
Abstract
Aging is associated with increasing impairments in brain homeostasis and represents the main risk factor across most neurodegenerative disorders. Melatonin, a neuroendocrine hormone that regulates mammalian chronobiology and endocrine functions is well known for its antioxidant potential, exhibiting both cytoprotective and chronobiotic abilities. Age-related decline of melatonin disrupting mitochondrial homeostasis and cytosolic DNA-mediated inflammatory reactions in neurons is a major contributory factor in the emergence of neurological abnormalities. There is scattered literature on the possible use of melatonin against neurodegenerative mechanisms in the aging process and its associated diseases. We have searched PUBMED with many combinations of key words for available literature spanning two decades. Based on the vast number of experimental papers, we hereby review recent advancements concerning the potential impact of melatonin on cellular redox balance and mitochondrial dynamics in the context of neurodegeneration. Next, we discuss a broader explanation of the involvement of disrupted redox homeostasis in the pathophysiology of age-related diseases and its connection to circadian mechanisms. Our effort may result in the discovery of novel therapeutic approaches. Finally, we summarize the current knowledge on molecular and circadian regulatory mechanisms of melatonin to overcome neurodegenerative diseases (NDDs) such as Alzheimer’s, Parkinson’s, Huntington’s disease, and amyotrophic lateral sclerosis, however, these findings need to be confirmed by larger, well-designed clinical trials. This review is also expected to uncover the associated molecular alterations in the aging brain and explain how melatonin-mediated circadian restoration of neuronal homeodynamics may increase healthy lifespan in age-related NDDs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biological aging is a complicated process that involves modification in cellular functioning, deterioration of physiological integrity, increasing vulnerability, senescence, and ultimately death (McHugh and Gil 2018). These changes affect every function of the organism ranging from structural ambiguities in macro features of the cell, abnormalities in the synthesis of cellular proteins, to multiple disruptions in the cellular physiochemical environment and a complete functional loss of an organ or systemic performance in a time-dependent manner (Li et al. 2021). These age-related modifications often result in the development and progression of neurodegenerative diseases (NDDs) including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) (Kubben and Misteli 2017). In the aging population, NDDs induce progressive loss of brain function which becomes a major factor leading to severe disability and death (Hou et al. 2019).
Neurodegeneration is characterized by progressive neurological disorders which are identified by distinct clinical, morphological, and biochemical aspects (Lananna and Musiek 2020). Due to high levels of polyunsaturated fatty acids (PUFAs) and heavier oxygen demand, high levels of ascorbate and iron in specific areas, and low content of antioxidants, the human brain is particularly susceptible to ROS-induced damage (Cobley et al. 2018). The mechanism of redox homeodynamics plays an important role in the regulation of biochemical activities including cell cycle regulation, activation of enzymes, DNA synthesis, and apoptotic responses. The development of an excessive amount of reactive oxygen species (ROS) inhibits the electron transport chain (ETC) which dysregulates mitochondrial dynamics in brain cells during aging and in the condition of neurodegeneration (Gudenschwager et al. 2021). Free radical-induced mitochondrial damage and neuroinflammation are pathological signs of aging and NDDs (Jiang et al. 2021). Understanding the mechanisms involved in age-related changes in brain structure and function is crucial to the identification of new potential therapeutic approaches and establishing multimodal health-care services to address the requirements of an aging population. Although several antioxidant defense mechanisms have been suggested to prevent neurodegeneration, their usefulness has often been questioned (Davies et al. 2017).
A growing body of evidence suggests that melatonin is a multifaceted homeostatic agent that exhibits protective functions in different cells and tissues through its ability to regulate numerous components of cell biology (Cardinali 2019). Melatonin is a unique phylogenetically sustained molecule found in all identified aerobic organisms and possesses cytoprotective competence in addition to its chronobiotic effects (Cardinali 2019). Besides its function in sleep and circadian regulatory mechanisms, melatonin is well known to have antioxidative, anti-inflammatory, neuroprotective, and anti-apoptotic properties in different cellular systems and animal studies (Tan et al. 2020; Kumar Verma et al. 2022; Xu et al. 2022). Melatonin has also been demonstrated to have immunomodulatory, antiangiogenic, and antiaging properties (Majidinia et al. 2018). Despite the absence of any conclusive data, melatonin has been suggested to possibly increase longevity (Hardeland 2013). Melatonin synthesis declines with age and also in certain neurological conditions such as AD and PD, suggesting that dysregulation in melatonin synthesis may contribute to the onset or progression of age-related NDDs. An understanding of how damaged neuronal cells are influenced by melatonin may explain the complicated nature of aging brain in both healthy and pathological conditions.
In this review, we have investigated the gaps in understanding of how melatonin affects neuronal redox signaling, circadian clock genes, and mitochondrial homeostasis during aging. We further investigate the therapeutic ability of melatonin and its possible use in restoring circadian rhythms in individuals experiencing age-associated NDDs. The detailed molecular mechanisms by which melatonin exerts protection against NDDs as well as the clinical applicability of melatonin in these disease conditions have also been discussed in this review.
Melatonin and nervous system aging
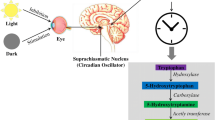
The mammalian suprachiasmatic nucleus (SCN) also known as a central endogenous clock governs the rhythmicity of nearly all biological processes. The SCN sends a neuronal signal that causes the pineal gland to start producing melatonin at night. Several organs and tissues other than the pineal gland are also capable of producing melatonin (Markus et al. 2021). SCN expresses melatonin receptor (MT1) and the circulatory melatonin is responsible for the regulation of this endogenous clock function. The age-related deficit in melatonin synthesis is linked to accelerated aging, and vice versa (Hardeland 2019). Furthermore, extra-pineal and pineal melatonin have been both recently recognized as an important aging biomarkers due to the substantial correlation between melatonin and aging (Martín Giménez et al. 2022).
The most detrimental alterations in the neurological system that occurs with age are a considerable loss in brain size notably in the forebrain (Lemaitre et al. 2012), midline temporal lobe (Du et al. 2006), a bilateral expansion in ventricular size, loss of grey matter in medial frontal cortical areas, white matter compression and decrease in the number of spines and dendritic branches (Duan et al. 2003). Furthermore, the hippocampal synaptic density is decreased during aging (Lister and Barnes 2009). The cognitive decline caused by reduced neuronal activity in the hippocampus is a recurring problem of aging. These and other changes eventually contribute to neurodegeneration, neurogenesis impairment, cognitive deficits, and sensory-motor deficiencies in aged individuals (Majidinia et al. 2018). A growing body of research indicates that age-associated changes in the neuronal cells are primarily caused by an imbalance between pro-oxidant and antioxidant, a potential enhancement in innate immunological response and an intensified increase in inflammation, and mitochondrial dysregulation (Tan et al. 2018). In terms of redox status, it was shown that the brains of longer-lived animals produce much less free radicals and reactive oxygen species (Buffenstein et al. 2008). Based on these findings and the strong antioxidative and anti-inflammatory actions, melatonin is a powerful contender for exerting anti-aging activities in the brain.
The significance of melatonin in the aging process is studied from various perspectives. For example, melatonin is an essential molecule involved in maintaining circadian amplitude, entrainment, and oscillator coherence of biological rhythms as well as sleep commencement (Zisapel 2018; Luo et al. 2020). The metabolic function of melatonin is accomplished by the regulation of key signal transduction pathways including Sirtuin 1, AMP-dependent protein kinase (AMPK), phosphatidylinositol 3-kinase (PI3K), and Akt (Ramis et al. 2015; Mayo et al. 2017). Besides SIRT1, its different isoforms from SIRT1 to SIRT7 are also involved in the maintenance of energy homeostasis and mitochondrial biogenesis (Afzaal et al. 2022). SIRT1 and AMPK are shown to collaborate in the aspect of metabolic sensing during aging because they both simultaneously respond to increases in AMP concentration, a sign of ATP insufficiency (Fulco and Sartorelli 2008). It is important to note that AMPK is a prominent stimulator for mitochondrial biogenesis and a component that extends longevity as well (Ramirez Reyes et al. 2021). Melatonin regulates mitochondrial complexes of the electron transport chain to improve respiration effectiveness, reduced electron leakage, cardiolipin peroxidation avoidance, and anti-apoptotic functions (Reiter et al. 2017). Melatonin-activated nuclear factor erythroid 2-related factor (Nrf) 2 via AMPK signaling mechanism. This results in the activation of antioxidative and detoxification enzymes such as superoxide dismutase (SOD), heme oxygenase 1 (HMOX-1), and NAD(P)H quinone dehydrogenase 1 (NQO1), which collectively suppress ROS production (Kang et al. 2022). Further, melatonin increases growth hormone (GH) levels by acting at the downstream component in the GH-signaling mechanism (Nassar et al. 2007). Inhibition of the insulin growth factor-1 (IGF-1) signaling mechanism, which shares its function with insulin, through PI3K and Akt pathway, considerably extends longevity in mouse (Guarente 2008). However, the nuclear factor kappa-B (NF-κB) signaling pathway is activated by GH and IGF-1 later in life, which speeds up the aging process by inducing inflammatory responses. The consequences of NF-κB signaling are counteracted by well-known age-suppressors including SIRTs and FoxOs in addition to drops in GH and IGF-1 concentrations during aging (Salminen and Kaarniranta 2010). Melatonin has a variety of immune-modulating properties. This function is critical in immunological remodeling and inflammation (Hardeland 2017a; Majidinia et al. 2018) (Fig. 1).

Protective effects of melatonin on aging: Circadian disruption is linked to aging and morbidity. Melatonin is a strong contender to reset the circadian rhythm which controls healthy aging and longevity. Melatonin increases growth hormone (GH) levels by acting at the downstream component in the GH-signalling mechanism. It counteracts the deleterious effects of aging via the regulation of inflammatory processes. During stressful conditions, sirtuins (SIRTs) and 5′ AMP-activated protein kinase (AMPK) have been shown to work in coordination. AMPK upregulates Forkhead box O (FoxO) transcription factor and SIRT1-7 which are involved in the maintenance of energy homeostasis, coupled with mitochondrial proliferation and extending longevity
Melatonin is demonstrated to attenuate DNA damage response (DDR) in humans (Majidinia et al. 2017) and in neuronal cells of senescence-accelerated female mice (Morioka et al. 1999). Chronic melatonin treatment in SAMP8 mice suppresses oxidative damage and calpain/Cdk5 signaling as well as hyperphosphorylation of GSK3 beta and tau markers of cerebral aging and neurodegeneration. These findings support the anti-aging and neuroprotective properties of melatonin (Martín Giménez et al. 2022). In another study, melatonin was found to slow down the aging of the brain, especially in the temporal cortex by lowering the level of nitric oxide (Akbulut et al. 2013). Melatonin has also been demonstrated to reduce oxidative damage and neurological impairments in the hippocampus of female golden hamsters with hyperthyroidism during aging (Rao et al. 2016). Our recent research has reported that the administration of melatonin lowered the extent of oxidative injury and neurodegeneration in both young and old chronodisrupted models of rats (Verma et al. 2021a, b). In comparison to SAMR1 mice, SAMP8 animals have higher concentrations of the transcriptional factor NF-κB, revealing that oxidative stress is a major factor in the aging process (Caballero et al. 2008). Melatonin exerts an inhibitory action on NF-κB, which helps to avoid age-related neurodegeneration.
Melatonin regulates oxidative stress: the antioxidant machinery
Redox homeostasis and melatonin
An equilibrium between the production and elimination of ROS is crucial for maintaining cellular redox homeostasis, which controls a variety of physiological activities by regulating signaling mechanisms and enabling the activation of redox-sensitive proteins and enzymes. While cellular signaling pathways and subcellular integrity can be damaged by the accumulation of oxidative products, this can also interfere with redox signaling mechanisms, which may ultimately lead to cellular aging and death (Cardoso and Moreira 2022). Melatonin can neutralize both ROS and reactive nitrogen species (RNS) (Wang et al. 2019). The ability of melatonin to cross cell membranes due to its amphipathic nature enables it to shield subcellular compartments from damage caused by free radicals (Mannino et al. 2021). Melatonin-induced redox control is achieved by its direct scavenging action against ROS with enzymatic and non-enzymatic antioxidant defense mechanisms (Ding et al. 2014; Verma et al. 2019, 2020b). The aromatic indole ring of melatonin which is enriched with electrons makes it a powerful electron donor to effectively neutralize free radicals in its direct antioxidant role (Verma et al. 2020a; Mannino et al. 2021). Through activation of antioxidant enzymes (e.g. SOD, GSH-Px, and CAT) (Liu and Ng 2000) and the stimulation of glutathione synthesis (Limón-Pacheco and Gonsebatt 2010), melatonin exhibits indirect antioxidant activity that enhances the effectiveness of other antioxidants with synergistic effects (Montilla et al. 2001).
