
Abstract
Objectives
To enhance the efficiency of influenza virosome-mediated gene delivery by engineering this virosome.
Results
A novel chimeric influenza virosome was constructed containing the glycoprotein of Vesicular stomatitis virus (VSV-G), along with its own hemagglutinin protein. To optimize the transfection efficiency of both chimeric and influenza cationic virosomes, HEK cells were transfected with plasmid DNA and virosomes and the transfection efficiency was assessed by FACS analysis. The chimeric virosome was significantly more efficient in mediating transfection for all amounts of DNA and virosomes compared to the influenza virosome.
Conclusions
Chimeric influenza virosome, including VSV-G, is superior to the conventional influenza virosome for gene delivery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
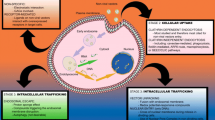
The influenza virosome is a membranous nanoparticle devoid of viral nucleocapsid and genetic material and has hemagglutinin (HA) glycoprotein on its surface. This acts as a bridge between viral and non-viral vectors. A virosome is similar to virus-like particles (VLP) and the applications of both are almost the same except that the former does not contain the capsid protein of the virus (Machida and Imataka 2015). This system is ideal for delivery of biological molecules to cells and, so far, it has been used extensively for this purpose. It has been used to deliver pDNA attached to its cationic surface (Schoen et al. 1999) and encapsulated in its lumen (de Jonge et al. 2007) to cells in vitro. In other studies, the influenza virosome was used to deliver siRNA (de Jonge et al. 2006a) to cells. Furthermore, influenza virosome is a promising tool to deliver DNA vaccines to antigen-presenting cells (APCs) more efficiently (Irvine et al. 2013). Having such wide array of applications, it is worthwhile to manipulate the influenza virosome in order to confer higher transfection efficiency.
VSV-G is the glycoprotein of Vesicular stomatitis virus (VSV). It binds to negatively-charged phospholipids (Carneiro et al. 2002) and, after binding, induces receptor-mediated endocytosis. VSV-G has been used routinely to pseudotype various viruses that are of use in gene therapy to heighten their transduction efficiency (Park et al. 2014; Gurusinghe et al. 2015). Expansion of host range and elevation of transduction efficiency were observed when retroviruses were pseudotyped by VSV-G (Mizuarai et al. 2001).
In this study, with the aim of improving the transfection efficiency of the influenza virosome, we constructed chimeric influenza virosome nanoparticles containing VSV-G by solubilizing influenza and VSV envelope using 1,2-dicaproyl-sn-glycero-3-phosphocholine (DCPC) and removing DCPC via dialysis. (DCPC is a detergent-like agent that is much less aggressive than most other detergents.) We characterized the influenza and chimeric virosomes by SDS-PAGE and conducted a fusion assay to determine that our virosomes are fusogenic, and that both HA and VSV-G proteins exist on the same membrane in our chimeric virosome. We also compared the transfection efficiency of our influenza and chimeric virosome and then optimized the transfection conditions concerning the proper amount of DNA and virosome to be used for transfection.
Materials and methods
Cell culture and virus preparation
HEK cells and influenza virus (H1N1) were from Influenza Research Lab (Pasteur Institute, Tehran, Iran) and VSV virus was obtained from Rabies Unit, Pasteur Institute of Iran. Cells were grown in Dulbecco’s minimum essential medium (DMEM) + 10 % (v/v) calf serum with antibiotics (100 IU penicillin ml−1 and 100 μg streptomycin ml−1), incubated at 37 °C + 5 % CO2. Virus propagation and purification were performed as described elsewhere.
Virosome preparation
Two sets of virosomes [(a) influenza cationic virosome and (b) chimeric (VSV + Influenza) cationic virosome] were prepared based on the method of de Jonge et al. (2006b). Viruses [(a) 1.5 μmol membrane phospholipids of influenza virus and (b) 1.5 μmol 50 % (w/w) VSV + influenza virus preparation] were sedimented (a discontinuous sucrose gradient of 10/60 %(w/v) sucrose in HEPES-buffered saline (HBS), 100,000×g for 1 h at 4 °C) and the resuspended pellets (pellet + 375 μl HBS) were dissolved in 100 mM 1,2-dicaproyl-sn-glycero-3-phosphocholine (DCPC) [375 μl DCPC stock (200 mM) + 375 μl of each suspension]. This mixture was held on ice for 30 min. After sedimentation of the nucleocapsid (100,000×g for 30 min at 4 °C), the supernatants (lipid + protein) were added to 1,2-dioleoyloxy-3-trimethylammonium propane chloride (DOTAP) preparation and held on ice for 30 min.
