed as mean ± SD obtained from the 3 independent experiments. The differences between all treatment and control groups were statistically significant according to ANOVA results (P < 0.001). *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001
Morbidity and mortality rate
Animals immunized with the chimeric protein individually and supplemented with 2 different adjuvants were challenged with LD90 of influenza A virus (H1N1/PR8), and their survival rate and body weight loss were measured.
Results revealed that bodyweight decreased in all groups 2 days after the PR8 challenge. From day 3, the rate of body weight was found to be different between the groups. As shown in Fig. 6a, the mice received the chimeric protein supplemented with Alum or MF59 endured few weight changes compared to those received chimeric protein alone. Also, results showed that the survival rate increased significantly in all chimeric-receiving mice compared to the vehicle and negative control groups (P < 0.001) in which all mice died at 9–12 days after the challenge date (Fig. 6b). The trend of mortality rate sharply increased in chimeric and chimeric-adjuvant -receiving mice 9 days after challenge date reaching to a maximum on day 13, followed by a steady-state in all chimeric -receiving mice.

Protection over a period of 15 days in mice challenged intranasally with one LD90 of A/H1N1/PR/8/34 virus: Weight loss (a) and survival rate (b). Error bars show standard deviations. The control group was considered as a mean of the vehicle and negative control groups. Mean weights are shown where n > 2
Considering results obtained regarding body weight loss and mortality, it can be assumed that, the chimeric protein supplemented with adjuvants or even alone has been partially protective in mice against a lethal dose of the Influenza virus. The mice immunized with the chimeric protein alone were protected by 75%, and those received the chimeric protein plus Alum or MF5 were protected by 67 or 50%, respectively. Different survival rates observed in this study were not statistically significant (P < 0.5).
Discussion
Annual influenza vaccination, as an effective strategy is used to prevent the disease and its complications (De Filette et al. 2006). However, because of the antigenic drift of the influenza viruses, there is a need for annual updating of the vaccines (Tompkins et al. 2007). Therefore, it is necessary to develop a vaccine with constant formulation. In this regard, subunit vaccines containing conserved regions of the influenza virus can be useful (Andersson et al. 2012). Subunit vaccines are constituted of one or more protective antigens (Ebrahimi et al. 2010; Xin et al. 2013). In this context, M2e, HA2, and NP antigens of the influenza A virus, as conserved domains seem to be promising (Rao et al. 2010; Lee et al. 2015; Deng et al. 2017).
The extracellular domain of M2 protein induces very poor immune responses in the case of a natural infection. Various carrier vehicles or adjuvants have been used to improve its immunogenicity (Lee et al. 2015). In this regard, hepatitis virus core particles (Ravin et al. 2015), keyhole limpet hemocyanin (Deng et al. 2015) and virus-like particles have been used as carriers (Kim et al. 2017), and cholera toxin and liposome-based cationic agents have been considered as adjuvants (Smith et al. 2010; Li et al. 2014). However, none of these researches led to the development of an M2e-based licensed vaccine.
In addition, NP is the main antigen generated after infection with an influenza virus distinguished by the host Cytotoxic T Lymphocytes (CTLs). The CTLs recognize NP antigen peptides presented with MHC-I molecules on the surface of a virus-infected cell, destroying the cell and eliminating the virus yield (Zheng et al. 2014). So far, various NP-based vaccine candidates have been developed, which could induce a certain level of effective immunity against heterologous subtypes; however, cross-protection has not been satisfied yet and further optimization is needed (Roose et al. 2009; Guo et al. 2010).
HA is the main protein of the virus that allows the fusion of viral receptors to target cells. Globular head of HA protein is very variable that undergoes constant changes, while HA2 as a highly conserved stem-like structure can be used as a universal vaccine not only within a subtype but also among different types of the virus (Sautto et al. 2018).
