
Abstract
Objective
To explore the glycerol utilization pathway in Corynebacterium glutamicum for succinate production under O2 deprivation.
Result
Overexpression of a glycerol facilitator, glycerol dehydrogenase and dihydroxyacetone kinase from Escherichia coli K-12 in C. glutamicum led to recombinant strains NC-3G diverting glycerol utilization towards succinate production under O2 deprivation. Under these conditions, strain NC-3G efficiently consumed glycerol and produced succinate without growth. The recombinant C. glutamicum utilizing glycerol as the sole carbon source showed higher intracellular NADH/NAD+ ratio compare with utilizing glucose. The mass conversion of succinate increased from 0.64 to 0.95. Using an anaerobic fed-batch fermentation process, the final strain produced 38.4 g succinate/l with an average yield of 1.02 g/g.
Conclusions
The metabolically-engineered strains showed an efficient succinate production using glycerol as sole carbon source under O2 deprivation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Succinic acid, is an important platform chemical being used in the pharmaceutical, agricultural and food industries (Zeikus et al. 1999). Its production has attracted considerable interest. Anaerobiospirillum succiniciproducens, Actinobacillus succinogenes and Mannheimia succiniciproducens can be used to produce succinic acid by fermentation under anaerobic conditions (Glassner and Datta 1992; Guettler et al. 1996; Lee et al. 2002). Glycerol is a main by-product of biodiesel and bioethanol production (Yazdani and Gonzalez 2007). By utilizing glycerol for the production of succinate, the economic efficiency of these biofuel production processes can be significantly increased.
Bio-based succinate production from glycerol has been described for a number of natural succinate producers, such as Basfia succiniciproducens (Scholten and Dagele 2008) and A. succiniciproducens (Lee et al. 2001), as well as for metabolically-engineered Escherichia coli strains (Blankschien et al. 2010; Zhang et al. 2010).
Metabolic engineering can increase succinate production in bacteria. Corynebacterium glutamicum, which is regarded as safe (GRAS status), has a long history in the fermentation industry for producing amino acids and nucleic acids (Vertès et al. 2005). The genome sequences of several C. glutamicum strains have been published (Yukawa et al. 2007; Ikeda and Nakagawa 2003; Kalinowski et al. 2003). C. glutamicum performs a mixed acid fermentation with succinate as one of the products besides lactate, and acetate (Okino et al. 2008). Thus, C. glutamicum is potentially useful for succinate production based on pentoses derived from lignocellulose hydrolysates or waste glycerol accumulated in biodiesel industry.
In this study, engineering a glycerol utilization pathway in C. glutamicum for succinate production under O2 deprivation, is presented. Firstly, the glycerol utilization pathway enzymes were overexpressed, then the NADH/NAD+ ratio in the cells using glycerol as the sole carbon under O2 deprivation was analyzed. Finally, anaerobic fed-batch fermentations were carried out.
Materials and methods
Bacterial strains, plasmids, and media
All strains and plasmids, their sources and relevant characteristics, are given in Supplementary Table 1. The parent strain of C. glutamicum was ATCC 13032. For genetic manipulations, E. coli JM109 strains were grown at 37 °C in lysogeny broth. C. glutamicum strains were routinely cultivated at 30 °C. Plasmid DNA transfer into C. glutamicum was carried out by electroporation. If appropriate, the final antibiotic concentrations were as follows: for E. coli 50 µg chloramphenicol ml−1 and 50 µg kanamycin ml−1; for C. glutamicum 10 µg chloramphenicol ml−1 and 10 µg kanamycin ml−1. The nutrient-rich medium (A medium) was used for aerobic growth (Wang et al. 2014a). The mineral salts medium (BT medium) was used for anaerobic fermentation (Wang et al. 2014b).