We prepared DOTAP as follows: 0.808 µmol DOTAP (35 % total phospholipids) was dissolved in chloroform, treated with a stream of N2 and kept under vacuum for 2–4 h. Removing DCPC by dialysis resulted in reconstitution of crude virosomes. This was done in Slide-A-Lyzer cassettes against 2 l HBS at 4 °C overnight and new HBS for another 4 h. Non-incorporated material from the reconstituted virus membranes was removed by ultracentrifugation [100/60 %(w/v) discontinuous sucrose gradient, 1.5 h, 100,000×g at 4 °C] and purified virosomes were dialyzed against 2 l HBS overnight at 4 °C to remove the sucrose. Samples from pellets and supernatants and viruses were subjected to SDS-PAGE electrophoresis on 12 % (w/v) gels. Protein was determined by the Lowry method.
Sandwich ELISA
Sandwich ELISA was employed alongside with SDS-PAGE to confirm the simultaneous presence of HA and VSV-G proteins on the surface of chimeric virosome. A 96-well ELISA plate was coated with 100 μl anti-HA monoclonal antibody (Abcam) as the capture antibody. After overnight incubation at 4 °C, the plate was washed three times with phosphate-buffered saline (PBS) containing 0.05 % Tween 20 (PBST). 150 μl blocking solution [5 % (v/v) skim milk in PBS] was added for 1 h at 37 °C and the plate was again washed four times with PBST. 100 μl 1:5000 diluted chimeric virosome was added in triplicates. After incubation at 37 °C for 90 min, the wells were washed three times in PBST. 100 μl 1:5000 diluted anti-VSV-G antibody conjugated with horseradish-peroxidase (Sigma–Aldrich) was added to each well as the detection antibody. After the plate was incubated for 1 h at 37 °C, it was washed three times. 100 µl substrate solution (3,3′,5,5′-tetramethylbenzidine, TMB) was added for 15 min, then the reaction was stopped by addition of 2 M H2SO4, and the absorbancy (A) values were measured at 450 nm for each well. The cut-off values were measured using mean value of negative control plus three times the standard deviation.
Nanoparticle size distribution analysis
Particle size distribution of virosome nanoparticles was determined by dynamic light scattering (DLS) using Zetasizer Nano-ZS90 (Malvern Instruments). Virosome samples were diluted 1:6 (v/v) in PBS and 1 ml from each was analyzed at 25 °C.
Fluorescent labeling
Virosomes were labeled by incubation with Octadecyl Rhodamine B (R18) fluorescent probe (1 h, rotary shaker, in the dark). The amount of R18 was calculated to be about 2–10 % of the total number of lipids in virosomes. Unincorporated R18 was removed by centrifugation on 1 ml Sephadex G75 columns.
Fusion assay
Fusion assay procedure on HEK cell line was adapted from Paternostre et al. (1989). To measure the kinetics of fusion in low pH, R18-Virosomes (10 μg protein) were injected into 1 ml × 106 HEK cells in HBS buffer (pH 7.4) in separate assays and incubated for 1 h in 4 °C. Unbound particles were removed by centrifugation at 300×g. Pellets were resuspended in HBS and kept on ice. Then 30 μl was pipetted into 800 μl pre-warmed HBS (37 °C, pH 7.4). Four equal aliquots were pipetted into wells of a 96-well flat-bottom microtiter plate. The pH was adjusted to 5.5 with citric acid, and the change of fluorescence was recorded with a Synergy4 BioTeK microplate fluorescence reader (excitation/emission 560/590 nm). The following equation was used for calculating the percentage of fusion:
where F0 and Ft are fluorescence at the time zero and fluorescence at a given time, respectively. FT is the total fluorescence of disrupted membranes as the result of adding sodium dodecyl sulfate (SDS).
In another assay, R18-labeled chimeric and influenza virosomes were pre-treated at pH 5.5 for 20 min. The pH was adjusted to neutral and interfering aggregates were removed (600×g, 3 min) and the supernatant was kept at neutral pH for 2 h to allow the VSV-G proteins on the surface of the chimeric virosomes to return to their natural pre-fusion conformation. The fusion assay was then conducted as mentioned above.
Another fusion assay was performed in which cells were incubated with 40 mM NH4Cl for 20 min. Subsequently, R18-labeled chimeric virosomes were directly added to cells at neutral pH, and the change in fluorescence was recorded for 1 h.