Cells can respond to melatonin through receptor-dependent or independent pathways. Melatonin directly crosses cell membranes without the help of receptors and scavenges ROS in the cytosol, mitochondria, and nucleus of the cell. Higher levels of melatonin are presented in the mitochondria, where it counteracts ROS and RNS (Reiter et al. 2016). In addition, melatonin prevents cardiolipin peroxidation (Petrosillo et al. 2009) and protects respiratory chain complexes and mtDNA from free radical-mediated oxidative injury. The anti-apoptotic property of melatonin inhibited Bax activity, which reduced mPTP opening and consequently lowered membrane permeabilization (Fang et al. 2019; Mehrzadi et al. 2021). Further, melatonin attenuates mitophagy and activates mitochondrial biogenesis. The primary receptor-dependent signaling mechanism induced by MT1 and MT2 is a reduction in adenylyl cyclase action, which lowers intracellular concentrations of cyclic adenosine monophosphate (cAMP). There have been reports that as a response to melatonin, a variety of molecules, such as phospholipase C (PLC), diacylglycerol (DAG), the second messengers inositol trisphosphate (IP3), and calcium ions (Ca2+), and kinases such as PKA, PKC, and MAP kinases, are assembled, activated, or produced (Cecon et al. 2018). Melatonin signaling generally controls transcription negatively for genes regulated by CREB (cAMP-responsive element binding) transcription factor and positively for genes regulated by ERK-mediated transcription factors. As a result, pro-oxidant enzymes are downregulated while antioxidant enzymes are unregulated (Sarti et al. 2013; Oishi et al. 2018) (Fig. 2).

Redox signaling mechanisms through which melatonin reduce oxidative damage. Melatonin can interact with cells by both receptor-dependent and/or independent mechanisms. On the left side, melatonin inhibits free radicals and reduces oxidative injury. Melatonin directly scavenges mitochondrial ROS/RNS in a receptor-independent manner hence avoiding mtDNA damage. In response to oxidative stress, melatonin regulates calcium ion (Ca2+) release into the cytosol and protects mitochondria. Interaction of Bax and Bak two pre-apoptotic proteins causes mitochondrial permeability opening of transition pores (mPTP) and release of Cyt c which is involved in the activation of the apoptotic mechanism, this is effectively inhibited by melatonin. Further melatonin (on the right side) acts via membrane receptors (MT1/MT2) which contain G protein-coupled seven transmembrane helices. Interaction of melatonin with the MT1 receptor activates Gi protein, which inhibits adenylyl cyclase (AC) action and lowers intracellular contents of cyclic adenosine monophosphate (cAMP). In turn, this inhibits protein kinase A (PKA) and cAMP-response element binding protein (CREB) transcription factor resulting in the inhibition of gene transcription. MT2 binding to Gq encompasses phospholipase C (PLC) activation, an increase in diacylglycerol (DAG) synthesis, protein kinase C (PKC) stimulation, and a rise in intracellular calcium (Ca2+) concentrations to activate a series of processes that boost transcriptional activity. The binding of melatonin to calmodulin regulates nitric oxide synthesis. Nuclear binding sites (RoRα and RZR) may also be used in some of these processes
Mitochondrial aging and melatonin
Aging is a major contributory factor that reduces the efficiency of the respiratory chain, increases electron leakage, and lowers ATP synthesis, all of which ultimately lead to mitochondrial dysfunction (Tjahjono et al. 2022). In consequence, the defective mitochondrial function triggers the production of ROS, which further encourages mitochondrial damage and cellular death (Lee et al. 2021). Melatonin levels in mitochondria appear to be higher than those in the blood (Venegas et al. 2012), it is especially fortunate because these organelles serve as a prime source of free radical generation. In addition to effectively neutralizing ROS in these organelles, melatonin may diminish electron leakage from mitochondrial respiratory chain complexes resulting in the stimulation of ATP synthesis (Hardeland 2017b). The action of minimizing electron leakage also reduces the production of free radicals, a process known as radical avoidance (Reiter et al. 2010). Melatonin effectively repairs the damage, especially at the level of Complex I and IV. Additionally, it protects from the detrimental effects carried out by mitochondrial Ca2+ and depolarization of mitochondrial membrane driven by H2O2, both of which may associate with caspase-mediated apoptosis (Atayik and Çakatay 2022a). According to the mechanism, melatonin-dependent prooxidant activity may speed up the formation of ROS by interacting with calmodulin. The interaction of melatonin with the transition pore opening of mitochondria or its complex III also triggers ROS production (Zhang and Zhang 2014; Wang et al. 2019).
Nrf2 and melatonin
Nrf2 is recognized as the first line of defense against oxidative stress, it plays a central role in the transcriptional regulation of genes that produce antioxidant enzymes, anti-apoptotic proteins, detoxifying agents, and drug transporters (Rattan 2022). Further, NRF2 controls the transcriptional activity of more than 500 cytoprotective genes, most of which are involved in antioxidative defense mechanisms against the rise in intracellular ROS. NRF2-KEAP1 (Kelch-like ECH-associated protein 1) is a key signaling mechanism that controls oxidative stress. It has been demonstrated that melatonin increases the expression of genes and proteins connected to Nrf2 signaling and peroxisomal activity. By eliminating damaged organelles, such as mitochondria, Nrf2-mediated autophagy improves the efficiency of adaptations (Calabrese and Kozumbo 2021). Melatonin prevents Nrf2 from being degraded and activates the Keap1-ARE pathway (Vriend and Reiter 2015; Bona et al. 2022). Melatonin interacts with Nrf2 to improve cardioprotective, neuroprotective, antioxidant, anti-inflammatory, anti-tumor properties and also strengthens bone microstructure and oocyte maturation (Kilic et al. 2013; Gupta et al. 2021).
The so-called “NRF2 activators” should instead be named “KEAP1 inhibitors,” as KEAP1 is their actual molecular target (Fabrizio et al. 2018). For the treatment of various chronic disorders, the NRF2 activators are currently being pharmacologically developed at different stages. Mostly new developed molecules activate NRF2 by blocking it from being degraded by KEAP1-dependent processes. Sulforaphane (SFN) and melatonin (ITH12674) were combined to produce a hybrid compound with a dual “drug-prodrug” mode of action for the treatment of brain ischemia (Egea et al. 2015). To reduce the development of oxidative stress and inflammation in neurodegenerative and neuropsychiatric diseases (AD, PD, HD, autism, schizophrenia, and depression), NRF2 activators have also been recommended as antioxidants. (Saso et al. 2020). In contrast, NRF2 inhibitors, render tumor cells more susceptible to cancer treatments. NRF2 inhibitors are still a long way from being used from the bench to the bedside since this mechanism of inhibition is either unexplained or not precise in all conditions.
Aging, circadian clock genes, and melatonin
Since circadian rhythmicity is essential for regular health, any disturbances in the functioning of the cellular clock can have harmful implications. Although these circadian oscillations change with age, it is the vulnerability of the circadian system, that results in age-related diseases (Welz and Benitah 2020). Besides having a role in aging processes, the circadian clock also governs other age-related mechanisms like DNA repair, oxidative stress balance, and metabolic regulation (Lananna and Musiek 2020). Brain and muscle Arnt-like protein-1 (Bmal1) is an indispensable clock gene that regulates numerous fundamental physiological activities. Extensive research has revealed that melatonin regulates Bmal1 and has beneficiary physiological effects on the treatment of age-related disorders including AD (Li et al. 2020), and PD (Delgado-Lara et al. 2020). The underlying processes through which Bmal1 deficiency induces AD and associated age-related disorders include oxidative stress and inflammation (Fan et al. 2022). Melatonin activates the Bmal1 gene through PI3K/AKT signaling and increases Bmal1 protein expression (Beker et al. 2019). A study has found that administration of melatonin enhanced hippocampus Bmal1 expression, rapid remodeling of hippocampus neurons, and modified the synaptic structure of the CA1 and dentate gyrus (DG) regions, indicating that melatonin can influence the structural reforms in hippocampus neuronal cells through the involvement of Bmal1 clock gene (Ikeno and Nelson 2015).
Bmal1-deficient mice showed signs of accelerated aging, including multiple age-related abnormalities and a nearly threefold reduction in longevity (Kondratov et al. 2006). Clock−/− mice seemed to have a greater rate of cataracts and inflammation, as well as a 15% shorter longevity (Dubrovsky et al. 2010). Age-dependent changes in the SCN such as diminished neuronal activity often result in circadian misalignment (Nakamura et al. 2015). Additionally, aging alters periodic rhythmicity in the expression of circadian clock genes (Cry1, Cry2, Per1, Per2, and Bmal1) in the SCN. Furthermore, in animal models, Bmal1 and Per2 impairment is connected to behavioral abnormalities, metabolic complications, malignant tumors, and accelerated aging (Kondratov et al. 2009). The daily rhythmic oscillations in clock genes mRNA expression were found to be changed in the SCN. For 3, 12, and 24 months, the mRNA expression of distinct clock genes demonstrated that aging was the primary driver of circadian instability. Melatonin has been shown to protect the clock genes against age-related circadian disruption. At 12 months, melatonin treatment for 11 days synchronized the rhythmicity of Cry1, Cry2, Bmal1, and Per2 genes, while alterations in Bmal1, Cry1, and Cry2 were resumed at 24 months. A decrease in the total number of melatonin receptors of the SCN might be the possible explanation for variations in the amplitude of the circadian gene (Mattam and Jagota 2014). Therefore, the effect of melatonin on circadian genes and aging needs to be investigated.
Natural and synthetic melatonin derivatives
Melatonin and its metabolites collectively act as the most effective first line of defense against oxidative stress through a wide range of processes, such as electron transfer, hydrogen transfer, metal chelation, and addressing pharmaceutical targets (Galano and Reiter 2018). Melatonin as well as its natural and synthetic derivatives possess tremendous antioxidant activities to counteract different ROS and reactive nitrogen species (RNS) involved in the oxidative damage to cellular biomolecules like proteins, lipids, and DNA (Zhang and Zhang 2014; Wang et al. 2019). Cyclic 3-hydroxymelatonin (C3OHM), N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), N1-acetyl-5-methoxykynuramine (AMK), 6-hydroxymelatonin (6OHM) and 5-methoxytryptamine (5MTA) are among some of the most important metabolites of melatonin (Reiter et al. 2021). C3OHM reacts with other free radicals to generate AFMK. AMK is produced as a result of an interaction between the radicals and AFMK (Reiter et al. 2014). Neuronal injury caused by lipid peroxidation and oxidative stress is inhibited by AFMK (Burkhardt et al. 2001). Further, neurotoxicity caused by quinolinic acid is inhibited by 6OHM (Maharaj et al. 2005). Natural derivatives of melatonin and several of its precursors, including serotonin (5HT) (Azouzi et al. 2017), 5-hydroxytryptophan (5HTP), and 5-methoxytryptamine (5MTA), are also effective antioxidants.
By using a wide range of methods and computer-assisted techniques, systematic rational research was carried out to synthesize novel melatonin substitutes. It has been possible to synthesize novel distinct molecules by adding various simple functional groups, such as –OH, –NH2, –SH, and –COOH to the melatonin (Reina et al. 2018). 5-MLT (5-melatonin) derivatives have been identified to act as free radical scavengers through electron transfer or H transfer. Therefore, the protective effect of melatonin against oxidative stress is an outcome of the cascadic interaction it has with its metabolites to exhibit a multipurpose and unique antioxidant defense system.
Melatonin and its analogs
The antioxidant defense mechanism is a complex process that involves a diverse range of chemical and nonchemical processes. The intriguing features of melatonin prompt to design and synthesis of melatonin-like substances is a fast-expanding field of research (Suzen 2013; Galano 2016). Some melatonin analogs with a sulfhydryl group exhibit antioxidant effects that are more potent than those of melatonin itself (Ng et al. 2000). Furthermore, in vitro analysis revealed that indole-based melatonin hydrazine analogs comprising 2-phenylindole (Suzen et al. 2006), indole-3-propionamides (Ateş-Alagöz et al. 2005) and N-methylindole (Shirinzadeh et al. 2010) derivatives were effective in the removal of free radicals. Modification in the 5-methoxy and 3-acylaminoethyl side chains of melatonin has demonstrated promising features to develop new molecules with stronger antioxidant potential in comparison to melatonin (Gürkök et al. 2009). Indole amino acid derivatives have also been developed and examined to mimic the antioxidant activity of indole-based melatonin. They have similar DPPH (2,2-diphenyl-1-picrylhydrazyl) scavenging efficiency to melatonin while their inhibitory action on lipid peroxidation was quiet improved (Suzen et al. 2012).
Since it is impossible to prevent aging, it is possible to prevent the changes brought about by aging from disrupting the homeostasis of individuals. Several experimental and clinical research gave important data regarding the safety and effectiveness of therapeutic interventions using melatonin individually or as a combination (Ferlazzo et al. 2020). Meanwhile, very few randomized clinical trials have addressed the minor side effects of melatonin administration, such as agitation, dizziness, headache, nausea, paresthesia of the lip, arm, or leg, numbness, sedation, and daytime sleepiness (Seabra et al. 2000; Wasdell et al. 2008). Aside from the treatment of sleep problems and as a pre-operative anxiolytic, melatonin cannot replace any conventional therapeutic method due to its unproven clinical utility in many pathological conditions (Andersen et al. 2016).
