
Abstract
Objectives
To develop a new vector for constitutive expression in Pichia pastoris based on the endogenous glycolytic PGK1 promoter.
Results
P. pastoris plasmids bearing at least 415 bp of PGK1 promoter sequences can be used to drive plasmid integration by addition at this locus without affecting cell growth. Based on this result, a new P. pastoris integrative vector, pPICK2, was constructed bearing some features that facilitate protein production in this yeast: a ~620 bp PGK1 promoter fragment with three options of restriction sites for plasmid linearization prior to yeast transformation: a codon-optimized α-factor secretion signal, a new polylinker, and the kan marker for vector propagation in bacteria and selection of yeast transformants.
Conclusions
A new constitutive vector for P. pastoris represents an alternative platform for recombinant protein production and metabolic engineering purposes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
High-level production of heterologous proteins using biological expression platforms is widely used for both academic and commercial purposes. One of the most widely used microorganisms for this purpose is the methylotrophic yeast, Pichia pastoris, with more than 600 heterologous proteins having been expressed (Zhang et al. 2009). Most expression vectors developed for this system are based on the regulated promoter of the alcohol oxidase 1 gene (P AOX1 ) that requires methanol for full induction (Cereghino et al. 2001). However, methanol causes safety concerns and during the fermentation process it may become toxic if O2 levels in the medium are not precisely controlled (Macauley-Patrick et al. 2005). Alternatively, constitutive expression may be employed with the advantage that a greater range of carbon sources may be used. Furthermore, the use of constitutive promoters is particularly attractive for the production of proteins on a large scale because it does not require the addition of inducers nor medium exchange thus allowing protein purification under continuous fermentation conditions.
The glycolytic pathway genes are highly expressed and their encoded enzymes may represent up to 30–40 % of total cellular soluble proteins (Fraenkel 1982). Because of this feature, these genes have been widely studied for the isolation of strong constitutive promoters in order to develop new expression vectors. Although few constitutive promoters have been described in P. pastoris the promoter of glycolytic glyceraldehyde-3-phosphate dehydrogenase gene (P GAP ) has been widely used for the production of several heterologous proteins (Zhang et al. 2009).
The P. pastoris 3-phosphoglycerate kinase gene (PGK1) has been isolated and its ~2 kb constitutive promoter (P PGK1 ) described (Almeida et al. 2005). Although not as strong as the more commonly used P GAP, P PGK1 should be considered for other uses such as in the development of new engineered pathways in Pichia or when hyperexpression is not desired. The objective of this study was to develop an expression vector based on a reduced version of P PGK1 and to study the effect on cell growth and heterologous protein production after vector integration at this locus.
Materials and methods
Strains and cultivation conditions
Escherichia coli DH5α (Gibco BRL) and TOP10 (Invitrogen) strains were routinely used for DNA manipulations. P. pastoris GS115 (his4) (Invitrogen) was used as a host for heterologous expression. Bacteria were grown in lysogeny broth (LB) with appropriate antibiotic (25 µg zeocin ml−1 or 100 µg ampicillin ml−1). The yeast was grown at 28 °C either on YPD (2 % peptone, 1 % yeast extract, 2 % glucose) or MDH (1.34 % YNB, 4 × 10−5 % biotin, 100 mM phosphate buffer, pH 6.0 and 2 % glucose). Screening for amylolytic clones was performed on plates containing MDHA (MDH supplemented with 1.5 % w/v solubilized starch). After 2 days growth plates were stained with I2 vapor.
DNA manipulation and DNA synthesis
Restriction enzymes, Klenow DNA polymerase, and T4 DNA ligase were used according to the manufacturers’ guidelines. DNA fragments were isolated and purified using the QIAquick Gel Extraction kit (Qiagen). DNA fragment used to remove restriction sites within the kan gene was chemically synthesized by Integrated DNA Technologies.