Genetic methods
Oligonucleotides used are given in Supplementary Table 2. Plasmid pk18mobsacB-Δpqo::P tac -glpF replacing the chromosomal pqo gene with the glpF gene from C. glutamicum under the control of the tac promoter from plasmid pXMJ19. First, the regions up and downstream regions (approx. 0.6 kb each) of the Δpqo deletion region were amplified with the oligonuclecotide pairs pqoF1/pqoR1, pqoF2/pqoR2, respectively. The two PCR products served as the templates for an overlap extension PCR with oligonucleotide pair pqoF1/pqoR2. The PCR product of about 1.2 kb, which carried XbaI cloning sites at the fusion site, was digested with EcoRI and HindIII and cloned into pK18mobsacB cut with the same enzymes. The resulting plasmid was named pK18mobsacB-Δpqo. Plasmid pXMJ19-glpF was constructed for cloning P tac -glpF fragment. The glpF gene was amplified using the oligonucleotide pair glpF1/glpF2 and chromosomal DNA of E. coli K-12. The PCR product of 0.8 kb was digested with PstI/BamHI and cloned into pXMJ19 cut with the same enzymes. The DNA fragment covering P tac -glpF was constructed by amplifying the gene from plasmid pXMJ19-glpF, using the oligonucleotide pair tac-glpF1/tac-glpR1. The resulting PCR product of 0.9 kb was digested with XbaI and cloned into pK18mobsacB-Δpqo::XbaI cut with the same enzymes.
Plasmid pXMJ19-gldA was constructed for overexpressing the gldA gene. The gldA gene was amplified using the oligonucleotide pair gldA1/gldA2 and chromosomal DNA of E. coli K-12. The PCR product of 1.1 kb was digested with XbaI/KpnI and cloned into pXMJ19 cut with the same enzymes.
Plasmid pEC-dhaKLM was constructed for overexpressing the dhaKLM gene. The dhaKLM gene was amplified using the oligonucleotide pair dhaKLM1/dhaKLM 2 and chromosomal DNA of E. coli K-12. The PCR product of 3.4 kb was digested with EcoRI/XbaI and cloned into pEC-XK99E cut with the same enzymes.
Resultant plasmids were introduced into each cell by electroporation. C. glutamicum NC-2 was transformed by electroporation with plasmids pK18mobsacB-Δpqo::P tac -glpF. The transfer of the resulting deletion plasmids into C. glutamicum and selection for the first and second recombination events were performed as described previously (Wang et al. 2014b). The results strain named NC-3.
Enzyme assay
The activities of glycerol dehydrogenase and dihydroxyacetone kinase were measured according to Shams and Gonzalez. (2008). The activity of glycerol dehydrogenase was measured in a reaction mixture (1 ml) containing 2 mM MgCl2, 500 mM NADH, 100 mM hydroxyacetone, 30 μl crude cell extract, and 100 mM of the appropriate buffer according to the pH of the assay. The activity of dihydroxyacetone kinase was recorded as the amount of NADH oxidized per unit of time in a coupled reaction with excess glycerol-3-phosphate dehydrogenase, where the reaction was started by adding 4 mM DHA (Molin et al. 2003). One unit of the overall glycerol dehydrogenase and dihydroxyacetone kinase activity was defined as the amount of enzyme required to produce 1 µmol NAD+ per min from the NADH. (IU, the amount of enzyme required to convert 1 μmol product per min from the substrate, 1 IU = 1 μmol/min.)
Culture conditions for cell growth and succinate production
For aerobic growth, 5 ml of an overnight culture was inoculated into 50 ml medium A containing 30 g glucose/l and grown at 30 °C for 18 h.
For organic acid production in serum bottle, the cells grown in aerobic-phase cultures were harvested by centrifugation at 4 °C (3000×g, 10 min). The cell pellets were subsequently washed twice with mineral salts medium. Following the second wash, the cells were resuspended in 25 ml BT medium containing 40 g carbon source/l (glycerol or glucose) and 300 mM NaHCO3. Organic acid production was started by adding glucose in a rotary shaker (150 rpm) at 30 °C.
Fed-batch fermentation was carried out as follows. The strains were precultured in 500 ml shake-flasks containing 100 ml A medium supplemented with 20 g glucose l−1 on a rotary shaker at 30 °C and 200 rpm. A 10 % (v/v) inoculum was used to inoculate a 5 l bioreactor with 3 l medium A supplemented with 30 g glucose/l. The culture was incubated on an agitation speed of 400 rpm for 12 h at 30 °C. The pH was automatically controlled at 7 by addition of 20 % NH4OH. After 12 h of aerobic culture, the cells were then harvested and inoculated into BT medium supplemented with 50 g glycerol/l and 300 mM NaHCO3. O2 was removed by introducing CO2 into bioreactor for 5 min. O2 deprivation conditions were maintained by preventing aeration and gentle agitation in the hermetically sealed bioreactor. The speed of agitation was 150 rpm, and the cultures were incubated at 30 °C. The pH was kept at pH 7.5 by automated addition of 20 % NH4OH.