Various researchers have studied the development of an M2e-based vaccine (Ebrahimi et al. 2010). In our previous study, we evaluated protective immunity of 3M2e in mice challenged with the influenza A virus and observed no protective immunity in the presence of the peptide alone. However, after fusion with Leishmania's major HSP70, 60% protective immunity was observed in virus-challenged mice (Shokouhi et al. 2018). Jalili et al. in their study, cloned, expressed, and purified a recombinant chimeric protein composed of the influenza A virus HA and ectodomain matrix 2 protein (3M2e-HA2) (Jalili et al. 2016). In another study, Sadeghi Neshat et al., constructed and purified HA2 peptide in a prokaryotic system and evaluated its immunogenic response and protective immunity in a complex with various adjuvants in virus-challenged mice. They showed proper induction of HA2 specific antibodies in mice, however, the peptide could induce protective immunity in only 20% of virus-challenged mice (Sadeghi Neshat et al. 2015).
Regarding NP peptide, there is a study which its results showed 33% protective immunity in virus-challenged mice (Data not published yet). Therefore, the combination of virus-conserved proteins together can be effective for induction of protective immunity against the influenza virus.
In the present study, the potency of a recombinant chimeric protein containing conserved regions of matrix protein, hemagglutinin, and a nucleoprotein derived from influenza A virus was evaluated as a vaccine candidate. Zhang et al. showed that 3 copies of the M2e gene induced the highest protective immunity in virus-challenged chickens compared to other different copies. In addition, they showed that virus-challenged mice that received 3 copies of M2e protein had a slow weight loss and recovered faster than other groups (Zhang et al. 2011). Herein, 3 copies of the M2e gene were used to mediate immune responses. The recombinant 3M2e-HA2-NP chimeric protein was produced in a prokaryotic system and was purified successfully. Constructability for the induction of specific antibody responses was evaluated and compared to that of the control groups. Results of measuring antibody responses showed that both types of Th1 and Th2 cells were involved in generating total antibody responses against M2e, HA2, and NP, indicating that the dominant role of HA2 has been more biased to Th2 immune cells for induction of specific antibody responses. However, anti-M2e and anti-NP antibodies mostly belonged to the IgG2a subclass in all immunized mice. Thus, the purified recombinant protein could induce simultaneous and comparable immune responses against individual components of the chimeric protein.
The Virus Neutralization Test (VNT) is a very specific and sensitive assay for influenza viruses that evaluates neutralizing antibody titer. Considering that, influenza virus attaches to the host cell through the globular head of HA, the VNT is not expected to neutralize viral particles according to immune sera antibodies against internal proteins and stalk domain of HA. However, anti-stalk mAbs are typically neutralizing (DiLillo et al. 2014; Kosik et al. 2019). It has been found that T cells are important in terms of protective responses against influenza infection. T cells play a critical role in virus clearance, reducing symptom severity, duration of the disease, and viral shedding (Choi et al. 2019).
Results of the present study showed that the chimeric protein containing conserved regions of Influenza proteins could potently stimulate memory CD4 cells and the importance and priority of NP protein for induction of cellular response was highlighted in lymphocyte proliferation assay, as expected.
Results of cytokine assay showed the potency of 3M2e-HA2-NP protein for the induction of robust IFN-γ responses. IFN-γ production demonstrates Th1-related immune responses (Rapoport and McLachlan 2014). Also, IFN-γ function is required to mediate cell immunity and enhance cell cytotoxicity, leading to priming and mature lymphocytes (Bhat et al. 2017; Popa-Fotea 2017).
It has been shown that aluminum salts mainly induce Th2 cells and rarely stimulate cellular immune responses (Spickler and Roth 2003). Interestingly, in our study, the chimeric protein supplemented with Alum stimulated IFN-γ production strongly. On the other hand, an increased level of IL-4 in all immunized groups indicated that the 3M2e-HA2-NP chimeric protein could generate balanced cellular and humoral immune responses.