Transfection
pEGFP-N1 plasmids in Escherichia coli DH5a were extracted by AccuPrep Nano-Plus Plasmid Mini Extraction Kit (Bioneer) based on manufacturer’s instructions. HEK cells [105 per well in a 24-well plate in Dulbecco’s minimum essential medium (DMEM) + 10 % (v/v) fetal bovine serum] were kept in 37 °C + 5 % (v/v) CO2 for 18–24 h. To find the optimum amount of DNA, 1, 2.5, 5 and 10 μg were mixed with 10 μg each virosome (method described as follows). 5 μg DNA was chosen as the best amount of DNA for transfection. Subsequently, 5 μg DNA with 5, 10, 20 and 40 μg virosomes were used in different assays for comparison. Before each transfection assay, the certain amount of plasmid and cationic virosome were incubated for 30 min in HEPES-buffered saline buffer. Then, the medium was removed from the cells and mixtures of plasmid/virosome were added to separate wells with serum-free medium. After 4 h, the medium was removed and replaced by 1 ml DMEM + 5 % (v/v) serum. Each well was observed by fluorescent microscopy after 24 h. Subsequently, cells were washed with fresh medium, pipetted and analyzed for enhanced green fluorescent protein (EGFP) expression by flow cytometry with Partec Particle Analyzing System (PAS).
Results and discussion
Figures 1 and 2 show the protein content of various steps in virosome preparation.

Gel electrophoresis of influenza virus and different preparation stages of influenza virosome. a Ladder, b intact influenza virus, c pellet, d purified influenza virosome. HA1, HA2, HA0 and NA membrane proteins are dominant in the virosome

Gel electrophoresis of VSV and influenza virus and different stages of chimeric virosome preparation. a Ladder, b intact influenza virus, c intact VSV virus, d pellet of chimeric preparation, e purified chimeric virosome. The protein content of the purified virosome is dominated by VSV-G and hemagglutinin membrane proteins. The pellet lane shows that the bulk of M1 proteins of influenza and P proteins of VSV virus were sedimented during ultracentrifugation
The result of the sandwich ELISA is depicted in Fig. 3.

Double-antibody sandwich ELISA. The cut-off value was measured to be 0.42 while the OD value for chimeric virosome was 1.92. The result provides conclusive evidence that both hemagglutinin and VSV-G proteins are present in our chimeric virosome preparation and also they exist on the same membrane simultaneously
Particle size distribution
The chimeric virosome mean size was 106 (±3.6) nm with a polydispersity index (PDI) of 0.132 (±0.023) and the influenza virosome mean size was 98.5 (±2.4) nm with PDI of 0.143 (±0.015). All the numbers are mean ± SD of three independent measurements.
Fusion assay
For the virosomes to be used as a DNA delivery system, the surface glycoproteins must remain functional after reconstitution of the membrane. Labeled R18 was incorporated in the membrane of the virosomes. An increase in fluorescence emission indicates the fusion of the two membranes and, in turn, confirms the functionality of the surface proteins. The fluorescence increase was observed for both influenza and chimeric virosomes after lowering the pH to 5.5, indicating that the HA and VSV-G proteins remained fusogenic (Fig. 4).

Results of the fusion assays. Chimeric (filled circle) and influenza (filled square) virosomes fused with cell membranes immediately as pH was lowered to 5.5, indicating that hemagglutinin and VSV-G proteins on their surfaces were fusogenic and functional. Chimeric virosome exposed to NH4Cl pre-treated cells (filled triangle) did not fuse with cells even after 1 h (only 5 min is shown in the figure), implying that chimeric virosome enters the cell via endocytosis and depends on endosomal acidification to fuse with endosomal membrane. Chimeric virosome pre-incubated in low pH followed by 2 h incubation in neutral pH (empty circle), fused with cells when the pH was lowered to 5.5 and reached the final extent of fusion like the untreated chimeric virosome, but in a much longer time; providing a compelling evidence that both hemagglutinin and VSV-G exist on the same membrane on the chimeric virosome. The influenza virosome pre-incubated in low pH (×) did not fuse with cells
To infer the entrance route of the virosomes, we preincubated the cells in 40 mM NH4Cl before conducting the fusion assay. NH4Cl inhibits endosomal acidification (Ohkuma and Poole 1978). After exposing chimeric virosomes to cells incubated with NH4Cl, no fluorescence increase was observed after 1 h (Fig. 4). Since HA and VSV-G perform the fusion in acidic environments and NH4Cl blocks the acidification of endosomes, we can conclude that chimeric virosome induces receptor-mediated endocytosis, and after maturation of the endosome and declining of pH, chimeric virosome performs fusion with the envelope of an endosome.