Serial deletion of P PGK1
Plasmid pPGKAMYB (de Almeida et al. 2005) was used as a backbone for analysis of promoter deletion and heterologous expression. Promoter deletions were accomplished either by double digestion of pPGKAMYB with specific restriction enzymes or by PCR. Promoter fragment P PGKΔ1 was generated after a BglII-KpnI double digestion of pPGKAMYB followed by Klenow treatment and vector re-ligation with T4 DNA ligase. The resulting plasmid was named pPGKΔ1AMY. The P PGKΔ3 deletion was obtained likewise after a BglII-PvuI double digestion of pPGKAMYB generating plasmid pPGKΔ3AMY. P PGKΔ2 was obtained by PCR amplification with primers PGK-D8 (5´-AGATCTCGAGCAAGTGTCCTATGCTG, BglII site underlined) and PGK-RZ (5′- GTTCGAATTTCGTAATCAATTGGGCTATG, BstBI site underlined). PCR was carried out with Taq DNA polymerase (Invitrogen) using pPGKAMYB as template and the following program: 30 cycles of 94 °C/30 s; 55 °C/15 s; 72 °C/30 s followed by a final extension of 72 °C/5 min. The ~0.73 kb amplicon was cloned into pGEM-T (Promega) and then used to replace P PGK1 after digestion of pPGKAMYB with BglII and BstBI, creating plasmid pPGKΔ2AMY. Plasmid pPZα is pPGKAMYB without the α-amylase gene.
P. Pastoris transformation
All vectors were linearized with SacI and introduced into P. pastoris GS115 following a standard electroporation protocol (“Pichia Expression Kit”—Invitrogen).
Southern blot
Total yeast DNA was extracted according to the protocol described on the Invitrogen. Approximately 10 μg yeast total DNA were digested with either BglII or EcoRI prior to transfer to nitrocellulose membrane. A 415-bp BglII-PvuI fragment derived from P PGK1 present in pPGKAMYB was used as probe. Probe labeling, pre-hybridization, washing and detection were performed according to AlkPhos Direct Kit manual (Amershan Bioscience). For the generation of the chemiluminescent signal, the CDP-Star reagent (Amersham Biosciences) was used.
Expression of α-amylase
A single yeast colony was inoculated in 25 ml YPD in a 250 ml Erlenmeyer flask and grown for 24 h at 28 °C at 200 rpm. These cells were inoculated in 100 ml YPD in a 1 l baffled Erlenmeyer flask to reach OD600 of 0.4 after 14 h when aliquots of 2 ml were removed every 2 h for detection of amylase activity, glucose consumption and growth measurements. Glucose analysis was performed by HPLC using an Aminex HPX-87H column (300 × 7.8 mm-Bio-Rad) coupled to a refractive index detector. Water was used as the mobile phase at 0.6 ml min−1. The column was set to 80 °C. A standard curve for glucose was used for sample quantification. Growth tests were performed in triplicate.
Protein analysis in denaturing polyacrylamide gel (SDS-PAGE)
A single yeast colony was inoculated in 20 ml YPD in a 125 ml Erlenmeyer flask and grown for 24 h at 200 rpm. Cells were then inoculated in 100 ml YPD in a 1 l Erlenmeyer flask to an initial OD600 of 0.03. The culture was grown for up to 96 and, at 12 h intervals, aliquots of 3 ml were removed and reserved. After determination of culture growth (OD600), samples were centrifuged at 5000×g for 5 min. Cell pellet was discarded and 1 ml supernatant was precipitated with TCA and resuspended in 20 µL SDS-PAGE sample buffer with or without β-mercaptoethanol. Protein electrophoresis was performed in denaturing 12.5 % (w/v) polyacrylamide gel.
Determination of amylase activity
Enzyme activity was measured according to the method of Fuwa (1954). Starch solution was prepared by adding 0.5 g soluble starch (Merck) to 100 ml cold water, followed by heating until solubilization. The solution was kept at 40 °C prior to use. Iodine reagent was prepared by mixing 1 vol 0.5 % I2, 1 vol 5 % (w/v) KI and 3 vol distilled water. Assay reactions containing 10–60 µl culture supernatant, 40 µl 0.5 M sodium acetate buffer (pH 6.0), 100 µl starch solution and water to make 200 µl, were incubated at 40 °C for 10 min. The reaction was stopped by adding 200 µl 1 M acetic acid, 200 µl iodine reagent and 4.4 ml distilled water followed by measurement at 660 nm. One unit of a-amylase activity was defined as the quantity of enzyme necessary to hydrolyze 0.1 mg starch per minute at 40 °C.