Mitochondrial neurodegeneration and melatonin
Mitochondrial activation of the cGAS/STING pathway
ROS-induced oxidation and impairment in DNA repair cause damage to the mitochondrial DNA (mtDNA) of neurons during neurodegeneration (Madabhushi et al. 2014). Reduced mitochondrial membrane potential (MMP) and enhanced mitochondrial permeability, determine the transfer of mtDNA into the cytoplasm (West and Shadel 2017) followed by activation of cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS) signaling pathway. cGAS detects cytosolic DNA, whether it is self—or foreign DNA (Sun et al. 2013). cGAS stimulates innate immunity by producing cyclic GMP-AMP, which binds to and activates STING (Chen et al. 2016). Through activation of IRF3 and NF-B, STING induces the expression of interferon (IFN) and pro-inflammatory interleukin (Li and Chen 2018). Mitochondrial ROS triggers the release of pro-inflammatory cytokine (IL-1β and IL-18) and caspase-1 activation (van de Veerdonk et al. 2011). These processes are very crucial in regulating neurodegeneration as gradual decline in synaptic neurotransmission underpins neuronal death in aging and neurodegenerative disorders (Wishart et al. 2006). Therefore, mitophagy, a particular type of autophagy, is essential for the clearance of damaged mitochondria in neurons (Chen et al. 2020b).
Mitophagy and melatonin
Mitophagy is a key regulating process, which maintains the redox integrity of mitochondria and also reduces the generation of excess ROS. In response to oxidative stress, mitophagy restricts the aggregation of damaged mitochondria and decreases the negative consequences of redox state failure. Down-regulation of mitophagy has been linked to a wide range of diseases (Garza-Lombó et al. 2020). Mitophagy is particularly governed by moderate ROS levels to enhance the activity of ROS signaling cascades (Frank et al. 2012). Melatonin-induced mitophagy has demonstrated promising abilities to treat mitochondrial dysregulation (Süzen et al. 2022). Since mitochondrial damage is thought to be both the origin and the target of ROS, impaired mitophagy is directly related to reduced longevity (Chen et al. 2020b; Lin et al. 2021). Mitophagy failure is associated with pathogenic conditions including diabetes, cancer, cardiovascular disease, and neurodegenerative diseases.
Melatonin is widely distributed in mitochondria as its uptake is facilitated by oligopeptide transporters namely PEPT1/2, in addition to its intra-mitochondrial biosynthesis of melatonin (Reiter et al. 2020). Melatonin decreases mitochondrial electron leakage and ROS generation and inhibits the opening of the mitochondrial permeability transition pore (MPTP) to retain mitochondrial membrane potential (Δψ) in detrimental bioenergetic settings, and also stimulates the uncoupling proteins (UCPs) to gradually decrease Δψ in a normal situation (Atayik and Çakatay 2022b). Melatonin plays a variety of mitoprotective roles besides its capacity to modulate redox status, especially in upregulating the transcription of genes involved in stress response mechanisms, antioxidant enzymes, and inhibiting the release of Cyt-c. Melatonin stimulates intermembrane heterotrimeric G protein and prevents the release of cytochrome c in response to stress (Suofu et al. 2017).
Emerging mitochondrial-focused senotherapeutic approaches which strive to activate melatonin-mediated mitophagy may be able to improve mitochondrial dysfunctions. Several feasible interventions (e.g., melatonin) to delay aging have been studied in senescence-accelerated mouse prone 8 (SAMP8) strain, albeit they have produced some contradictory findings (Okatani et al. 2002; Cheng et al. 2008; Caballero et al. 2009). Melatonin administration can regulate senescence-related molecular and morphological features. Melatonin has also been demonstrated to promote oxidative phosphorylation in the mitochondria of hepatic and neuronal cells (Manchester et al. 2015; Reiter et al. 2017, 2018). Replicative senescence in mesenchymal stem cells (MSCs) lowered mitophagy by preventing mitofission which increased mitochondrial dysfunction. Melatonin administration prevented replicative senescence by improving mitophagy and mitochondrial functionality by upregulating heat shock 70 kDa protein 1L (HSPA1L) (Lee et al. 2020).
Therapeutic potential of melatonin in neurodegenerative diseases
Although the pathogenesis of neurodegenerative diseases (NDDs) is multifaceted and still poorly understood, however, redox homeodynamics is identified as an important aspect in the development and progression of nearly all age-related NDDs (Leyane et al. 2022). Numerous researchers have recently focused their attention on the possible use of melatonin as a neuroprotective molecule (Arribas et al. 2018; Chen et al. 2020a; Monayo and Liu 2022). According to research, all disorders of the central and peripheral neurological systems, including AD, PD, HD, and ALS, have a diminished capacity to maintain the equilibrium between the production of free radicals and the activity of their defense mechanisms (Hacışevki and Baba 2018). Therefore, antioxidative and anti-inflammatory actions of melatonin may thus be playing an important role in suppressing higher levels of oxidative damage and associated mechanisms to counteract age-associated NDDs (Chen et al. 2020a).
Melatonin and Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disorder, identified as the most frequent cause of dementia with the prevalence increasing in part due to an aging global population. One of the well-known theories used to explain disease etiology is the development and aggregation of β-Amyloid plaques (Abeysinghe et al. 2020). One of the most prominent signs of AD is the disruption in regular circadian rhythm, which is shown by an increase in daytime sleepiness and nighttime awakenings (Mattis and Sehgal 2016). Previous research has shown that melatonin level is dramatically reduced while the expression of melatonin receptors is substantially increased in the brains of AD patients (Shukla et al. 2017; Alghamdi 2018), suggesting that melatonin abnormality is one of the possible leading causes of the disease (Cardinali et al. 2014; Galano et al. 2018). Further, melatonin treatment has been demonstrated to improve cognition and reduce circadian rhythm disruption in AD patients (Musiek et al. 2015; Sumsuzzman et al. 2021). The antioxidative activities of melatonin have been found to prevent the excessive production of harmful free radicals generated from pathogenic processes in AD as well as reduce the consequent neuronal damage in the cells (Huang et al. 2016).
Importantly, the efficacy of melatonin to mitigate both A and Tau diseases was demonstrated by a decline in senile plaque formation (Olcese et al. 2009; Peng et al. 2013) and Aβ oligomerization (Olcese et al. 2009), a reduction in Tau hyperphosphorylation (Peng et al. 2013), and a decrease in intracellular argyrophilic substance accumulation (presumably neurofibrillary tangles) (Peng et al. 2013). Studies have shown that melatonin has a positive influence on AD symptoms due to its ability to restore mitochondrial function, and regulates the autophagic responses in the AD brain (Dragicevic et al. 2011; Luo et al. 2020). Non-transgenic senescence-accelerated OXYS rats, which are used as a model of sporadic AD, have shown that given low doses of melatonin (0.04 mg/kg/day; the total amount of 7.2 mg/kg) for 6 months results in significant improvements in anxiety, spatial and working memory (Rudnitskaya et al. 2015). Melatonin had a stronger positive effect on improving the learning capability of metabolic and senescence-associated AD models and reversed the memory impairments in the toxin-induced AD model (Zhai et al. 2022). Finally, and most importantly, melatonin has been shown to have a beneficial influence on clinical symptoms, as animals given melatonin treatments had higher survival rates (Matsubara et al. 2003) and improved cognitive abilities (Olcese et al. 2009).
It has also been suggested that reduced levels of melatonin in AD cause dysregulation in neurogenesis (Song 2019). Melatonin plays a function in the stimulation of neurogenesis and synaptic plasticity via neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor (GDNF) (Niles et al. 2004). These neurotrophic factors are important for neurogenesis. As a result, a decrease in melatonin may lead to a decrease in BDNF and GDNF production.
Additionally, melatonin has anti-amyloid properties that are effective in defending neuronal cells against cytotoxicity caused by amyloid-beta (Vincent 2018). Melatonin inhibits the production of amyloidogenic peptides by suppressing β- and γ-secretases, which are implicated in the cleavage of amyloid-beta proteins from amyloid precursor protein (APP) (Shukla et al. 2017), and it also inhibits the accumulation of amyloid aggregates by having interaction with amyloid-beta proteins (Wang and Wang 2006). Melatonin can also inhibit APP maturation by obstructing APP secretion at basal values into different cell lines and APP mRNA levels (Lahiri 1999). This mechanism is thought to involve protein kinase C, which is an upstream regulator of GSK3, by stimulating the PI3K/Akt and phospholipase C/diacylglycerol (PLC/DAG) pathways (Jeong et al. 2014). Activation of glycogen synthase kinase 3 beta (GSK3β) is involved in amyloid-beta diseases, while activation of protein kinase C phosphorylates and inactivates GSK3β. Melatonin can prevent neurotoxicity by inhibiting the accumulation and production of amyloid fibrils by binding with amyloid-beta proteins (Li et al. 2020) (Fig. 3).

Melatonin regulates key signaling pathways in neurogenesis. A diagrammatic representation of pathways by which melatonin regulates neurogenesis. Melatonin stimulates the Wnt/β-catenin signaling, which in turn enhances the transcription of survival-related genes. Further, melatonin causes inhibition of GSK3 which protects catenin from being phosphorylated and degraded, hence avoiding apoptosis. Since GSK3 participates in a cross-talk between both the Wnt/β-catenin and Notch signaling pathways, melatonin-induced GSK3 regulation also affects the underlying processes (blue dashed arrows). Melatonin controls both extracellular and intracellular disintegration of Notch via regulating and modulating the APP cleaving secretases (ADAM10, BACE1, and secretase), which in turn influences the production of NICD and the transcriptional activity of Notch effector genes. Through its influence on the Notch pathway, melatonin also inhibits the aggregation of Aβ. Additionally, leucine-rich repeat kinase 2 (LRRK2), which is implicated in the modification of Notch signaling, is also controlled by melatonin. Melatonin controls a self-renewal transcription factor SOX2, which is taken into account in APP metabolism to influence the SHH signaling pathway. Furthermore, melatonin promptly activates genes and proteins involved in the SHH pathway. Melatonin regulates the development and processing of APP inside the cell and inhibits the accumulation of Aβ and α-synuclein through the SHH-induced activation of GRP78. By facilitating PI3K/Akt signaling, SHH activation promotes neuroprotection. It may also prevent cell death by increasing Bcl-2 via a GRP78-dependent mechanism. Melatonin may modulate the Hippo signaling mechanism by inhibiting MST1 and regulating the downstream transcriptional factor YAP implicated in oxidative stress-mediated neuronal death, which in turn influences cellular proliferation and inhibits apoptosis. Aβ amyloid beta, ADAM10 a disintegrin and metalloproteinase domain-containing protein 10, AP amyloid precursor protein, Bcl-2 B cell lymphoma 2, GSK3β glycogen synthase kinase 3 beta, BACE1 beta-secretase 1, GRP78 glucose-regulated protein, MST1 mammalian sterile 20 (STE20)–like kinase 1, LRRK2 leucine-rich repeat kinase 2, NICD Notch intracellular domain, SHH sonic hedgehog, SOX2 PI3K/Akt PI3 kinase/protein kinase B, YAP yes-associated protein, SRY (sex-determining region Y)-box 2
Melatonin and Parkinson’s disease
Parkinson’s disease (PD) is an accelerating neurodegenerative disorder that mostly affects elderly individuals worldwide, and is characterized by the loss of dopaminergic neurons that emerge from the substantia nigra pars compacta (Pajares et al. 2020). The destruction of dopaminergic neurons and Lewy pathology accompany the characteristic motor symptoms, which include bradykinesia, stiffness, and postural instability. Additionally, PD patients exhibit several non-motor symptoms, including sleep disturbances, cognition, sensory impairments, and autonomic dysfunctions (Poewe et al. 2017). Previous research has revealed a correlation between the motor symptoms of PD and the amount of melatonin in the CSF (Leston et al. 2010). Neuropathological and corresponding imaging reports have demonstrated an alteration in melatonin levels and a reduction in hypothalamic areas in PD patients (Breen et al. 2016). Melatonin can also suppress the production of α-synuclein hazardous oligomers, α-synuclein fibrils, and oxidative stress, as well as control the expression of genes and proteins involved in the apoptotic signal transduction pathway (Ono et al. 2012).
SCN and various CNS regions, including the cerebral and cerebellar cortex and the midbrain, have been found to contain MT1 and MT2 receptors (Ng et al. 2017). In substantia nigra and amygdala, the density of MT1 and MT2 melatonin receptors was found to be lower in PD patients (Adi et al. 2010), resulting in the development of a hypothesis that the breakdown of the melatonergic system contributes to the disrupted sleep/wake cycle identified in PD. Additionally, the rhythmic profile of circulatory melatonin was reduced in PD patients, specifically those who demonstrated excessive daytime sleepiness (Videnovic et al. 2014). The administration of melatonin significantly restored the expression of the circadian clock gene in the rotenone-induced experimental rat model of PD (Mattam and Jagota 2015). The reasons behind the application of melatonin strategies were chosen because PD patients have low levels of PER1 and BMAL1 in the respective circadian morning and night schedules (Cai et al. 2010; Breen et al. 2014). Another explanation of circadian disruption in Bmal1 expression is that dopamine can control the functioning of the BMAL1/CLOCK complex, therefore its absence in PD impairs this essential part of the molecular clock (Breen et al. 2014). After supplementation of melatonin Bmal1 expression improved in PD patients (Delgado-Lara et al. 2020), suggesting a close association between Bmal1 and PD. The combined effect of melatonin with dopaminergic neurons can reduce free radicals, prolong the survivability of transplanted neurons, and improve motor impairments in PD patients (Asemi-Rad et al. 2022).