Puri et al. (1990) showed that HA protein undergoes an irreversible conformational change in acidic environment and becomes dysfunctional. However, we know that the conformational change induced in VSV-G by low pH is reversible (Gaudin et al. 1991). We used these properties of HA and VSV-G to understand whether both proteins exist in the same membrane. After incubation of chimeric and influenza virosomes in low pH and returning the pH back to neutral for 2 h, we conducted the fusion assay as described. For the influenza virosome, as expected, no fusion was observed. For the chimeric virosome the fusion occurred, and the final extent of the fusion reached the same amount as chimeric virosome that was not pre-incubated at low pH, but in a longer time (Fig. 4). Arguably this is evidence that both proteins are in the same membrane simultaneously. Because if they were on different membranes and our chimeric virosome was comprised of two separate populations of HA and VSV-G virosomes, the final fusion result would not reach the final degree we observed in our experiment, since in such a condition, half of the virosomes would be incapable of performing the fusion.
Optimization of cationic virosome-mediated gene delivery
To optimize the conditions of virosome-mediated transfection, we mixed 10 µg of both cationic influenza and chimeric virosomes with four different amounts of pEGFP-N1 plasmid (1, 2.5, 5 and 10 µg) and ran the transfection assay as described in Materials and methods. 5 µg plasmid yielded the highest efficiency for both type of virosomes (Fig. 5); hence, we chose it to proceed with our optimization procedure. For all amounts of the plasmid used in our experiment, chimeric virosome exhibited much higher transfection efficiency.

Effect of DNA concentration on virosome-mediated transfection. 10 µg chimeric (filled circle) and influenza (filled square) cationic virosomes were incubated with four different amounts of pEGFP-N1 plasmid. Transfection efficiency increased up to 5 µg plasmid DNA, after that it decreased slightly for both virosomes. All values are means (±SD) of three independent measurements, each ran in triplicate
For both types of virosomes, we incubated them at 5, 10, 20 and 40 µg with 5 µg plasmid and conducted the transfection assay as described. The transfection efficiency increased with the amount of virosomes and, for each amount, the chimeric virosome demonstrated significantly higher transfection efficiency than the influenza virosome (Student’s t test was used to compare the significance of the results) (Fig. 6).

Effect of virosome envelope protein concentration on transfection efficiency. a 5 µg pEGFP-N1 plasmid was incubated with 4 different amounts of both chimeric (black bar) and influenza (grey bar) cationic virosomes. Transfection efficiency increased with each higher amount of virosome protein. All values are the mean (±SD) of three independent experiments, each ran in triplicate. Statistical analysis was by the Student’s t test: *, ** P < 0.01 versus value of transfection by the influenza virosome for each concentration. b % increase of transfection with chimeric virosome compared to the influenza virosome for each concentration
As indicated in Fig. 6b, the ratio of transfection efficiency of chimeric virosome to conventional virosome was the highest when 10 μg virosomes were used. Figure 7 shows the FACS results of transfection efficacy for both virosomes using 10 μg of each.

Fluorescence-activated cell sorting (FACS) analysis of virosome-mediated DNA transfection. a HEK cells transfected with PBS (Negative control). Cells were gated and the threshold of fluorescence intensity for positive answer was set according to negative control. b, c transfection of 5 μg DNA with 10 μg influenza virosome (b) and chimeric virosome (c)
By optimizing the conditions of transfection, using 5 µg plasmid and 40 µg of each virosome, we reached up to 75 % of transfection efficiency for chimeric virosome and 61 % for influenza virosome. The values of transfection efficiencies for all amounts of virosomes were significantly higher for the cationic chimeric virosome.
We have two possible explanations for the higher transfection efficacy of chimeric virosome compared to conventional influenza virosome. One is that VSV-G has a ubiquitous receptor on cell membranes. Non-specific electrostatic and hydrophobic interactions and, in general, negatively-charged phospholipids (Carneiro et al. 2002) are involved in binding of VSV-G to membranes. Since the receptor of VSV-G is so widely distributed on biological membranes, the existence of VSV-G along with HA in the chimeric virosome substantially increases the potential binding sites of the virosome, while for the influenza virosome the only receptor is sialic acid.