NADH/NAD+ assays
The intracellular concentrations of NADH and NAD+ were measured using the NAD+/NADH assay kit (AmpliteTM Colorimetric NAD/NADH Assay Kit, AAT Bioquest, USA).
Analytical methods
Cell growth was monitored from the OD600 value and converted into the dry cell weight (DCW) using DCW(g/l) = 0.4 × OD600. The mass yield of succinic acid was defined as the amount of succinic acid from 1 g carbon source (glucose or glycerol) consumed and expressed in g/g.
Culture samples were centrifuged (18,000×g, 4 °C,1 min) and the supernatants were analyzed for glucose and glycerol by HPLC using an Aminex HPX-87H column (Bio-Rad) operating at 45 °C with 5 mM H2SO4 as the mobile phase at 0.6 ml/min and detection with an RI detector.
Acetate, succinate, and pyruvate were quantified by HPLC equipped with a UV detector and conductivity meter and Grace Prevai™ column operating at 215 nm with 25 mM KH2PO4 (pH 2.5) as mobile phase at 1 ml/min.
Results and discussion
Utilization of glycerol for anaerobic succinate production by C. glutamicum NC-3G
C. glutamicum does not utilize glycerol as a carbon source. Unlike in E. coli, which can grow on glycerol as a sole carbon source, no glycerol metabolic pathway is present in any of C. glutamicum sequenced to date. To give these strains the ability to use glycerol as carbon source under O2 deprivation, we explored a anaerobic glycerol utilization pathway for succinate production (Fig. 1). The E. coli glpF gene coding a glycerol facilitator was integrated into C. glutamicum NC-2 chromosome, resulting in strain NC-2G. The E. coli gldA and dhaKLM genes were isolated by PCR and precisely subcloned under the control of the strong constitutive promoter (tac or trc) that is present in vector pXMJ19 and pEC-xk99E respectively. The resulting strain named NC-3G. The specific activity of GLDH was 1.3 U/mg and the specific activity of DAK was 2.1 U/mg, indicating their functional expression. Using NC-3G cells incubated under O2 deprivation conditions, we evaluated the productivity of organic acids from glycerol by comparing the production of organic acid achieved from glucose. In both cases, predominantly succinate and acetate were produced, with trace amounts of pyruvate (Table 1). The consumption rate of glycerol (1.2 g/l.h) was approx. half that of glucose (2.2 g/l.h). Moreover, the production rates of succinic acid were comparable in both cases, reaching approx. 1.1 g/l.h, whereas the production rate of acetate from glucose (4.2 g/l) was higher than that from glycerol (3.1 g/l). The yield of succinic acid was higher from glycerol (1.02 g/g) than from glucose (0.8 g/g). These results suggest that the utilization of glycerol favours succinate production and lowers acetate production.

Engineered pathway for anaerobic glycerol utilization to succinate. glpF glycerol facilitator, gldA glycerol dehydrogenase, dhaKLM dihydroxyacetone kinase, glpK glycerol kinase, tpiA triosephosphate isomerase, ldhA lactate dehydrogenase, pta-ackA phosphate acetyltransferase-acetate kinase, DHAP dihydroxyacetone 3-phosphate, DHA dihydroxyacetone, PEP phosphoenolpyruvate, G3P glycerol 3-phosphate, GA3P glyceraldehyde 3-phosphate, PYR pyruvate, LA lactate, Ace acetate, OAA oxaloacetate, MAL malate, FUM fumarate, SUC succinate. Dotted lines represent enzymes and reactions so far not found in C. glutamicum
Effect of NaHCO3 addition on glycerol utilization during succinate production
In the absence of NaHCO3, the glycerol consumption rate was 0.31 ± 0.02 g/l.h and the yield of succinate was 0.91 ± 0.02 g/g. 200 mM NaHCO3 was added to the medium and led to a 3.5-fold increase in succinate production. These results may be attributable to anaplerotic enzymes, which include phosphoenolpyruvate carboxylase (PEPC) and/or pyruvate carboxylase (PC), fixing CO2 to produce C4 dicarboxylic acids from phosphoenolpyruvate (PEP) or pyruvate (Fig. 1). Glycerol consumption and succinate production rates were also increased 3.2-fold and 3.5-fold, respectively, in the presence of bicarbonate (Table 2). Glycerol is metabolized to PEP or pyruvate via the glycerol utilization pathway along with NADH generation by the activity of GLDH and GAPDH. To our knowledge, GAPDH is inhibited by the accumulation of intracellular NADH (Inui et al. 2004). Similarly, accumulation of intracellular NADH also inhibited GLDH led to glycerol utilization decreased. Bicarbonate may mediate activation of NAD+ regeneration in vivo. Thus, addition of bicarbonate led to the conversion of PEP/pyruvate to OAA accompanied by CO2 fixation mediated by either PEPC or PC.