Furthermore, protective potency of the chimeric protein was evaluated in mice challenged with influenza A virus and results showed that the chimeric protein individually or in combination with different adjuvants increased survival rate of challenged mice significantly compared to the control group. Interestingly, lower body weight loss and the highest survival rate were achieved when the chimeric protein was used without any adjuvants, even though the differences were not significant. In other words, although, Alum improved the chimera specific immune responses potently and MF59 somehow, they could not enhance its protectivity. Maybe, further immune system promotion is not necessarily better for animal protection against the influenza virus. Adjuvants play important roles in enhancing desired antibodies against low immunogenicity of purified protein-based antigens. However, material accumulation of the adjuvants in the body can be toxic, and in the case of aluminum, it may influence the brain and bone tissue potentially resulting in the development of the Alzheimer's disease (Aguilar and Rodríguez 2007). Although the benefits of using adjuvants in vaccines outweigh the adverse effects, under specific conditions, the use of a vaccine not requiring adjuvant is desirable in veterinary medicine as well as in sensitive individuals (Spickler and Roth 2003).
There were some limitations and drawbacks to this study. First, there were a few animals in each group, which is insufficient to conclude statistical and clinical significances regarding the assumption that, whether the use of chimeric protein without any adjuvants has the superiority. Moreover, the other immune system factors involved in influenza infection such as other cytokines secreted from Natural Killer (NK) cells shown to play an essential role in the immune response against influenza infection were not investigated in this study (Guo et al. 2011).
Conclusion
In this study, the construct of the chimeric protein containing conserved regions of human influenza A virus proteins was prepared and characterized, and its efficacy was evaluated against influenza infection in vitro and in vivo. Results showed sufficient immunogenicity and protectivity of the chimeric protein. In addition, its production is economically and temporally cost-effective.
A complex of the Alum as adjuvant along with the chimeric protein improved immunogenicity significantly, while it did not influence its protectivity. According to the findings of the study, it is suggested to consider the chimeric protein as a promising universal influenza vaccine candidate. To reach this milestone, we are going to evaluate the extent of protection provided by the chimeric protein in a challenge with heterologous subtypes of the influenza virus in our future studies.
References
Aguilar JC, Rodríguez EG (2007) Vaccine adjuvants revisited. Vaccine 25(19):3752–3762
Andersson A-MC, Håkansson KO, Jensen BAH, Christensen D, Andersen P, Thomsen AR, Christensen JP (2012) Increased immunogenicity and protective efficacy of influenza M2e fused to a tetramerizing protein. PLoS ONE 7(10):e46395
Bhat P, Leggatt G, Waterhouse N, Frazer IH (2017) Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis 8(6):e2836–e2836
Calabro S et al (2013) The adjuvant effect of MF59 is due to the oil-in-water emulsion formulation, none of the individual components induce a comparable adjuvant effect. Vaccine 31(33):3363–3369
CDC (2018) Vaccine effectiveness—how well does the flu vaccine work? https://www.cdc.gov/flu/professionals/vaccination/effectiveness-studies.htm.
Choi A et al (2019) Chimeric hemagglutinin-based influenza virus vaccines induce protective stalk-specific humoral immunity and cellular responses in mice. ImmunoHorizons 3(4):133–148
De Filette M, Ramne A, Birkett A, Lycke N, Löwenadler B, Jou WM, Saelens X, Fiers W (2006) The universal influenza vaccine M2e-HBc administered intranasally in combination with the adjuvant CTA1-DD provides complete protection. Vaccine 24(5):544–551
Deng L, Cho KJ, Fiers W, Saelens X (2015) M2e-based universal influenza A vaccines. Vaccines 3(1):105–136