Another possible explanation for the higher transfection efficiency is that endosomal release is improved by VSV-G. VSV-G is an extremely fusogenic protein at low pH. Therefore, it might aid HA to perform endosomal escape more quickly. One study attributed the enhancement of gene transfer to augmented endosomal release contributed by VSV-G (Barsoum et al. 1997). In that study, a gene was transferred to cells once by a VSV-G pseudotyped baculovirus and once with the wild-type baculovirus. The bulk of the genes transferred by pseudotyped virus ended up in a condensed form in the nucleus but, in the case of wild-type baculovirus, they mostly remained in the endosomes. This is because VSV-G in the pseudotyped virus induced membrane fusion more efficiently than the native baculovirus glycoprotein, gp64.
In conclusion, chimeric virosome nanoparticle are more efficient in gene delivery than the conventional influenza virosome. Another advantage of this chimeric virosome is that, unlike the influenza virosome, it is resilient to acidic pH which increases its stability in various storage conditions. This study sets the stage for further development of this novel delivery system. More studies are required to investigate the potential of this chimeric virosome to perform in vivo gene delivery and to be used as a vaccine adjuvant carrier.
References
Barsoum J, Brown R, McKee M, Boyce FM (1997) Efficient transduction of mammalian cells by a recombinant baculovirus having the vesicular stomatitis virus G glycoprotein. Hum Gene Ther 8:2011–2018
Carneiro FA, Bianconi ML, Weissmüller G et al (2002) Membrane recognition by vesicular stomatitis virus involves enthalpy-driven protein-lipid interactions. J Virol 76:3756–3764
De Jonge J, Holtrop M, Wilschut J, Huckriede A (2006a) Reconstituted influenza virus envelopes as an efficient carrier system for cellular delivery of small-interfering RNAs. Gene Ther 13:400–411
De Jonge J, Schoen P, terVeer W et al (2006b) Use of a dialyzable short-chain phospholipid for efficient solubilization and reconstitution of influenza virus envelopes. Biochim Biophys Acta 1758:527–536
De Jonge J, Leenhouts JM, Holtrop M et al (2007) Cellular gene transfer mediated by influenza virosomes with encapsulated plasmid DNA. Biochem J 405:41–49
Gaudin Y, Tuffereau C, Segretain D et al (1991) Reversible conformational changes and fusion activity of rabies virus glycoprotein. J Virol 65:4853–4859
Gurusinghe S, Young P, Michelsen J, Strappe P (2015) Suppression of dedifferentiation and hypertrophy in canine chondrocytes through lentiviral vector expression of Sox9 and induced pluripotency stem cell factors. Biotechnol Lett 37:1495–1504
Irvine DJ, Swartz MA, Szeto GL (2013) Engineering synthetic vaccines using cues from natural immunity. Nat Mater 12:978–990
Machida K, Imataka H (2015) Production methods for viral particles. Biotechnol Lett 37:753–760
Mizuarai S, Ono K, Yamaguchi K et al (2001) Production of transgenic quails with high frequency of germ-line transmission using VSV-G pseudotyped retroviral vector. Biochem Biophys Res Commun 286:456–463
Ohkuma S, Poole B (1978) Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci USA 75:3327–3331
Park S, Pyo C-W, Choi S-Y (2014) High-efficiency lentiviral transduction of primary human CD34+ hematopoietic cells with low-dose viral inocula. Biotechnol Lett 37:1–8
Paternostre MT, Lowy RJ, Blumenthal R (1989) pH-dependent fusion of reconstituted vesicular stomatitis virus envelopes with vero cells: measurement by dequenching of fluorescence. FEBS Lett 243:251–258
Puri A, Booy FP, Doms RW et al (1990) Conformational changes and fusion activity of influenza virus hemagglutinin of the H2 and H3 subtypes: effects of acid pretreatment. J Virol 64:3824–3832
Schoen P, Chonn A, Cullis PR et al (1999) Gene transfer mediated by fusion protein hemagglutinin reconstituted in cationic lipid vesicles. Gene Ther 6:823–832
Acknowledgment
This work was supported by a grant from Pasteur Institute, Tehran, Iran.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that there are no conflicts of interest
Additional information
Yahya Mohammadzadeh and Narges Rasouli are equal first authors.
Rights and permissions
About this article

Cite this article
Mohammadzadeh, Y., Rasouli, N., Aref, M.H.S. et al. A novel chimeric influenza virosome containing Vesicular stomatitis G protein as a more efficient gene delivery system. Biotechnol Lett 38, 1321–1329 (2016). https://doi.org/10.1007/s10529-016-2108-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-016-2108-1