Stimulation of mitochondrial autophagy triggered by PINK1 and Parkin was crucial for preserving robust mitochondrial homeostasis and reducing the brain damage imposed by mitochondrial dysfunction in PD (Mammucari and Rizzuto 2010). Melatonin inhibited MPTP-induced stress against free radical deposition, DNA damage, and disruption of proton potential in mitochondria (Srinivasan et al. 2011). Lewy bodies, which are recognized as cytopathological biomarkers of parkinsonism, consist of improper distribution of tubulin, MAP1, and MAP2 microtubule-associated proteins. Melatonin stimulates cytoskeletal realignments and is believed to be therapeutically relevant for the treatment of parkinsonism and, most likely, dementias with Lewy bodies (Chen et al. 2005). The protective effect of melatonin is ascribed to normalizations of complex I activity in unilaterally 6- hydroxydopamine (6-OHDA) treated hemiparkinsonian rats (Benitez-King et al. 2004). Melatonin and its metabolite AMK decrease NO production and scavenge reactive nitrogen species which should also assist in cell survival, as well as other protective properties such as stimulation of antioxidant enzymes Cu-ZnSOD, MnSOD, GPx which has been established in cultured dopaminergic cells (Dabbeni-Sala et al. 2001).
Melatonin is also involved in the mitochondrial-hyperphosphorylation-neuronal apoptotic pathway. Melatonin inhibited not only 1-methyl-4-phenylpyridinium (MPP+) induced cell death in cerebellar granular neurons, but also activation of Cdk5 and cleavage of p35 to p25 (Hanagasi et al. 2005), kinases all of which are implicated in neuronal function and plasticity (Alvira et al. 2006). However, it is unclear whether melatonin at therapeutic doses in the MPP+ -induced PD investigation affects p25 and Cdk5 activity indirectly through mitochondrial actions and/or directly through receptor-dependent signaling mechanisms.
Melatonin and Huntington disease
Huntington’s disease (HD) is a dominantly inherited neurodegenerative disorder that results in progressive motor deficits, psychiatric symptoms, and cognitive impairment (Tabrizi et al. 2019). It is caused by an abnormally expanded CAG repeat expansion in the huntingtin gene (HTT), which confers a predominant toxic gain of function in the mutant huntingtin protein. The clinical manifestations of HD have been largely attributed to neurodegeneration, particularly in the striatum and cortex region of the brain (Tabrizi et al. 2020).
The plasma levels of melatonin in Huntington disease (HD) patients are reduced when compared to healthy individuals (Kalliolia et al. 2014). Melatonin protects neurons from kainic acid-induced neuronal loss, which can result in HD-related disease in both in vitro and in vivo experiments (Tan et al. 1998; Xue et al. 2017). In another study, melatonin reduced the oxidative damage and neurological injury caused by 3-nitropropionic acid (3-NP), which has been used to simulate the pathogenesis of HD both in vitro and in vivo experimental setups (Túnez et al. 2004; Nam et al. 2005; Mu et al. 2014). According to reports, mitochondrial dysfunction is one of the primary abnormalities of HD (Mochel and Haller 2011). Wang et al. discovered that melatonin lowers neuronal cell death while maintaining and activating MT1 in an HTT mutant cell model (Wang et al. 2011). Furthermore, the study found that HD mice have lower MT1 levels than wild-type mice (Wang et al. 2011). Conversely, a nonselective melatonin receptor antagonist prevents mitochondrial dysfunction and neuronal death by blocking the protective effect of melatonin (Zhang et al. 2019). Therefore, melatonin prevents neuronal cell injury caused by mitochondrial abnormalities in HD, through an MT1-dependent regulatory mechanism.
cGAS STING signaling and HD
Cytosolic DNA and inflammatory responses have been attributed to HD with signs of activation in the cGAS-STING-IRF3 signaling in postmortem striata of HD patients (Jauhari et al. 2020). In the cell-cultured model of HD, higher cytosolic mtDNA content has been detected, while DNase I transfection in these cells reduced inflammation. Remarkably, melatonin plays a direct role in mediating the escape of cytosolic mtDNA in both a neurodegenerative model of HD and an accelerated model of aging. Melatonin deficits in healthy cells as well as the existence of mHTT in a neurodegenerative model of mouse both promote ROS injury, mtDNA release, stimulation of the cGAS network, and pathogenic inflammatory processes, which results in synaptic damage and neurodegeneration. Melatonin may therefore be a possible treatment for conditions associated with aging since melatonin deficits alter neuronal and synaptic susceptibility (Jauhari et al. 2020).
Another molecular explanation is a rise of intracellular Ca2+ level mediated by Ca2+ import through the channel of N-methyl-d-aspartate (NMDA) receptor for mitochondrial-dependent cell death in HD (Ruiz et al. 2010). Research has shown that activation of the mPTP opening results in cellular death (Jonas et al. 2015). It has been found that in mouse primary striatal neurons, melatonin reduces the NMDA receptor-induced rise in Ca2+ by suppressing mPTP activity (Andrabi et al. 2004). The antioxidative, neuroprotective, and antiapoptotic properties of melatonin have also been proven in research to have an impact on HD, but further detailed investigations are required (Wang 2009).
Melatonin and amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that affects the first and second motor neurons. The loss of motor neurons, which is accompanied by significant demyelination in the anterior horn of the spinal cord, is a characteristic feature of ALS. From a pathophysiological perspective, three primary mechanisms in ALS are examined: (a) Superoxide dismutase 1 (SOD1) gene alterations, which lead to a deleterious augmentation in functionality along with enhanced reactivity toward aberrant substrates (tyrosine nitration) and a reduced capability for zinc binding results in a lowered capacity for antioxidants. (b) alterations in neurofilament genes, attributed to oxidative changes or hyperphosphorylation of cytoskeletal proteins, destroying particular motor axon; (c) excitotoxicity triggered by increasing glutamic acid levels in the cerebrospinal fluid and the deterioration of excitatory amino acid transporters (Van Den Bosch et al. 2006; Bald et al. 2021). People having a genetic variant related to the ALS, i.e., familial type of ALS, and who have early signs of motor neuronal dysfunction, such as impaired motor coordination are examples of such patients (Polimeni et al. 2014).
Since oxidative stress has a central role in the etiology of ALS, it is acceptable that being a potential antioxidant, melatonin could be beneficial to those suffering from this disease (Pollari et al. 2014).. Melatonin has been attributed to a delay in disease commencement, reduced motor neuron loss, and extended lifespan in two different studies of SOD1G93A-transgenic mouse models (Weishaupt et al. 2006; Zhang et al. 2013). However other studies in SOD1G93A-transgenic mouse models reported no benefit from melatonin, and one related it to a shorter lifespan, higher motor neuron loss, and increased expression of SOD1 (Dardiotis et al. 2013; Jaiswal 2016).
Melatonin not only halts disease progression and premature death but also substantially inhibits motor neuronal loss by interrupting the Rip2/caspase-1 and caspase-3 pathways, as well as inactivating mitochondrial release of cytochrome C in a mutant superoxide dismutase 1 (SOD1) (G93A) transgenic mouse model of ALS (Zhang et al. 2013). The anti-apoptotic activity of melatonin in ALS has been linked to the suppression of caspase-1, cytochrome C, and caspase-3 signaling pathways. Furthermore, it has been demonstrated that the concentration of melatonin and its receptor MT1 is significantly decreased in the spinal cords of ALS mice in comparison with wild-type mice (Zhang et al. 2013). Therefore, antiapoptotic actions of melatonin in ALS are carried out by the MT1-dependent signaling pathway.
Melatonin inhibits disease progression by reducing cellular loss in cultured motor neurons triggered by glutamate and extends the longevity in a mouse model of ALS (SOD1 (G93A)-transgenic mice). Melatonin is expected to prevent neuronal degeneration and the development of ALS by decreasing oxidative stress (Weishaupt et al. 2006). It has been discovered that ALS patients have higher levels of circulatory serum protein carbonyls, an oxidative stress marker, compared to healthy people. However, after the treatment of melatonin, the level of circulatory serum protein carbonyls was restored to normal in ALS patients. Therefore, melatonin may have antioxidant properties that minimize motor neuron death by inhibiting the production of NO. Further investigations are required to uncover the molecular basis of the neuroprotective effects of melatonin on ALS.
Role of melatonin in neurogenesis regulation
Melatonin regulates Wnt/β-catenin signaling
Adult neurogenesis is strictly controlled by both internal genetic elements and external environmental variables. The functional interaction among growing neurons in the adult brain is regulated by several important signaling mechanisms (Fig. 3). Neuronal growth, cell fate determination, mobility, synapse formation, and integration into neuronal pathways are all important functions of the Wnt/β -catenin signaling pathway in adult neurogenesis (Oliva et al. 2018). β-Catenin is the decisive factor for precursors in proliferation or differentiation throughout neuronal development in mammals and it eventually regulates the genesis of neural precursor cells (NPC) to determine the cerebral cortical size (Chenn and Walsh 2002). These findings reveal that Wnt signaling is important for synaptic functions, memory consolidation, and cognitive functions in the aging process. Interestingly research has shown that melatonin inhibits apoptosis by regulating mitochondrial membrane potential and activating the β-catenin pathway (Jeong and Park 2015).
In AD patients, activated GSK3β has been linked to tau hyperphosphorylation and reduced β-catenin expression. Activation of Wnt signaling reduced amyloidosis by decreasing both APP cleavage and Aβ synthesis in a mouse model of AD (Huang et al. 2018). The beneficiary effect of melatonin on GSK3β and restoration of dysregulated β-catenin levels enhances and widens its mechanistic neuroprotective approach against AD (Shukla et al. 2017). Further, the Wnt/β-catenin signaling mechanism is also examined in the cellular model of HD. The homeostasis of the β-catenin complex is adversely affected in the mutant HD model. Upregulation in β-catenin expression protected against the HD mutations by replenishing β-catenin equilibrium, which was specifically disrupted in mutant HD (Godin et al. 2010).
Melatonin and notch signaling pathway
Notch signaling enhances proliferative transmission and has been linked to cellular fate determinations in the early stages of brain development. Activation of the Notch intracellular domain (NICD) causes accumulation of Aβ and impairment in blood–brain barrier (BBB) permeability (Marathe et al. 2017). The amyloid precursor protein (APP) cleaving secretases are also implicated in both intra- and extracellular breakdown of Notch, where changes in the proteolytic degradation of Notch by γ-secretase are included in the pathophysiology of AD. In addition to being essential for the production of Aβ peptides from APP, beta-secretase 1 (BACE1) controls the breakdown of Notch ligands which are attached to membranes. The embryonic mortality of mice lacking the disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), resulting from the absence of Notch receptor-mediated signaling (Saftig and Lichtenthaler 2015), confirms the significance of this pathway in the pathogenesis of AD. Additionally, Notch 1 receptors serve as the alternate substrate for γ-secretase, which is implicated in Aβ production. Any interference in Notch signaling has irreversible harmful effects; hence specific therapeutic suppression of γ-secretase to restrict the synthesis of Aβ should be taken into consideration to avoid inhibiting the processing of Notch1 receptors (Mendiola-Precoma et al. 2016).
In the PD model, GSK3β is further involved in the communication between the Wnt/β-catenin and Notch signaling pathways which are responsible to govern NSC dynamics (Singh et al. 2018). Melatonin inhibits α-synuclein accumulation (Chang et al. 2012), inhibits GSK3 activation (Chinchalongporn et al. 2018), and modulates Wnt/β-catenin, indicating that melatonin affects the Notch system via many methods. Additionally, melatonin activates leucine-rich repeat kinase 2 (LRRK2) (Sun et al. 2016), an important protein that regulates Notch signaling and is involved in the pathophysiology of familial and idiopathic PD (Imai et al. 2015, p. 2).
Melatonin, sonic hedgehog, and hippo signaling pathways
The establishment and flexibility in neuronal networks of the hippocampus are significantly influenced by the sonic hedgehog (SHH) signaling system, which controls adult stem cell division and the production of growth and angiogenic components. Melatonin is known to regulate the self-renewal transcription factor sex-determining region Y (SRY)-box 2 (SOX2) (Sarlak et al. 2016) and also control the proliferation of stem cells (Sotthibundhu et al. 2010; Tocharus et al. 2014). Importantly, SOX2-mediated regulation of the SHH pathway is essential for maintenance of neuronal stem cells (NSCs) (Favaro et al. 2009). The association of SOX2 with APP confirms its role in the etiology of AD (Sarlak et al. 2016). Additionally, Aβ disrupts the SHH signaling mechanism (Vorobyeva and Saunders 2018), and the fact that APP colocalizes with the SHH constituent validates its crucial role in the pathogenesis of AD.