Deglycosylation test and zymogram
Samples used for deglycosylation test and zymogram analysis were derived from a 48 h-culture supernatant which was collected, dialyzed, lyophilized, dissolved in 50 μl 0.5 M sodium acetate buffer (pH 5.5) and stored at −20 °C prior to use. Deglycosylation was performed with 5 μl sample incubated with PNGase F (peptide-N-glycosidase F, Sigma-Aldrich). A zymogram was carried out with 1 μl supernatant sample resolved on SDS-PAGE under native conditions. After electrophoresis, the gel was incubated in a 50 mM sodium acetate buffer (pH 5.5) for 1 h and then transferred to a 0.5 % starch solution dissolved in the same buffer following incubation for 12 h at 4 °C. Subsequently, the gel was incubated at 37 °C for 4 h and developed in iodine reacent to visualize starch degradation.
Construction of expression vector pPICK2
To construct the expression vector based on P PGK1 , we used as a platform pPICKα, a plasmid in which the Sh ble marker present in pPICZαA was replaced by kan (Reis et al. 2012). The promoter fragment used in the construction of the expression vector was named P PGKΔ4 , and was obtained by PCR using primers PGK-D10 (5′-CAGATCTGCGAGGCAAGCATCTACT, BglII site underlined) and PGK-RZ. A new S. cerevisiae α-factor signal was chemically synthesized with codon adaptation for P. pastoris. This 276 pb secretion signal was amplified with primers 5PROPP-F (5′-CTTCGAAACGATGAGATTCCCATC, BstBI site underline) and 3PROPP-R (5′-GTCGACGCGGCCGCACTAGTCCCGGGGAATTCGGCTTCAGCTTCTCTCTTCTC) which has a polylinker (EcoRI-SmaI-SpeI-NotI-SalI) at its 5´-end (underlined). The two amplicons (promoter and signal sequence) were digested with BstBI, ligated, and re-amplified with Phusion DNA polymerase (Thermo Scientific) using primers PGK-D10 and 3PROPP-R following manufacturer’s instructions. The resulting ~930-pb amplicon was cloned into pPCV-B (Janner et al. 2013). After double digestion with BglII-SalI, the insert was subcloned into pPICKα digested with the same enzymes to replace P AOX1 and the original α-factor secretion sequence. The resulting vector was named pPICKα-PRO. To remove the PvuI and SmaI sites present in kan, a 270-pb DNA fragment bearing silent point mutations in these sites was chemically synthesized (Supplementary Fig. 1) and subcloned into pPICKα-PRO digested with NsiI. The resulting vector was named pPICK2. The sequences of the promoter, optimized α-factor signal and polylinker present on pPICK2 are shown on Supplementary Fig. 2.

Deletion analysis of P PGK1 . a Physical map of pPGKAMYB b schematic representation of the fragments generated after P PGK1 deletions c α-amylase activity halos detected on starch plates seeded with the following yeast clones: (1) clone Zα (negative control), (2) clone AMYB, (3) clone Δ1, (4) clone Δ3

Southern blotting analysis of genomic DNA extracted from different P. pastoris clones. Total DNA was either digested with BglII (a) or EcoRI (b) and probed with a 415-bp fragment derived from P PGK1 c schematic representation of the PGK1 locus after integration of different plasmids. P. pastoris GS115 DNA was used as a control. The expected sizes of the fragments generated after genomic DNA digestion with EcoRI (E) or BglII (b) are shown. The annealing position of the probe is shown as a horizontal bar
Results and discussion
Deletion analysis of P PGK1
In order to determine the smallest functional fragment of the PGK1 promoter, we used plasmid pPGKAMYB (Fig. 1a) to create three other plasmids bearing different promoter fragments (Fig. 1b) which were placed upstream of amyE, a truncated version of the B. subtilis α-amylase gene. The resulting vectors were used to transform P. pastoris GS115 after linearization with SacI in order to direct integration into the PGK1 locus. Selected transformants were plated on starch plates and the resulting hydrolysis halos visualized after iodine staining were of similar sizes (Fig. 1c). This result shows that a reduced promoter size of ~410 bp (P PGKΔ3 ) was long enough to promote vector integration into the P. pastoris genome. Similarly, P GAP , which is present in the commercial pGAPZ system developed by Invitrogen for constitutive expression in P. pastoris, only has 483 bp and this system has been used efficiently to produce several heterologous proteins (Zhang et al. 2009). A small promoter length has the added advantage of facilitating the construction of PCR-derived mutant promoter libraries for fine tune expression in yeast (Qin et al. 2011). Stadlmayr et al. (2010) compared 26 different P. pastoris promoters using gene expression profile data and heterologous protein production under different conditions. Microarray data analysis placed PGK1 in a set of 15 genes that are highly expressed in P. pastoris. However, a comparison of promoter activity based on heterologous protein production showed that P PGK1 is weaker than constitutive P GAP and P TEF1. This study also pointed out that promoter strength was directly influenced by different experimental factors such as media composition and cell growth phase, and in most cases there is a correlation between gene dosage and protein production. It has been argued that promoter selection should be based on the products and characteristics of the fermentation process (Çalık et al. 2015). For metabolic engineering purposes it is thus important to have a repertoire of different promoters with various strengths.