Deng L, Kim JR, Chang TZ, Zhang H, Mohan T, Champion JA, Wang B-Z (2017) Protein nanoparticle vaccine based on flagellin carrier fused to influenza conserved epitopes confers full protection against influenza A virus challenge. Virology 509:82–89
DiLillo DJ, Tan GS, Palese P, Ravetch JV (2014) Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat Med 20(2):143–151
Ebrahimi SM, Tebianian M, Toghyani H, Memarnejadian A, Attaran HR (2010) Cloning, expression and purification of the influenza A (H9N2) virus M2e antigen and truncated Mycobacterium tuberculosis HSP70 as a fusion protein in Pichia pastoris. Protein Express Purif 70(1):7–12
Farahmand B, Akbari A, Akbari K, Fotouhi Chahouki F, Mehrbod P, Jalili N (2017) Cloning, expression and purification of hemagglutinin conserved domain (HA2) of influenza A virus, to be used in broad-spectrum subunit vaccine cocktails. Vacres 4(1):34–40
Grant E, Wu C, Chan K-F, Eckle S, Bharadwaj M, Zou QM, Kedzierska K, Chen W (2013) Nucleoprotein of influenza A virus is a major target of immunodominant CD8+ T-cell responses. Immunol Cell Biol 91(2):184–194
Grohskopf LA, Sokolow LZ, Broder KR, Walter EB, Bresee JS, Fry AM, Jernigan DB (2017) Prevention and control of seasonal influenza with vaccines: recommendations of the advisory committee on immunization practices—United States, 2017–2018 influenza season. MMWR 66(2):1–20
Guo L, Zheng M, Ding Y, Li D, Yang Z, Wang H (2010) Protection against multiple influenza A virus subtypes by intranasal administration of recombinant nucleoprotein. Arch Virol 155(11):1765–1775
Guo H, Kumar P, Malarkannan S (2011) Evasion of natural killer cells by influenza virus. J Leukoc Biol 89(2):189–194
Hatami S, Fotouhi F, Hashemi M, Saleh M, Nazeri E, Shokohi H, Farahmand B (2017) Prokaryotic expression of chimeric trimer protein (3M2e-HA2-NP) derived from conserved domains of influenza virus A/H1N1 as a promising universal subunit vaccine. SJKUMS 91:111–120
Impagliazzo A et al (2015) A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 349(6254):1301
Jalili N, Taheri N, Tavakoli R, Fotoohi F, Akbari A, Farahmand B (2016) Expression and purification of a recombinant chimeric protein (3M2e-HA2) composed of influenza virus hemagglutinin and matrix protein conserved domain for universal subunit vaccine development. JMUMS 26(137):12–22
Kim EH, Han G-Y, Nguyen H (2017) An adenovirus-vectored influenza vaccine induces durable cross-protective hemagglutinin stalk antibody responses in mice. Viruses 9(8):234
Kosik I et al (2019) Neuraminidase inhibition contributes to influenza A virus neutralization by anti-hemagglutinin stem antibodies. J Exp Med 216(2):304
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Lee VJ et al (2018) Advances in measuring influenza burden of disease. Influenza Other Respir. Viruses 12(1):3–9
Lee YN, Kim MC, Lee YT, Kim YJ, Kang SM (2015) Mechanisms of cross-protection by influenza virus M2-based vaccines. Immune Netw 15(5):213–221
Li J, Arévalo MT, Chen Y, Posadas O, Smith JA, Zeng M (2014) Intranasal immunization with influenza antigens conjugated with cholera toxin subunit B stimulates broad spectrum immunity against influenza viruses. Hum Vaccin Immunother 10(5):1211–1220
Maamary J, Wang TT, Tan GS, Palese P, Ravetch JV (2017) Increasing the breadth and potency of response to the seasonal influenza virus vaccine by immune complex immunization. Proc Natl Acad Sci USA 114(38):10172–10177. https://doi.org/10.1073/pnas.1707950114
Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, Peters B (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform 9:514–514
Popa-Fotea N-M (2017) Influenza vaccination-old and new emerging challenges. Roum Arch Microbiol Immunol 76(3–4):183–187
Rao SS, Kong W-P, Wei C-J, Hoeven NV, Gorres JP, Nason M, Andersen H, Tumpey TM, Nabel GJ (2010) comparative efficacy of hemagglutinin, nucleoprotein, and matrix 2 protein gene-based vaccination against H5N1 influenza in mouse and ferret. PLoS Biol 5(3):e9812
Rapoport B, McLachlan SM (2014) Graves' hyperthyroidism is antibody-mediated but is predominantly a Th1-type cytokine disease. J Clin Endocr Metab 99(11):4060–4061
Ravin NV, Blokhina EA, Kuprianov VV, Stepanova LA, Shaldjan AA, Kovaleva AA, Tsybalova LM, Skryabin KG (2015) Development of a candidate influenza vaccine based on virus-like particles displaying influenza M2e peptide into the immunodominant loop region of hepatitis B core antigen: insertion of multiple copies of M2e increases immunogenicity and protective efficiency. Vaccine 33(29):3392–3397