Furthermore, Aβ disrupts the SHH signaling system (Vorobyeva and Saunders 2018). SHH enhances neuroprotection and antiapoptotic actions by reducing oxidative stress (Chen et al. 2017). The production of SHH activates Bcl-2 via a glucose-regulated protein 78 (GRP78)-dependent mechanism, which inhibits cell death (Chen et al. 2015). GRP78 regulates intracellular formation and processing of APP and inhibits Aβ and α-synuclein accumulation, which suggests that activation of GRP78 would undoubtedly lower the neurodegenerative alterations involved in the progression of both AD and PD (Casas 2017). Additionally, in the adult nigrostriatal circuit, SHH promotes cellular as well as neurochemical homeostatic balance and its stimulation rescues dopaminergic neurons by regulating PI3K/Akt signaling in the PD model of rodents (Shao et al. 2017). A crosstalk between mammalian sterile 20-like kinase 1 (MST1)/Hippo signaling pathway and additional fundamental signaling mechanisms which preserve cellular homeostasis and cell death mechanisms have been linked to neurodegenerative diseases (Fallahi et al. 2016). Particularly, MST1 activation lowers the activity of a transcriptional regulator yes-associated protein (YAP) which results in oxidative stress-mediated neuronal cell death.
Concluding remarks
Aging deteriorates the rhythmicity of circadian oscillations resulting in a decline in melatonin synthesis. Mitochondrial disruption, low immunity, and reduced melatonin content make older individuals more vulnerable to altered redox regulation and age-dependent NDDs. Melatonin as a key aspect in the regulation of chronobiology and endocrine physiology enjoys a strong reputation of being an anti-inflammatory, neuroprotective, immune enhancer, and endocrine-modulating substance. The processes which involve the utilization of melatonin to promote neuroprotection by regulating, modifying, and inhibiting neuronal signaling mechanisms during aging and neurodegeneration have been discussed at length in this review article.
In addition, mitochondrial dysfunction is a prominent aspect of neurodegeneration. ROS-induced damage in mtDNA activates the cGAS-STING mechanism in neuronal cells resulting in neuroinflammatory and neurodegenerative processes. This cascading response is effectively neutralized by melatonin which stimulates mitophagy and augments mitochondrial function to improve lifespan and healthspan. Melatonin has been shown in numerous studies to be able to combat the deleterious effects of aging and NDDs including AD, PD, HD, and ALS by protecting against oxidative damage, mitochondrial impairments, and inflammatory responses. Furthermore, the ability of melatonin to promote neurogenesis via Wnt/β-catenin, Notch, sonic hedgehog, and Hippo-like signaling pathways add to its effectiveness in the treatment and prevention of NDDs. Given the favorable results obtained from several experimental models of NDDs, as well as melatonin’s low toxicity pharmacological profile, we propose that these results may be used to develop clinical protocols to investigate the therapeutic effects of melatonin in human neurological disorders. An important aspect of this review is to emphasize that melatonin receives more attention in the context of its potential benefits as a therapeutic agent for the treatment of age-related NDDs. Careful investigation in this field could have a substantial impact on the management of the most prevalent and socially destructive neurological conditions and open the way for an interesting new era in gerontology.
References
Abeysinghe AADT, Deshapriya RDUS, Udawatte C (2020) Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci 256:117996. https://doi.org/10.1016/j.lfs.2020.117996
Adi N, Mash DC, Ali Y et al (2010) Melatonin MT1 and MT2 receptor expression in Parkinson’s disease. Med Sci Monit 16:BR61–BR67
Afzaal A, Rehman K, Kamal S, Akash MSH (2022) Versatile role of sirtuins in metabolic disorders: from modulation of mitochondrial function to therapeutic interventions. J Biochem Mol Toxicol 36:e23047. https://doi.org/10.1002/jbt.23047
Akbulut K, Güney Ş, Çetin F et al (2013) Melatonin delays brain aging by decreasing the nitric oxide level. Neurophysiology 45. https://doi.org/10.1007/s11062-013-9368-3
Alghamdi BS (2018) The neuroprotective role of melatonin in neurological disorders. J Neurosci Res 96:1136–1149. https://doi.org/10.1002/jnr.24220
Alvira D, Tajes M, Verdaguer E et al (2006) Inhibition of the cdk5/p25 fragment formation may explain the antiapoptotic effects of melatonin in an experimental model of Parkinson’s disease. J Pineal Res 40:251–258. https://doi.org/10.1111/j.1600-079X.2005.00308.x
Andersen LPH, Gögenur I, Rosenberg J, Reiter RJ (2016) The safety of melatonin in humans. Clin Drug Investig 36:169–175. https://doi.org/10.1007/s40261-015-0368-5
Andrabi SA, Sayeed I, Siemen D et al (2004) Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J 18:869–871. https://doi.org/10.1096/fj.03-1031fje
Arribas RL, Romero A, Egea J, de los Ríos C (2018) Modulation of serine/threonine phosphatases by melatonin: therapeutic approaches in neurodegenerative diseases: modulation of Ser/Thr phosphatases by melatonin. Br J Pharmacol 175:3220–3229. https://doi.org/10.1111/bph.14365
Asemi-Rad A, Moafi M, Aliaghaei A et al (2022) The effect of dopaminergic neuron transplantation and melatonin co-administration on oxidative stress-induced cell death in Parkinson’s disease. Metab Brain Dis. https://doi.org/10.1007/s11011-022-01021-5
Atayik MC, Çakatay U (2022a) Mitochondria-targeted senotherapeutic interventions. Biogerontology 23:401–423. https://doi.org/10.1007/s10522-022-09973-y
Atayik MC, Çakatay U (2022b) Melatonin-related signaling pathways and their regulatory effects in aging organisms. Biogerontology 23:529–539. https://doi.org/10.1007/s10522-022-09981-y
Ateş-Alagöz Z, Coban T, Suzen S (2005) A comparative study: evaluation of antioxidant activity of melatonin and some indole derivatives. Med Chem Res 14:169–179. https://doi.org/10.1007/s00044-005-0132-0
Azouzi S, Santuz H, Morandat S et al (2017) Antioxidant and membrane binding properties of serotonin protect lipids from oxidation. Biophys J 112:1863–1873. https://doi.org/10.1016/j.bpj.2017.03.037
Bald EM, Nance CS, Schultz JL (2021) Melatonin may slow disease progression in amyotrophic lateral sclerosis: findings from the Pooled Resource Open-Access ALS Clinic Trials database. Muscle Nerve 63:572–576. https://doi.org/10.1002/mus.27168
Beker MC, Caglayan B, Caglayan AB et al (2019) Interaction of melatonin and Bmal1 in the regulation of PI3K/AKT pathway components and cellular survival. Sci Rep 9:19082. https://doi.org/10.1038/s41598-019-55663-0
Benitez-King G, Ramírez-Rodríguez G, Ortíz L, Meza I (2004) The neuronal cytoskeleton as a potential therapeutical target in neurodegenerative diseases and schizophrenia. Curr Drug Targets CNS Neurol Disord 3:515–533. https://doi.org/10.2174/1568007043336761
Bona S, Fernandes SA, Moreira ACJ et al (2022) Melatonin restores zinc levels, activates the Keap1/Nrf2 pathway, and modulates endoplasmic reticular stress and HSP in rats with chronic hepatotoxicity. WJGPT 13:11–22. https://doi.org/10.4292/wjgpt.v13.i2.11
Breen DP, Vuono R, Nawarathna U et al (2014) Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol 71:589–595. https://doi.org/10.1001/jamaneurol.2014.65
Breen DP, Nombela C, Vuono R et al (2016) Hypothalamic volume loss is associated with reduced melatonin output in Parkinson’s disease. Mov Disord 31:1062–1066. https://doi.org/10.1002/mds.26592
Buffenstein R, Edrey YH, Yang T, Mele J (2008) The oxidative stress theory of aging: embattled or invincible? Insights from non-traditional model organisms. Age 30:99–109. https://doi.org/10.1007/s11357-008-9058-z
Burkhardt S, Reiter RJ, Tan DX et al (2001) DNA oxidatively damaged by chromium(III) and H(2)O(2) is protected by the antioxidants melatonin, N(1)-acetyl-N(2)-formyl-5-methoxykynuramine, resveratrol and uric acid. Int J Biochem Cell Biol 33:775–783. https://doi.org/10.1016/s1357-2725(01)00052-8
Caballero B, Vega-Naredo I, Sierra V et al (2008) Favorable effects of a prolonged treatment with melatonin on the level of oxidative damage and neurodegeneration in senescence-accelerated mice. J Pineal Res 45:302–311. https://doi.org/10.1111/j.1600-079X.2008.00591.x
Caballero B, Vega-Naredo I, Sierra V et al (2009) Melatonin alters cell death processes in response to age-related oxidative stress in the brain of senescence-accelerated mice. J Pineal Res 46:106–114. https://doi.org/10.1111/j.1600-079X.2008.00637.x
Cai Y, Liu S, Sothern RB et al (2010) Expression of clock genes Per1 and Bmal1 in total leukocytes in health and Parkinson’s disease: clock genes in PD. Eur J Neurol 17:550–554. https://doi.org/10.1111/j.1468-1331.2009.02848.x
Calabrese EJ, Kozumbo WJ (2021) The hormetic dose-response mechanism: Nrf2 activation. Pharmacol Res 167:105526. https://doi.org/10.1016/j.phrs.2021.105526
Cardinali DP (2019) Melatonin: clinical perspectives in neurodegeneration. Front Endocrinol 10:480. https://doi.org/10.3389/fendo.2019.00480
Cardinali DP, Vigo DE, Olivar N et al (2014) Melatonin therapy in patients with Alzheimer’s disease. Antioxidants 3:245–277. https://doi.org/10.3390/antiox3020245
Cardoso S, Moreira PI (2022) Targeting mitochondria and redox dyshomeostasis in brain ageing: an update. In: Çakatay U (ed) Redox signaling and biomarkers in ageing. Springer, Cham, pp 147–183
Casas C (2017) GRP78 at the centre of the stage in cancer and neuroprotection. Front Neurosci 11:177. https://doi.org/10.3389/fnins.2017.00177
Cecon E, Oishi A, Jockers R (2018) Melatonin receptors: molecular pharmacology and signalling in the context of system bias. Br J Pharmacol 175:3263–3280. https://doi.org/10.1111/bph.13950
Chang C-F, Huang H-J, Lee H-C et al (2012) Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and α-synuclein aggregation. J Pineal Res 52:312–321. https://doi.org/10.1111/j.1600-079X.2011.00945.x
Chen L-J, Gao Y-Q, Li X-J et al (2005) Melatonin protects against MPTP/MPP+ -induced mitochondrial DNA oxidative damage in vivo and in vitro. J Pineal Res 39:34–42. https://doi.org/10.1111/j.1600-079X.2005.00209.x
Chen K-Y, Cheng C-J, Wang L-C (2015) Activation of sonic hedgehog leads to survival enhancement of astrocytes via the GRP78-dependent pathway in mice infected with Angiostrongylus cantonensis. Biomed Res Int 2015:674371. https://doi.org/10.1155/2015/674371
Chen Q, Sun L, Chen ZJ (2016) Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17:1142–1149. https://doi.org/10.1038/ni.3558
Chen K-Y, Chiu C-H, Wang L-C (2017) Anti-apoptotic effects of Sonic hedgehog signalling through oxidative stress reduction in astrocytes co-cultured with excretory-secretory products of larval Angiostrongylus cantonensis. Sci Rep 7:41574. https://doi.org/10.1038/srep41574
Chen D, Zhang T, Lee TH (2020a) Cellular mechanisms of melatonin: insight from neurodegenerative diseases. Biomolecules 10:1158. https://doi.org/10.3390/biom10081158
Chen G, Kroemer G, Kepp O (2020b) Mitophagy: an emerging role in aging and age-associated diseases. Front Cell Dev Biol 8:200. https://doi.org/10.3389/fcell.2020.00200
Cheng S, Ma C, Qu H et al (2008) Differential effects of melatonin on hippocampal neurodegeneration in different aged accelerated senescence prone mouse-8. Neuro Endocrinol Lett 29:91–99
Chenn A, Walsh CA (2002) Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297:365–369. https://doi.org/10.1126/science.1074192
Chinchalongporn V, Shukla M, Govitrapong P (2018) Melatonin ameliorates Aβ42 -induced alteration of βAPP-processing secretases via the melatonin receptor through the Pin1/GSK3β/NF-κB pathway in SH-SY5Y cells. J Pineal Res 64:e12470. https://doi.org/10.1111/jpi.12470
Cobley JN, Fiorello ML, Bailey DM (2018) 13 reasons why the brain is susceptible to oxidative stress. Redox Biol 15:490–503. https://doi.org/10.1016/j.redox.2018.01.008