In the present work, only transformants derived from pPGKAMYB (clone AMYB; ~2 kb promoter), pPGKΔ3AMY (clone Δ3; 0.41 kb promoter) and pPZα (clone Zα; ~2.0 kb promoter without amyE) were further analyzed. Southern blotting analysis was carried out to confirm the site of plasmid integration into the P. pastoris chromosome. Based on the predicted sizes of the fragments obtained after genomic DNA digestion with either BglII (Fig. 2a) or EcoRI (Fig. 2b) we were able to confirm that integration occurred by homologous recombination at the PGK1 locus. All constructs presented only one integrated copy with the exception of clone Zα. The observation of three fragments in Fig. 2a and the presence of a stronger signal corresponding to the ~1.9 kb fragment in Fig. 2b suggest that clone Zα has two in tandem integrated copies of the plasmid as depicted in Fig. 2c.
Cell growth and heterologous protein production
The physiology of clones AMYB, Δ3 (both have 1 integrated copy of the expression cassette) and Zα (negative control) was analyzed in shake flasks under conditions for constitutive α-amylase expression. Enzyme activity was expected to be similar in both clones AMYB and Δ3 since P PGK1 controlling amyE expression was reconstituted upon plasmid integration (see Fig. 2c). On the other hand, the size of the endogenous P PGK1 is reduced to ~0.41 kb in clone Δ3, and this could impact cell physiology since PGK1 is considered an essential gene. Also, constitutive production of α-amylase could impair cell growth. Despite concerns involving possible cytotoxic effects due to constitutive production of heterologous proteins, Menéndez et al. (2004) showed that the levels of exo-levanase production in P. pastoris were similar when either P GAP or P AOX1 were used. As shown in Fig. 3a, all three clones presented similar growth profiles (μ = 0.34 h−1) and glucose consumption (Fig. 3b). Also, as expected, α-amylase activity was similar in clones AMYB and Δ3 (Fig. 3c) and reached ~43,000 U l−1 after 40 h flask cultivation. Yang et al. (2010) expressed a thermostable, raw starch-digesting α-amylase from Thermobifida fusca using the constitutive GAP promoter and obtained 510 U l−1 after 72 h cultivation in a flask. Liu et al. (2012) also using P GAP obtained about 125 mg barley α-amylase l−1 (510 U mg−1) after 54 h cultivation in a 50 l bioreactor under fed-batch conditions. In our work, we obtained higher enzyme activities without any additives such as YNB, biotin or glucose feeding. Also, unlike other works in which the α-amylase gene was codon-optimized for expression Pichia, we used the native B. subtilis amyE gene sequence without any codon adaptation.

Cell growth and amylolytic activity of P. pastoris clones transformed with different constructions. The assay was performed using 100 ml YPD medium in a 1 l baffled Erlenmeyer flask. The culture was incubated in an orbital shaker at 28 °C with an agitation of 200 rpm for up to 40 h. a Cell growth b glucose consumption c α-amylase activity
The analysis of culture supernatant revealed an induced protein species with an apparent molecular mass of ~60 kDa which was not visible in the control supernatant (Fig. 4a). The size of this secreted protein was bigger than the theoretical molecular mass expected for the B. subtilis α-amylase (~54 kDa). This could be due to glycosylation, a post-translational modification that may occur in secreted proteins (Daly and Hearn 2005). To test this a supernatant sample from clone Δ3 was treated with PNGase F, which is generally used to identify N-linked glycosylation. After the deglycosylation reaction, the size of the induced protein was reduced to a molecular mass consistent with that of AmyE (Fig. 4b, lane 2). To demonstrate that the protein that had shifted in size corresponded to an α-amylase we performed a zymogram analysis which confirmed the presence of an glycosylated amylase in culture supernatant from clone Δ3 (Fig. 4c).