Effects of glycerol metabolism on intracellular NADH/NAD+ ratio under O2 deprivation
The yield of succinate increased when the glycerol utilization pathway was activated under O2 deprivation (Table 1). To analyze the mechanism of glycerol metabolism on succinate production, the ratio of NADH/NAD+ was measured (Table 1). With glycerol as substrate, the NADH/NAD+ ratio was 0.95 compared to 0.64 when glucose was used under O2 deprivation. Thus more NADH is produced from glycerol metabolism than from glucose. One mol NADH is formed per glycerol converted to DHA. A higher ratio of NADH/NAD+ is beneficial for the generation of succinic acid because NADH must be consumed for NAD+ regeneration to maintain the NADH/NAD+ ratio at a suitable level. These results indicated that NAD+-dependent glycerol dehydrogenase supplies additional NADH in C. glutamicum NC-3G by utilizing glycerol under O2 deprivation. For this reason, more succinic acid was produced to consume the NADH and maintain the NADH/NAD+ ratio in a balanced state.
Fed-batch fermentation for succinate production
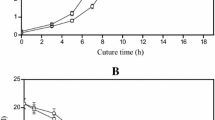
In order to evaluate the suitability of strain NC-3G as a host for the production of succinate, fed-batch fermentations were carried out. Since the growth of C. glutamicum was arrested and the resting cells retained the capability to metabolize carbon source (Litsanov et al. 2012; Okino et al. 2008), a high titer could be achieved by high cell-density fermentation. Thus, we performed a dual-phase fermentation: an aerobic growth phase in a 5 l bioreactor with 3 l medium followed by an anaerobic succinate production phase in a 1.5 l bioreactor. The anaerobic fermentation was carried out with initially 35.2 g dry cell/l. As described in Fig. 2, 37.6 g glycerol/l was consumed within 48 h and succinate increased continuously and reached 38.4 g/l with an average yield of 1.02 g/g. This represents 80 % of the maximum theoretical yield for glycerol under O2 deprivation. Related strains have produced 0.7 g succinate per g glucose = 72 % of the maximum theoretical yield (1.1 g succinate per g glucose). The succinate production rate (0.8 g/l.h) of NC-3G using glycerol was half that when using glucose (1.7 g/l.h) (Fig. 2). In addition to succinate, the cells excreted 2.4 g pyruvate/l, 3.9 g acetate/l as by-products. The glycerol consumption rate and the succinate productivity were decreased after the second feeding. This could be attributed to cell autolysis or osmotic stress (Litsanov et al. 2012).

Fed-batch fermentation for succinate production by NC-3G from glycerol (a) or glucose (b)
In a similar study, Litsanov et al. (2013) constructed a C. glutamicum strain, BL-1/pVWEx1-glpFKD, also containing a glycerol utilization pathway for aerobic succinate production. The strain formed 9.3 g succinate/l from 34.5 g glycerol/l, representing 42 % of the maximal theoretical yield under aerobic conditions. Our strain NC-3G was more robust in producing succinate with a higher succinate productivity and titer under O2 deprivation.
In conclusion, we describe for the first time the efficient use of glycerol as substrate for succinate production with metabolically-engineered C. glutamicum under O2 deprivation. The process shows two unique features: (i) the highest known volumetric productivity of all currently described microbial strains for anaerobic succinate production from glycerol and (ii) the first time constructing anaerobic glycerol utilization pathway in C. glutamicum. Taken together, glycerol supply extra reducing equivalents can favorably be converted to reduced product, thereby further increasing the value of the process.