Dabbeni-Sala F, Di Santonull S, Franceschini D et al (2001) Melatonin protects against 6-OHDA-induced neurotoxicity in rats: a role for mitochondrial complex I activity. FASEB J 15:164–170. https://doi.org/10.1096/fj.00-0129com
Dardiotis E, Panayiotou E, Feldman ML et al (2013) Intraperitoneal melatonin is not neuroprotective in the G93ASOD1 transgenic mouse model of familial ALS and may exacerbate neurodegeneration. Neurosci Lett 548:170–175. https://doi.org/10.1016/j.neulet.2013.05.058
Davies JMS, Cillard J, Friguet B et al (2017) The Oxygen Paradox, the French Paradox, and age-related diseases. GeroScience 39:499–550. https://doi.org/10.1007/s11357-017-0002-y
Delgado-Lara DL, González-Enríquez GV, Torres-Mendoza BM et al (2020) Effect of melatonin administration on the PER1 and BMAL1 clock genes in patients with Parkinson’s disease. Biomed Pharmacother 129:110485. https://doi.org/10.1016/j.biopha.2020.110485
Ding K, Wang H, Xu J et al (2014) Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2–ARE signaling pathway as a potential mechanism. Free Radic Biol Med 73:1–11. https://doi.org/10.1016/j.freeradbiomed.2014.04.031
Dragicevic N, Copes N, O’Neal-Moffitt G et al (2011) Melatonin treatment restores mitochondrial function in Alzheimer’s mice: a mitochondrial protective role of melatonin membrane receptor signaling: melatonin restores mito. function in AD mice. J Pineal Res 51:75–86. https://doi.org/10.1111/j.1600-079X.2011.00864.x
Du A-T, Schuff N, Chao LL et al (2006) Age effects on atrophy rates of entorhinal cortex and hippocampus. Neurobiol Aging 27:733–740. https://doi.org/10.1016/j.neurobiolaging.2005.03.021
Duan H, Wearne SL, Rocher AB et al (2003) Age-related dendritic and spine changes in corticocortically projecting neurons in macaque monkeys. Cereb Cortex 13:950–961. https://doi.org/10.1093/cercor/13.9.950
Dubrovsky YV, Samsa WE, Kondratov RV (2010) Deficiency of circadian protein CLOCK reduces lifespan and increases age-related cataract development in mice. Aging 2:936–944. https://doi.org/10.18632/aging.100241
Egea J, Buendia I, Parada E et al (2015) Melatonin-sulforaphane hybrid ITH12674 induces neuroprotection in oxidative stress conditions by a “drug-prodrug” mechanism of action. Br J Pharmacol 172:1807–1821. https://doi.org/10.1111/bph.13025
Fabrizio FP, Sparaneo A, Trombetta D, Muscarella LA (2018) Epigenetic versus genetic deregulation of the KEAP1/NRF2 axis in solid tumors: focus on methylation and noncoding RNAs. Oxid Med Cell Longev 2018:2492063. https://doi.org/10.1155/2018/2492063
Fallahi E, O’Driscoll NA, Matallanas D (2016) The MST/hippo pathway and cell death: a non-canonical affair. Genes 7:E28. https://doi.org/10.3390/genes7060028
Fan R, Peng X, Xie L et al (2022) Importance of Bmal1 in Alzheimer’s disease and associated aging-related diseases: mechanisms and interventions. Aging Cell 21:e13704. https://doi.org/10.1111/acel.13704
Fang Y, Zhao C, Xiang H et al (2019) Melatonin inhibits formation of mitochondrial permeability transition pores and improves oxidative phosphorylation of frozen-thawed ram sperm. Front Endocrinol 10:896. https://doi.org/10.3389/fendo.2019.00896
Favaro R, Valotta M, Ferri ALM et al (2009) Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat Neurosci 12:1248–1256. https://doi.org/10.1038/nn.2397
Ferlazzo N, Andolina G, Cannata A et al (2020) Is melatonin the cornucopia of the 21st century? Antioxidants 9:1088. https://doi.org/10.3390/antiox9111088
Frank M, Duvezin-Caubet S, Koob S et al (2012) Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 1823:2297–2310. https://doi.org/10.1016/j.bbamcr.2012.08.007
Fulco M, Sartorelli V (2008) Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle 7:3669–3679. https://doi.org/10.4161/cc.7.23.7164
Galano A (2016) Computational-aided design of melatonin analogues with outstanding multifunctional antioxidant capacity. RSC Adv 6:22951–22963. https://doi.org/10.1039/C6RA00549G
Galano A, Reiter RJ (2018) Melatonin and its metabolites vs oxidative stress: from individual actions to collective protection. J Pineal Res 65:e12514. https://doi.org/10.1111/jpi.12514
Galano A, Tan D-X, Reiter RJ (2018) Melatonin: a versatile protector against oxidative DNA damage. Molecules 23:E530. https://doi.org/10.3390/molecules23030530
Garza-Lombó C, Pappa A, Panayiotidis MI, Franco R (2020) Redox homeostasis, oxidative stress and mitophagy. Mitochondrion 51:105–117. https://doi.org/10.1016/j.mito.2020.01.002
Godin JD, Poizat G, Hickey MA et al (2010) Mutant huntingtin-impaired degradation of beta-catenin causes neurotoxicity in Huntington’s disease. EMBO J 29:2433–2445. https://doi.org/10.1038/emboj.2010.117
Guarente L (2008) Mitochondria—a nexus for aging, calorie restriction, and sirtuins? Cell 132:171–176. https://doi.org/10.1016/j.cell.2008.01.007
Gudenschwager C, Chavez I, Cardenas C, Gonzalez-Billault C (2021) Directly reprogrammed human neurons to understand age-related energy metabolism impairment and mitochondrial dysfunction in healthy aging and neurodegeneration. Oxid Med Cell Longev 2021:5586052. https://doi.org/10.1155/2021/5586052
Gupta A, Behl T, Sehgal A et al (2021) Therapeutic potential of Nrf-2 pathway in the treatment of diabetic neuropathy and nephropathy. Mol Biol Rep 48:2761–2774. https://doi.org/10.1007/s11033-021-06257-5
Gürkök G, Coban T, Suzen S (2009) Melatonin analogue new indole hydrazide/hydrazone derivatives with antioxidant behavior: synthesis and structure-activity relationships. J Enzyme Inhib Med Chem 24:506–515. https://doi.org/10.1080/14756360802218516
Hacışevki A, Baba B (2018) An overview of melatonin as an antioxidant molecule: a biochemical approach. In: Manuela Drăgoi C, Crenguţa Nicolae A (eds) Melatonin—molecular biology, clinical and pharmaceutical approaches. IntechOpen
Hanagasi HA, Ayribas D, Baysal K, Emre M (2005) Mitochondrial complex I, II/III, and IV activities in familial and sporadic Parkinson’s disease. Int J Neurosci 115:479–493. https://doi.org/10.1080/00207450590523017
Hardeland R (2013) Melatonin and the theories of aging: a critical appraisal of melatonin’s role in antiaging mechanisms. J Pineal Res 55:325–356. https://doi.org/10.1111/jpi.12090
Hardeland R (2017a) Melatonin in healthy aging and longevity. In: Rattan S, Sharma R (eds) Hormones in ageing and longevity. Springer, Cham, pp 209–242
Hardeland R (2017b) Melatonin and the electron transport chain. Cell Mol Life Sci 74:3883–3896. https://doi.org/10.1007/s00018-017-2615-9
Hardeland R (2019) Aging, melatonin, and the pro- and anti-inflammatory networks. Int J Mol Sci 20:1223. https://doi.org/10.3390/ijms20051223
Hou Y, Dan X, Babbar M et al (2019) Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 15:565–581. https://doi.org/10.1038/s41582-019-0244-7
Huang W-J, Zhang X, Chen W-W (2016) Role of oxidative stress in Alzheimer’s disease. Biomed Rep 4:519–522. https://doi.org/10.3892/br.2016.630
Huang M, Liang Y, Chen H et al (2018) The role of fluoxetine in activating Wnt/β-catenin signaling and repressing β-amyloid production in an Alzheimer mouse model. Front Aging Neurosci 10:164. https://doi.org/10.3389/fnagi.2018.00164
Ikeno T, Nelson RJ (2015) Acute melatonin treatment alters dendritic morphology and circadian clock gene expression in the hippocampus of Siberian hamsters. Hippocampus 25:142–148. https://doi.org/10.1002/hipo.22358
Imai Y, Kobayashi Y, Inoshita T et al (2015) The Parkinson’s disease-associated protein kinase LRRK2 modulates notch signaling through the endosomal pathway. PLoS Genet 11:e1005503. https://doi.org/10.1371/journal.pgen.1005503
Jaiswal MK (2016) Riluzole but not melatonin ameliorates acute motor neuron degeneration and moderately inhibits SOD1-mediated excitotoxicity induced disrupted mitochondrial Ca2+ signaling in amyotrophic lateral sclerosis. Front Cell Neurosci 10:295. https://doi.org/10.3389/fncel.2016.00295
Jauhari A, Baranov SV, Suofu Y et al (2020) Melatonin inhibits cytosolic mitochondrial DNA–induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J Clin Investig 130:3124–3136. https://doi.org/10.1172/JCI135026
Jeong J-K, Park S-Y (2015) Melatonin regulates the autophagic flux via activation of alpha-7 nicotinic acetylcholine receptors. J Pineal Res 59:24–37. https://doi.org/10.1111/jpi.12235
Jeong J-K, Lee J-H, Moon J-H et al (2014) Melatonin-mediated β-catenin activation protects neuron cells against prion protein-induced neurotoxicity. J Pineal Res 57:427–434. https://doi.org/10.1111/jpi.12182
Jiang J, Liang S, Zhang J et al (2021) Melatonin ameliorates PM2.5-induced cardiac perivascular fibrosis through regulating mitochondrial redox homeostasis. J Pineal Res 70:e12686. https://doi.org/10.1111/jpi.12686
Jonas EA, Porter GA, Beutner G et al (2015) Cell death disguised: the mitochondrial permeability transition pore as the c-subunit of the F1FO ATP synthase. Pharmacol Res 99:382–392. https://doi.org/10.1016/j.phrs.2015.04.013
Kalliolia E, Silajdžić E, Nambron R et al (2014) Plasma melatonin is reduced in Huntington’s disease. Mov Disord 29:1511–1515. https://doi.org/10.1002/mds.26003
Kang J-Y, Xu M-M, Sun Y et al (2022) Melatonin attenuates LPS-induced pyroptosis in acute lung injury by inhibiting NLRP3-GSDMD pathway via activating Nrf2/HO-1 signaling axis. Int Immunopharmacol 109:108782. https://doi.org/10.1016/j.intimp.2022.108782
Kilic U, Kilic E, Tuzcu Z et al (2013) Melatonin suppresses cisplatin-induced nephrotoxicity via activation of Nrf-2/HO-1 pathway. Nutr Metab 10:7. https://doi.org/10.1186/1743-7075-10-7
Kondratov RV, Kondratova AA, Gorbacheva VY et al (2006) Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev 20:1868–1873. https://doi.org/10.1101/gad.1432206
Kondratov RV, Vykhovanets O, Kondratova AA, Antoch MP (2009) Antioxidant N-acetyl-L-cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging 1:979–987. https://doi.org/10.18632/aging.100113
Kubben N, Misteli T (2017) Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat Rev Mol Cell Biol 18:595–609. https://doi.org/10.1038/nrm.2017.68
Kumar Verma A, Singh S, Srivastava P, Ibrahim Rizvi S (2022) Melatonin stabilizes age-dependent alterations in erythrocyte membrane induced by “Artificial Light at Night” in a chronodisrupted model of rat. Gen Comp Endocrinol 316:113960. https://doi.org/10.1016/j.ygcen.2021.113960
Lahiri DK (1999) Melatonin affects the metabolism of the beta-amyloid precursor protein in different cell types. J Pineal Res 26:137–146. https://doi.org/10.1111/j.1600-079x.1999.tb00575.x
Lananna BV, Musiek ES (2020) The wrinkling of time: aging, inflammation, oxidative stress, and the circadian clock in neurodegeneration. Neurobiol Dis 139:104832. https://doi.org/10.1016/j.nbd.2020.104832
Lee JH, Yoon YM, Song K et al (2020) Melatonin suppresses senescence-derived mitochondrial dysfunction in mesenchymal stem cells via the HSPA1L–mitophagy pathway. Aging Cell 19:e13111. https://doi.org/10.1111/acel.13111
Lee YH, Park JY, Lee H et al (2021) Targeting mitochondrial metabolism as a strategy to treat senescence. Cells 10:3003. https://doi.org/10.3390/cells10113003
Lemaitre H, Goldman AL, Sambataro F et al (2012) Normal age-related brain morphometric changes: nonuniformity across cortical thickness, surface area and gray matter volume? Neurobiol Aging 33:617. https://doi.org/10.1016/j.neurobiolaging.2010.07.013
Leston J, Harthé C, Brun J et al (2010) Melatonin is released in the third ventricle in humans. A study in movement disorders. Neurosci Lett 469:294–297. https://doi.org/10.1016/j.neulet.2009.12.008
Leyane TS, Jere SW, Houreld NN (2022) Oxidative stress in ageing and chronic degenerative pathologies: molecular mechanisms involved in counteracting oxidative stress and chronic inflammation. IJMS 23:7273. https://doi.org/10.3390/ijms23137273
Li T, Chen ZJ (2018) The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215:1287–1299. https://doi.org/10.1084/jem.20180139
Li Y, Zhang J, Wan J et al (2020) Melatonin regulates Aβ production/clearance balance and Aβ neurotoxicity: a potential therapeutic molecule for Alzheimer’s disease. Biomed Pharmacother 132:110887. https://doi.org/10.1016/j.biopha.2020.110887