Roose K, Fiers W, Saelens X (2009) Pandemic preparedness: toward a universal influenza vaccine. Drug News Perspect 22(2):80–92
Sadeghi Neshat S, Farahmand B, Kianmehr Z, Zamani S, Saleh M, Fotouhi F (2015) Immunogenicity enhancement of influenza virus stalk domain using Leishmania major heat shock protein, one step closer to universal vaccine. IUMS 33(349):1–12
Sautto GA, Kirchenbaum GA, Ross TM (2018) Towards a universal influenza vaccine: different approaches for one goal. Virol J 15:17
Shokouhi H, Farahmand B, Ghaemi A, Mazaheri V, Fotouhi F (2018) Vaccination with three tandem repeats of M2 extracellular domain fused to Leismania major HSP70 protects mice against influenza A virus challenge. Virus Res 251:40–46
Smith LR et al (2010) Phase 1 clinical trials of the safety and immunogenicity of adjuvanted plasmid DNA vaccines encoding influenza A virus H5 hemagglutinin. Vaccine 28(13):2565–2572
Spickler AR, Roth JA (2003) Adjuvants in veterinary vaccines: modes of action and adverse effects. J Vet Intern Med 17(3):273–281
Standard ACU (2016) Animal Care and Use Standard- Infection of mice with influenza virus. The University Of Melbourne, Melbourne, Office for Research Ethics & Integrity pp 1–5
Steel J, Lowen A (2014) Influenza A virus reassortment. Curr Top Microbiol Immunol 385:377–401
Tompkins SM et al (2007) Matrix protein 2 vaccination and protection against influenza viruses, including subtype H5N1. Emerg Infect Dis 13(3):426
Truelove S et al (2016) A comparison of hemagglutination inhibition and neutralization assays for characterizing immunity to seasonal influenza A. Influenza Other Respir Viruses 10(6):518–524
Xin Q et al (2013) Subunit vaccine consisting of multi-stage antigens has high protective efficacy against Mycobacterium tuberculosis infection in mice. PLoS ONE 8(8):e72745
Yassine HM et al (2015) Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat Med 21:1065
Zamani S, Fotouhi Chahouki F, Mehrbod P, Sadeghi Neshat S, Farahmand B (2017) Production and evaluation of polyclonal antibody against influenza A virus matrix 2 conserved protein for research and diagnosis purposes. Iran J Virol 11(4):31–38
Zhang X, Liu M, Liu C, Du J, Shi W, Sun E, Li H, Li J, Zhang Y (2011) Vaccination with different M2e epitope densities confers partial protection against H5N1 influenza A virus challenge in chickens. Intervirology 54(5):290–299
Zheng M, Luo J, Chen Z (2014) Development of universal influenza vaccines based on influenza virus M and NP genes. Infection 42(2):251–262
Acknowledgements
This study was performed in the Department of Influenza and other Respiratory Viruses, Pasteur Institute of Iran, Tehran (Grant Number 759) that hereby thanks to all colleagues for their valuable assistance.
Supporting information
Supplementary Table 1—demonstrated B-cell epitopes analysis of chimeric protein using ElliPro (http://tools.iedb.org/ellipro/): a) Predicted linear Epitope; b) Predicted Discontinuous Epitope
Supplementary Figure 1—demonstrated 3D structure of HA2 molecule and important antigenic regions as determined by ElliPro: a) Predicted linear Epitope; b) Predicted Discontinuous Epitope (In this ball-and-stick model, yellow balls indicated residues of predicted epitopes and white sticks showed structures of non-epitope and core residues).
Funding
This study was funded by Pasteur Institute of Iran, Tehran (Grant Number 759).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article contains studies with animals performed by the authors. All animal experiments were approved and performed based on the guidelines of the Ethics Committee of the Pasteur Institute of Iran (IR.PII.REC.1395.82).
Research involoving human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Saleh, M., Nowroozi, J., Farahmand, B. et al. An approach to the influenza chimeric subunit vaccine (3M2e-HA2-NP) provides efficient protection against lethal virus challenge. Biotechnol Lett 42, 1147–1159 (2020). https://doi.org/10.1007/s10529-020-02822-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-020-02822-3