References
Blankschien MD, Clomburg JM, Gonzalez R (2010) Metabolic engineering of Escherichia coli for the production of succinate from glycerol. Metab Eng 12:409–419
Glassner DA, Datta R (1992) Process for the production and purification of succinic acid. US Patent 5,143–834
Guettler MV, Jain MK, Rumler D (1996) Method for making succinic acid, bacterial variants for use in the process, and methods for obtaining variants. US Patent 5,573–931
Ikeda M, Nakagawa S (2003) The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol 62:99–109
Inui M, Murakami S, Okino S, Kawaguchi H, Vertès AA, Yukawa H (2004) Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under O2 deprivation conditions. J Mol Microbiol Biotechnol 7:182–196
Jakoby M, Ngouoto-Nkili CE, Burkovski A (1999) Construction and application of new Corynebacterium glutamicum vectors. Biotechnol Technol 13:437–441
Kalinowski J, Bathe B, Bartels D, Bischoff N et al (2003) The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol 104:5–25
Kirchner O, Tauch A (2003) Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J Biotechnol 104:287–299
Lee PC, Lee WG, Lee SY, Chang HN (2001) Succinic acid production with reduced by-product formation in the fermentation of Anaerobiospirillum succiniciproducens using glycerol as a carbon source. Biotechnol Bioeng 72:41–48
Lee PC, Lee SY, Hong SH, Chang HN (2002) Isolation and characterization of a new succinic acid-producing bacterium, Mannheimia succiniciproducens MBEL55E, from bovine rumen. Appl Microbiol Biotechnol 58:663–668
Litsanov B, Brocker M, Bott M (2012) Toward Homosuccinate Fermentation: metabolic engineering of Corynebacterium glutamicum for anaerobic production of succinate from glucose and formate. Appl Environ Microbiol 78:3325–3337
Litsanov B, Brocker M, Bott M (2013) Glycerol as a substrate for aerobic succinate production in minimal medium with Corynebacterium glutamicum. Microb Biotechnol 6:189–195
Molin M, Norbeck J, Blomberg A (2003) Dihydroxyacetone kinases in Saccharomyces cerevisiae are involved in detoxification of dihydroxyacetone. J Biol Chem 278(3):1415–1423
Okino S, Noburyu R, Suda M, Jojima T, Inui M, Yukawa H (2008) An efficient succinic acid production process in a metabolically engineered Corynebacterium glutamicum strain. Appl Microbiol Biotechnol 81:459–464
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73
Scholten E, Dagele D (2008) Succinic acid production by a newly isolated bacterium. Biotechnol Lett 30:2143–2146
Shams YS, Gonzalez R (2008) Engineering Escherichia coli for the efficient conversion of glycerol to ethanol and coproducts. Metab Eng 10:340–351
Vertès AA, Inui M, Yukawa H (2005) Manipulating corynebacteria, from individual genes to chromosomes. Appl Environ Microbiol 71:7633–7642
Wang C, Zhang HL, Cai H, Zhou ZH, Chen YL, Chen YL, Ouyang PK (2014a) Succinic acid production from corn cob hydrolysates by genetically engineered Corynebacterium glutamicum. Appl Biochem Biotechnol 172:340–350
Wang C, Cai H, Zhou ZH, Zhang K, Chen ZJ, Chen YL, Wan HG, Ouyang PK (2014b) Investigation of ptsG gene in response to xylose utilization in Corynebacterium glutamicum. J Ind Microbiol Biotechnol 41:1249–1258
Yazdani SS, Gonzalez R (2007) Anaerobic fermentation of glycerol: a path to economic viability for the biofuels industry. Curr Opin Biotechnol 18:213–219
Yukawa H, Omumasaba CA, Nonaka H, Kos P, Okai N, Suzuki N, Suda N, Tsuge Y, Watanabe J, Ikeda Y, Vertès AA, Inui M (2007) Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology 153:1042–1058
Zeikus JG, Jain MK, Elankovan P (1999) Biotechnology of succinic acid production and markets for derived industrial products. Appl Microbiol Biotechnol 51:545–552
Zhang X, Shanmuqam KT, Ingram LO (2010) Fermentation of glycerol to succinate by metabolically engineered strains of Escherichia coli. Appl Environ Microbiol 76:2397–2401
Acknowledgments
This work was supported by the 973 Program of China (Grant No. 2011CB707405).
Supporting information
Supplementary Table 1—Strains and plasmids used in this study.
Supplementary Table 2—Oligonucleotides used in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, C., Cai, H., Chen, Z. et al. Engineering a glycerol utilization pathway in Corynebacterium glutamicum for succinate production under O2 deprivation. Biotechnol Lett 38, 1791–1797 (2016). https://doi.org/10.1007/s10529-016-2166-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-016-2166-4