Analysis of α-amylase produced by clone Δ3. a Time course analysis of total protein produced by clone Δ3. The supernatant from clone Δ3 grown for 12, 24, 36, 72 and 96 h was analyzed on a 10 % SDS-PAGE. The supernatant of clone Zα which was grown for 96 h was used as a negative control. Arrow indicates the position of the ~60 kDa α-amylase. b N-glycosylation test: the supernatant of clone Δ3 was treated (2) or not (1) with PNGase F c zymogram analysis of (b)
Construction of expression vector
Based on the deletion analysis of P PGK1 we constructed a vector, pPICK2, for constitutive expression in P. pastoris (Fig. 5). This new vector is based on the pPICZα series developed by Invitrogen, that carries the Sh ble marker allowing selection on media containing zeocin, an expensive drug. We replaced the original Sh ble marker for kan a dominant marker used in several yeasts (Agaphonov et al. 2010). Although the use of kan increases the plasmid size it has the advantage of allowing the use of the much cheaper kanamycin and G418 antibiotics for bacterial vector propagation and yeast transformation, respectively. Also, yeast transformants bearing multiple integrated vectors can be identified by screening clones resistant to higher concentrations of G418 (Scorer et al. 1994). This is the basis for different gene amplification approaches available for expression optimization in P. pastoris (Sunga et al. 2008).

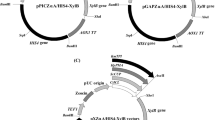
Physical map of pPICK2
Although we showed that a PGK1-promoter fragment of only 415 pb (P PGKΔ3 ) was sufficient to drive plasmid integration and heterologous expression in P. pastoris, we decided to use a longer fragment of ~620 pb (P PGKΔ4 ) because it contains a useful PvuI site (Fig. 1b) that could be used alternatively to SacI and XbaI for plasmid linearization. Plasmid linearization prior to transformation increases integration efficiency in yeast (Romanos et al. 1992). Also, the presence of 3 restriction sites on P PGKΔ4 allows a more versatile choice of restriction enzymes when different genes bearing these sites are cloned into pPICK2. To test the functionality of P PGKΔ4 in another genomic context we replaced the original AOX1 promoter present in pPIC9 by P PGKΔ4 and placed the eGFP (enhanced Green Fluorescent Protein) reporter gene under its transcriptional control (data not shown). The resulting vector was targeted to the HIS4 and PGK1 loci by linearization with SalI or SacI, respectively. The resulting transformants produced roughly the same amounts of eGFP (data not shown) thus showing that P PGKΔ4 may also be used to drive the expression of an heterologous gene when integrated into another locus other than PGK1.
The original S. cerevisiae α-factor sequence was adapted for optimized expression in P. pastoris but it can be easily replaced by alternative secretion signals. A new polylinker was constructed offering unique restriction sites representing common commercially available enzymes. When cloning into the XhoI site present in pPICK2, the α-factor secretion signal sequence between this site and the polylinker should be regenerated if one wishes the proper N-terminal processing of the secreted protein. The original His6x tag was maintained to facilitated protein purification when using nickel-charged affinity resins, as well as the BglII and BamHI sites for in vitro multimerization of expression cassettes. Finally, the fact that all functional elements present in pPICK2 are modular prompts this vector as a versatile platform for protein expression in P. pastoris.