Li Z, Zhang Z, Ren Y et al (2021) Aging and age-related diseases: from mechanisms to therapeutic strategies. Biogerontology 22:165–187. https://doi.org/10.1007/s10522-021-09910-5
Limón-Pacheco JH, Gonsebatt ME (2010) The glutathione system and its regulation by neurohormone melatonin in the central nervous system. Cent Nerv Syst Agents Med Chem 10:287–297. https://doi.org/10.2174/187152410793429683
Lin Q, Chen J, Gu L et al (2021) New insights into mitophagy and stem cells. Stem Cell Res Ther 12:452. https://doi.org/10.1186/s13287-021-02520-5
Lister JP, Barnes CA (2009) Neurobiological changes in the hippocampus during normative aging. Arch Neurol 66:829–833. https://doi.org/10.1001/archneurol.2009.125
Liu F, Ng TB (2000) Effect of pineal indoles on activities of the antioxidant defense enzymes superoxide dismutase, catalase, and glutathione reductase, and levels of reduced and oxidized glutathione in rat tissues. Biochem Cell Biol 78:447–453. https://doi.org/10.1139/o00-018
Luo F, Sandhu AF, Rungratanawanich W et al (2020) Melatonin and autophagy in aging-related neurodegenerative diseases. Int J Mol Sci 21:7174. https://doi.org/10.3390/ijms21197174
Madabhushi R, Pan L, Tsai L-H (2014) DNA damage and its links to neurodegeneration. Neuron 83:266–282. https://doi.org/10.1016/j.neuron.2014.06.034
Maharaj DS, Maharaj H, Antunes EM et al (2005) 6-Hydroxymelatonin protects against quinolinic-acid-induced oxidative neurotoxicity in the rat hippocampus. J Pharm Pharmacol 57:877–881. https://doi.org/10.1211/0022357056424
Majidinia M, Sadeghpour A, Mehrzadi S et al (2017) Melatonin: a pleiotropic molecule that modulates DNA damage response and repair pathways. J Pineal Res 63:e12416. https://doi.org/10.1111/jpi.12416
Majidinia M, Reiter RJ, Shakouri SK, Yousefi B (2018) The role of melatonin, a multitasking molecule, in retarding the processes of ageing. Ageing Res Rev 47:198–213. https://doi.org/10.1016/j.arr.2018.07.010
Mammucari C, Rizzuto R (2010) Signaling pathways in mitochondrial dysfunction and aging. Mech Ageing Dev 131:536–543. https://doi.org/10.1016/j.mad.2010.07.003
Manchester LC, Coto-Montes A, Boga JA et al (2015) Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J Pineal Res 59:403–419. https://doi.org/10.1111/jpi.12267
Mannino G, Pernici C, Serio G et al (2021) Melatonin and phytomelatonin: chemistry, biosynthesis, metabolism, distribution and bioactivity in plants and animals—an overview. Int J Mol Sci 22:9996. https://doi.org/10.3390/ijms22189996
Marathe S, Jaquet M, Annoni J-M, Alberi L (2017) Jagged1 is altered in Alzheimer’s disease and regulates spatial memory processing. Front Cell Neurosci 11:220. https://doi.org/10.3389/fncel.2017.00220
Markus RP, Sousa KS, da Silveira C-M et al (2021) Possible role of pineal and extra-pineal melatonin in surveillance, immunity, and first-line defense. Int J Mol Sci 22:12143. https://doi.org/10.3390/ijms222212143
Martín Giménez VM, de Las Heras N, Lahera V et al (2022) Melatonin as an anti-aging therapy for age-related cardiovascular and neurodegenerative diseases. Front Aging Neurosci 14:888292. https://doi.org/10.3389/fnagi.2022.888292
Matsubara E, Bryant-Thomas T, Pacheco Quinto J et al (2003) Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J Neurochem 85:1101–1108. https://doi.org/10.1046/j.1471-4159.2003.01654.x
Mattam U, Jagota A (2014) Differential role of melatonin in restoration of age-induced alterations in daily rhythms of expression of various clock genes in suprachiasmatic nucleus of male Wistar rats. Biogerontology 15:257–268. https://doi.org/10.1007/s10522-014-9495-2
Mattam U, Jagota A (2015) Daily rhythms of serotonin metabolism and the expression of clock genes in suprachiasmatic nucleus of rotenone-induced Parkinson’s disease male Wistar rat model and effect of melatonin administration. Biogerontology 16:109–123. https://doi.org/10.1007/s10522-014-9541-0
Mattis J, Sehgal A (2016) Circadian rhythms, sleep, and disorders of aging. Trends Endocrinol Metab 27:192–203. https://doi.org/10.1016/j.tem.2016.02.003
Mayo JC, Sainz RM, González Menéndez P et al (2017) Melatonin and sirtuins: a “not-so unexpected” relationship. J Pineal Res 62:e12391. https://doi.org/10.1111/jpi.12391
McHugh D, Gil J (2018) Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol 217:65–77. https://doi.org/10.1083/jcb.201708092
Mehrzadi S, Pourhanifeh MH, Mirzaei A et al (2021) An updated review of mechanistic potentials of melatonin against cancer: pivotal roles in angiogenesis, apoptosis, autophagy, endoplasmic reticulum stress and oxidative stress. Cancer Cell Int 21:188. https://doi.org/10.1186/s12935-021-01892-1
Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G (2016) Therapies for prevention and treatment of Alzheimer’s disease. Biomed Res Int 2016:2589276. https://doi.org/10.1155/2016/2589276
Mochel F, Haller RG (2011) Energy deficit in Huntington disease: why it matters. J Clin Invest 121:493–499. https://doi.org/10.1172/JCI45691
Monayo SM, Liu X (2022) The prospective application of melatonin in treating epigenetic dysfunctional diseases. Front Pharmacol 13:867500. https://doi.org/10.3389/fphar.2022.867500
Montilla P, Cruz A, Padillo FJ et al (2001) Melatonin versus vitamin E as protective treatment against oxidative stress after extra-hepatic bile duct ligation in rats. J Pineal Res 31(2):138–144. https://doi.org/10.1034/j.1600-079x.2001.310207.x
Morioka N, Okatani Y, Wakatsuki A (1999) Melatonin protects against age-related DNA damage in the brains of female senescence-accelerated mice. J Pineal Res 27:202–209. https://doi.org/10.1111/j.1600-079x.1999.tb00616.x
Mu S, Lin E, Liu B et al (2014) Melatonin reduces projection neuronal injury induced by 3-nitropropionic acid in the rat striatum. Neurodegener Dis 14:139–150. https://doi.org/10.1159/000365891
Musiek ES, Xiong DD, Holtzman DM (2015) Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp Mol Med 47:e148–e148. https://doi.org/10.1038/emm.2014.121
Nakamura TJ, Nakamura W, Tokuda IT et al (2015) Age-related changes in the circadian system unmasked by constant conditions. eNeuro 2(4):15. https://doi.org/10.1523/ENEURO.0064-15.2015
Nam E, Lee SM, Koh SE et al (2005) Melatonin protects against neuronal damage induced by 3-nitropropionic acid in rat striatum. Brain Res 1046:90–96. https://doi.org/10.1016/j.brainres.2005.03.053
Nassar E, Mulligan C, Taylor L et al (2007) Effects of a single dose of N-Acetyl-5-methoxytryptamine (Melatonin) and resistance exercise on the growth hormone/IGF-1 axis in young males and females. J Int Soc Sports Nutr 4:14. https://doi.org/10.1186/1550-2783-4-14
Ng TB, Liu F, Zhao L (2000) Antioxidative and free radical scavenging activities of pineal indoles. J Neural Transm 107:1243–1251. https://doi.org/10.1007/s007020070014
Ng KY, Leong MK, Liang H, Paxinos G (2017) Melatonin receptors: distribution in mammalian brain and their respective putative functions. Brain Struct Funct 222:2921–2939. https://doi.org/10.1007/s00429-017-1439-6
Niles LP, Armstrong KJ, Rincón Castro LM et al (2004) Neural stem cells express melatonin receptors and neurotrophic factors: colocalization of the MT1 receptor with neuronal and glial markers. BMC Neurosci 5:41. https://doi.org/10.1186/1471-2202-5-41
Oishi A, Cecon E, Jockers R (2018) Melatonin receptor signaling: impact of receptor oligomerization on receptor function. In: International Review of Cell and Molecular Biology. Elsevier, pp 59–77
Okatani Y, Wakatsuki A, Reiter RJ, Miyahara Y (2002) Melatonin reduces oxidative damage of neural lipids and proteins in senescence-accelerated mouse. Neurobiol Aging 23:639–644. https://doi.org/10.1016/s0197-4580(02)00005-2
Olcese JM, Cao C, Mori T et al (2009) Protection against cognitive deficits and markers of neurodegeneration by long-term oral administration of melatonin in a transgenic model of Alzheimer disease. J Pineal Res 47:82–96. https://doi.org/10.1111/j.1600-079X.2009.00692.x
Oliva CA, Montecinos-Oliva C, Inestrosa NC (2018) Wnt signaling in the central nervous system: new insights in health and disease. Prog Mol Biol Transl Sci 153:81–130. https://doi.org/10.1016/bs.pmbts.2017.11.018
Ono K, Mochizuki H, Ikeda T et al (2012) Effect of melatonin on α-synuclein self-assembly and cytotoxicity. Neurobiol Aging 33:2172–2185. https://doi.org/10.1016/j.neurobiolaging.2011.10.015
Pajares M, Rojo IA, Manda G et al (2020) Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells 9:E1687. https://doi.org/10.3390/cells9071687
Peng C-X, Hu J, Liu D et al (2013) Disease-modified glycogen synthase kinase-3β intervention by melatonin arrests the pathology and memory deficits in an Alzheimer’s animal model. Neurobiol Aging 34:1555–1563. https://doi.org/10.1016/j.neurobiolaging.2012.12.010
Petrosillo G, Moro N, Ruggiero FM, Paradies G (2009) Melatonin inhibits cardiolipin peroxidation in mitochondria and prevents the mitochondrial permeability transition and cytochrome c release. Free Radic Biol Med 47:969–974. https://doi.org/10.1016/j.freeradbiomed.2009.06.032
Poewe W, Seppi K, Tanner CM et al (2017) Parkinson disease. Nat Rev Dis Primers 3:17013. https://doi.org/10.1038/nrdp.2017.13
Polimeni G, Esposito E, Bevelacqua V et al (2014) Role of melatonin supplementation in neurodegenerative disorders. Front Biosci 19:429–446. https://doi.org/10.2741/4217
Pollari E, Goldsteins G, Bart G et al (2014) The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front Cell Neurosci 8:131. https://doi.org/10.3389/fncel.2014.00131
Ramirez Reyes JMJ, Cuesta R, Pause A (2021) Folliculin: a regulator of transcription through AMPK and mTOR signaling pathways. Front Cell Dev Biol 9:667311. https://doi.org/10.3389/fcell.2021.667311
Ramis MR, Esteban S, Miralles A et al (2015) Caloric restriction, resveratrol and melatonin: role of SIRT1 and implications for aging and related-diseases. Mech Ageing Dev 146–148:28–41. https://doi.org/10.1016/j.mad.2015.03.008
Rao G, Verma R, Mukherjee A et al (2016) Melatonin alleviates hyperthyroidism induced oxidative stress and neuronal cell death in hippocampus of aged female golden hamster, Mesocricetus auratus. Exp Gerontol 82:125–130. https://doi.org/10.1016/j.exger.2016.06.014
Rattan SIS (2022) Physiological hormesis and hormetins in biogerontology. Curr Opin Toxicol 29:19–24. https://doi.org/10.1016/j.cotox.2022.01.001
Reina M, Castañeda-Arriaga R, Perez-Gonzalez A et al (2018) A computer-assisted systematic search for melatonin derivatives with high potential as antioxidants. Melatonin Res 1:27–58. https://doi.org/10.32794/mr11250003
Reiter RJ, Manchester LC, Tan D-X (2010) Neurotoxins: free radical mechanisms and melatonin protection. Curr Neuropharmacol 8:194–210. https://doi.org/10.2174/157015910792246236
Reiter RJ, Tan D-X, Galano A (2014) Melatonin reduces lipid peroxidation and membrane viscosity. Front Physiol 5:377. https://doi.org/10.3389/fphys.2014.00377
Reiter RJ, Mayo JC, Tan D-X et al (2016) Melatonin as an antioxidant: under promises but over delivers. J Pineal Res 61:253–278. https://doi.org/10.1111/jpi.12360
Reiter RJ, Rosales-Corral S, Tan DX et al (2017) Melatonin as a mitochondria-targeted antioxidant: one of evolution’s best ideas. Cell Mol Life Sci 74:3863–3881. https://doi.org/10.1007/s00018-017-2609-7
Reiter RJ, Tan DX, Rosales-Corral S et al (2018) Mitochondria: central organelles for melatonin’s antioxidant and anti-aging actions. Molecules 23:E509. https://doi.org/10.3390/molecules23020509
Reiter RJ, Ma Q, Sharma R (2020) Melatonin in mitochondria: mitigating clear and present dangers. Physiology 35:86–95. https://doi.org/10.1152/physiol.00034.2019
Reiter RJ, Sharma R, Ma Q (2021) Switching diseased cells from cytosolic aerobic glycolysis to mitochondrial oxidative phosphorylation: a metabolic rhythm regulated by melatonin? J Pineal Res 70:e12677. https://doi.org/10.1111/jpi.12677