References
Agaphonov M, Romanova N, Choi ES, Ter-Avanesyan M (2010) A novel kanamycin/G418 resistance marker for direct selection of transformants in Escherichia coli and different yeast species. Yeast 27:189–195
Çalık P, Ata O, Güneş H, Massahi A, Boy E, Keskin A, Öztürk S, Zerze GH, Özdamar TH (2015) Recombinant protein production in Pichia pastoris under glyceraldehyde-3-phosphate dehydrogenase promoter: from carbon source metabolism to bioreactor operation parameters. Biochem Eng J 95:20–36
Cereghino LGP, Cereghino JL, Sunga AJ, Johnson MA, Lim M, Gleeson MA, Cregg JM (2001) New selectable marker/auxotrophic host strain combinations for molecular genetic manipulation of Pichia pastoris. Gene 263:159–169
Daly R, Hearn MT (2005) Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit 18:119–138
de Almeida JR, de Moraes LM, Torres FA (2005) Molecular characterization of the 3-phosphoglycerate kinase gene (PGK1) from the methylotrophic yeast Pichia pastoris. Yeast 22:725–737
Fraenkel DG (1982) Carbohydrate metabolism. In: Strathern JN, Jones EW, Broach JR (eds) The molecular biology of the yeast Saccharomyces: metabolism and gene expression. Cold Spring Harbor Laboratory Press, NY, pp 1–37
Fuwa H (1954) A new method for microdetermination of amylase activity by the use of amylose as the substrate. J Biochem 41:583–603
Janner CR, Brito AL, Moraes LM, Reis VC, Torres FA (2013) pPCV, a versatile vector for cloning PCR products. Springerplus 2:441
Liu ZW, Yin HX, Yi XP, Zhang AL, Luo JX, Zhang TY, Fu CY, Zhang ZH, Shen JC, Chen LP (2012) Constitutive expression of barley alpha-amylase in Pichia pastoris by high-density cell culture. Mol Biol Rep 39:5805–5810
Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM (2005) Heterologous protein production using the Pichia pastoris expression system. Yeast 22:249–270
Menéndez C, Hernández L, Banguela A, País J (2004) Functional production and secretion of the Gluconoacetobacter diazatrophicus fructose-releasing exo-levanase (LsdB) in Pichia pastoris. Enz Microb Technol 34:446–452
Qin X, Qian J, Yao G, Zhuang Y, Zhang S, Chu J (2011) GAP promoter library for fine-tuning of gene expression in Pichia pastoris. Appl Environ Microbiol 77:3600–3608
Reis VC, Nicola AM, de Neto OSO, Batista VD, de Moraes LM, Torres FA (2012) Genetic characterization and construction of an auxotrophic strain of Saccharomyces cerevisiae JP1, a Brazilian industrial yeast strain for bioethanol production. J Ind Microbiol Biotechnol 39:1673–1683
Romanos MA, Scorer CA, Clare JJ (1992) Foreign gene expression in yeast: a review. Yeast 8:423–488
Scorer CA, Clare JJ, McCombie WR, Romanos MA, Sreekrishna K (1994) Rapid selection using G418 of high copy number transformants of Pichia pastoris for high-level foreign gene expression. Biotechnology 12:181–184
Stadlmayr G, Mecklenbrauker A, Rothmuller M, Maurer M, Sauer M, Mattanovich D, Gasser B (2010) Identification and characterisation of novel Pichia pastoris promoters for heterologous protein production. J Biotechnol 150:519–529
Sunga AJ, Tolstorukov I, Cregg JM (2008) Posttransformational vector amplification in the yeast Pichia pastoris. FEMS Yeast Res 8:870–876
Yang CH, Huang YC, Chen CY, Wen CY (2010) Expression of Thermobifida fusca thermostable raw starch digesting alpha-amylase in Pichia pastoris and its application in raw sago starch hydrolysis. J Ind Microbiol Biotechnol 37:401–406
Zhang AL, Luo JX, Zhang TY, Pan YW, Tan YH, Fu CY, Tu FZ (2009) Recent advances on the GAP promoter derived expression system of Pichia pastoris. Mol Biol Rep 36:1611–1619
Acknowledgments
This work was financially supported by FAPDF (Grant# 193.000.582/2009) and CNPq. The authors are thankful to Alexsandro Sobreira Galdino for his assistance in the zymogram methodology.
Supporting Information
Supplementary Fig. 1–Sequence of the 270-bp synthetic DNA fragment used to remove the PvuI and SmaI sites present in the kan coding sequence
Supplementary Fig. 2–Relevant sequences required for heterologous gene expression present on pPICK2
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Arruda, A., Reis, V.C.B., Batista, V.D.F. et al. A constitutive expression system for Pichia pastoris based on the PGK1 promoter. Biotechnol Lett 38, 509–517 (2016). https://doi.org/10.1007/s10529-015-2002-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-015-2002-2