Rudnitskaya EA, Muraleva NA, Maksimova KY et al (2015) Melatonin attenuates memory impairment, amyloid-β accumulation, and neurodegeneration in a rat model of sporadic Alzheimer’s disease. J Alzheimers Dis 47:103–116. https://doi.org/10.3233/JAD-150161
Ruiz A, Matute C, Alberdi E (2010) Intracellular Ca2+ release through ryanodine receptors contributes to AMPA receptor-mediated mitochondrial dysfunction and ER stress in oligodendrocytes. Cell Death Dis 1:e54. https://doi.org/10.1038/cddis.2010.31
Saftig P, Lichtenthaler SF (2015) The alpha secretase ADAM10: a metalloprotease with multiple functions in the brain. Prog Neurobiol 135:1–20. https://doi.org/10.1016/j.pneurobio.2015.10.003
Salminen A, Kaarniranta K (2010) Insulin/IGF-1 paradox of aging: regulation via AKT/IKK/NF-kappaB signaling. Cell Signal 22:573–577. https://doi.org/10.1016/j.cellsig.2009.10.006
Sarlak G, Htoo HH, Hernandez J-F et al (2016) Sox2 functionally interacts with βAPP, the βAPP intracellular domain and ADAM10 at a transcriptional level in human cells. Neuroscience 312:153–164. https://doi.org/10.1016/j.neuroscience.2015.11.022
Sarti P, Magnifico M, Altieri F et al (2013) New evidence for cross talk between melatonin and mitochondria mediated by a circadian-compatible interaction with nitric oxide. IJMS 14:11259–11276. https://doi.org/10.3390/ijms140611259
Saso L, Gürer-Orhan H, Stepanić V (2020) Modulators of oxidative stress: chemical and pharmacological aspects. Antioxidants 9:657. https://doi.org/10.3390/antiox9080657
Seabra ML, Bignotto M, Pinto LR, Tufik S (2000) Randomized, double-blind clinical trial, controlled with placebo, of the toxicology of chronic melatonin treatment. J Pineal Res 29:193–200. https://doi.org/10.1034/j.1600-0633.2002.290401.x
Shao S, Wang G-L, Raymond C et al (2017) Activation of Sonic hedgehog signal by Purmorphamine, in a mouse model of Parkinson’s disease, protects dopaminergic neurons and attenuates inflammatory response by mediating PI3K/AKt signaling pathway. Mol Med Rep 16:1269–1277. https://doi.org/10.3892/mmr.2017.6751
Shirinzadeh H, Eren B, Gurer-Orhan H et al (2010) Novel indole-based analogs of melatonin: synthesis and in vitro antioxidant activity studies. Molecules 15:2187–2202. https://doi.org/10.3390/molecules15042187
Shukla M, Govitrapong P, Boontem P et al (2017) Mechanisms of melatonin in alleviating Alzheimer’s disease. Curr Neuropharmacol 15:1010–1031. https://doi.org/10.2174/1570159X15666170313123454
Singh S, Mishra A, Bharti S et al (2018) Glycogen synthase kinase-3β regulates equilibrium between neurogenesis and gliogenesis in rat model of Parkinson’s disease: a crosstalk with wnt and notch signaling. Mol Neurobiol 55:6500–6517. https://doi.org/10.1007/s12035-017-0860-4
Song J (2019) Pineal gland dysfunction in Alzheimer’s disease: relationship with the immune-pineal axis, sleep disturbance, and neurogenesis. Mol Neurodegener 14:28. https://doi.org/10.1186/s13024-019-0330-8
Sotthibundhu A, Phansuwan-Pujito P, Govitrapong P (2010) Melatonin increases proliferation of cultured neural stem cells obtained from adult mouse subventricular zone. J Pineal Res 49:291–300. https://doi.org/10.1111/j.1600-079X.2010.00794.x
Srinivasan V, Spence DW, Pandi-Perumal SR et al (2011) Melatonin in mitochondrial dysfunction and related disorders. Int J Alzheimer’s Dis 2011:1–16. https://doi.org/10.4061/2011/326320
Sumsuzzman DMd, Choi J, Jin Y, Hong Y (2021) Neurocognitive effects of melatonin treatment in healthy adults and individuals with Alzheimer’s disease and insomnia: a systematic review and meta-analysis of randomized controlled trials. Neurosci Biobehav Rev 127:459–473. https://doi.org/10.1016/j.neubiorev.2021.04.034
Sun L, Wu J, Du F et al (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. https://doi.org/10.1126/science.1232458
Sun X, Ran D, Zhao X et al (2016) Melatonin attenuates hLRRK2-induced sleep disturbances and synaptic dysfunction in a Drosophila model of Parkinson’s disease. Mol Med Rep 13:3936–3944. https://doi.org/10.3892/mmr.2016.4991
Suofu Y, Li W, Jean-Alphonse FG et al (2017) Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc Natl Acad Sci USA 114:E7997–E8006. https://doi.org/10.1073/pnas.1705768114
Suzen S (2013) Melatonin and synthetic analogs as antioxidants. Curr Drug Deliv 10:71–75. https://doi.org/10.2174/1567201811310010013
Suzen S, Bozkaya P, Coban T, Nebioğu D (2006) Investigation of the in vitro antioxidant behaviour of some 2-phenylindole derivatives: discussion on possible antioxidant mechanisms and comparison with melatonin. J Enzyme Inhib Med Chem 21:405–411. https://doi.org/10.1080/14756360500381210
Suzen S, Cihaner SS, Coban T (2012) Synthesis and comparison of antioxidant properties of indole-based melatonin analogue indole amino acid derivatives. Chem Biol Drug Des 79:76–83. https://doi.org/10.1111/j.1747-0285.2011.01216.x
Süzen S, Atayik MC, Sirinzade H et al (2022) Melatonin and redox homeostasis. Melatonin Res 5:304–324. https://doi.org/10.32794/mr112500134
Tabrizi SJ, Ghosh R, Leavitt BR (2019) Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron 101:801–819. https://doi.org/10.1016/j.neuron.2019.01.039
Tabrizi SJ, Flower MD, Ross CA, Wild EJ (2020) Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol 16:529–546. https://doi.org/10.1038/s41582-020-0389-4
Tan DX, Manchester LC, Reiter RJ et al (1998) Melatonin protects hippocampal neurons in vivo against kainic acid-induced damage in mice. J Neurosci Res 54:382–389. https://doi.org/10.1002/(SICI)1097-4547(19981101)54:3%3c382::AID-JNR9%3e3.0.CO;2-Y
Tan BL, Norhaizan ME, Liew W-P-P, Sulaiman Rahman H (2018) Antioxidant and oxidative stress: a mutual interplay in age-related diseases. Front Pharmacol 9:1162. https://doi.org/10.3389/fphar.2018.01162
Tan HY, Ng KY, Koh RY, Chye SM (2020) Pharmacological effects of melatonin as neuroprotectant in rodent model: a review on the current biological evidence. Cell Mol Neurobiol 40:25–51. https://doi.org/10.1007/s10571-019-00724-1
Tjahjono E, Kirienko DR, Kirienko NV (2022) The emergent role of mitochondrial surveillance in cellular health. Aging Cell 21:e13710. https://doi.org/10.1111/acel.13710
Tocharus C, Puriboriboon Y, Junmanee T et al (2014) Melatonin enhances adult rat hippocampal progenitor cell proliferation via ERK signaling pathway through melatonin receptor. Neuroscience 275:314–321. https://doi.org/10.1016/j.neuroscience.2014.06.026
Túnez I, Montilla P, Del Carmen MM et al (2004) Protective effect of melatonin on 3-nitropropionic acid-induced oxidative stress in synaptosomes in an animal model of Huntington’s disease. J Pineal Res 37:252–256. https://doi.org/10.1111/j.1600-079X.2004.00163.x
van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LAB (2011) Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol 32:110–116. https://doi.org/10.1016/j.it.2011.01.003
Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W (2006) The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochem Biophys Acta 1762:1068–1082. https://doi.org/10.1016/j.bbadis.2006.05.002
Venegas C, García JA, Escames G et al (2012) Extrapineal melatonin: analysis of its subcellular distribution and daily fluctuations. J Pineal Res 52:217–227. https://doi.org/10.1111/j.1600-079X.2011.00931.x
Verma AK, Singh S, Rizvi SI (2019) Redox homeostasis in a rodent model of circadian disruption: effect of melatonin supplementation. Gen Comp Endocrinol. https://doi.org/10.1016/j.ygcen.2019.04.016
Verma AK, Garg G, Singh S, Rizvi SI (2020a) Melatonin protects against membrane alterations affected by ‘Artificial Light at Night’ in a circadian-disrupted model of rat. Biol Rhythm Res. https://doi.org/10.1080/09291016.2020.1741265
Verma AK, Singh S, Rizvi SI (2020b) Age-dependent altered redox homeostasis in the chronodisrupted rat model and moderation by melatonin administration. Chronobiol Int. https://doi.org/10.1080/07420528.2020.1792483
Verma AK, Singh S, Garg G, Rizvi SI (2021a) Melatonin exerts neuroprotection in a chronodisrupted rat model through reduction in oxidative stress and modulation of autophagy. Chronobiol Int. https://doi.org/10.1080/07420528.2021.1966025
Verma AK, Singh S, Rizvi SI (2021b) Age-dependent effect of continuous ‘artificial light at night’ on circadian rhythm in male rats: neuroprotective role of melatonin. Biogerontology. https://doi.org/10.1007/s10522-021-09933-y
Videnovic A, Noble C, Reid KJ et al (2014) Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurol 71:463–469. https://doi.org/10.1001/jamaneurol.2013.6239
Vincent B (2018) Protective roles of melatonin against the amyloid-dependent development of Alzheimer’s disease: a critical review. Pharmacol Res 134:223–237. https://doi.org/10.1016/j.phrs.2018.06.011
Vorobyeva AG, Saunders AJ (2018) Amyloid-β interrupts canonical Sonic hedgehog signaling by distorting primary cilia structure. Cilia 7:5. https://doi.org/10.1186/s13630-018-0059-y
Vriend J, Reiter RJ (2015) The Keap1-Nrf2-antioxidant response element pathway: a review of its regulation by melatonin and the proteasome. Mol Cell Endocrinol 401:213–220. https://doi.org/10.1016/j.mce.2014.12.013
Wang X (2009) The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci Ther 15:345–357. https://doi.org/10.1111/j.1755-5949.2009.00105.x
Wang J, Wang Z (2006) Role of melatonin in Alzheimer-like neurodegeneration. Acta Pharmacol Sin 27:41–49. https://doi.org/10.1111/j.1745-7254.2006.00260.x
Wang X, Sirianni A, Pei Z et al (2011) The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. J Neurosci 31:14496–14507. https://doi.org/10.1523/JNEUROSCI.3059-11.2011
Wang J, Wang X, He Y et al (2019) Antioxidant and pro-oxidant activities of melatonin in the presence of copper and polyphenols in vitro and in vivo. Cells 8:E903. https://doi.org/10.3390/cells8080903
Wasdell MB, Jan JE, Bomben MM et al (2008) A randomized, placebo-controlled trial of controlled release melatonin treatment of delayed sleep phase syndrome and impaired sleep maintenance in children with neurodevelopmental disabilities. J Pineal Res 44:57–64. https://doi.org/10.1111/j.1600-079X.2007.00528.x
Weishaupt JH, Bartels C, Pölking E et al (2006) Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J Pineal Res 41:313–323. https://doi.org/10.1111/j.1600-079X.2006.00377.x
Welz P-S, Benitah SA (2020) Molecular connections between circadian clocks and aging. J Mol Biol 432:3661–3679. https://doi.org/10.1016/j.jmb.2019.12.036
West AP, Shadel GS (2017) Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17:363–375. https://doi.org/10.1038/nri.2017.21
Wishart TM, Parson SH, Gillingwater TH (2006) Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol 65:733–739. https://doi.org/10.1097/01.jnen.0000228202.35163.c4
Xu C, He Z, Li J (2022) Melatonin as a potential neuroprotectant: mechanisms in subarachnoid hemorrhage-induced early brain injury. Front Aging Neurosci 14:899678. https://doi.org/10.3389/fnagi.2022.899678
Xue F, Shi C, Chen Q et al (2017) Melatonin mediates protective effects against kainic acid-induced neuronal death through safeguarding ER stress and mitochondrial disturbance. Front Mol Neurosci 10:49. https://doi.org/10.3389/fnmol.2017.00049
Zhai Z, Xie D, Qin T et al (2022) Effect and mechanism of exogenous melatonin on cognitive deficits in animal models of Alzheimer’s disease: a systematic review and meta-analysis. Neuroscience 505:91–110. https://doi.org/10.1016/j.neuroscience.2022.09.012
Zhang H-M, Zhang Y (2014) Melatonin: a well-documented antioxidant with conditional pro-oxidant actions. J Pineal Res 57:131–146. https://doi.org/10.1111/jpi.12162
Zhang Y, Cook A, Kim J et al (2013) Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 55:26–35. https://doi.org/10.1016/j.nbd.2013.03.008
Zhang L, Li F, Su X et al (2019) Melatonin prevents lung injury by regulating apelin 13 to improve mitochondrial dysfunction. Exp Mol Med 51:1–12. https://doi.org/10.1038/s12276-019-0273-8
Zisapel N (2018) New perspectives on the role of melatonin in human sleep, circadian rhythms and their regulation: melatonin in human sleep and circadian rhythms. Br J Pharmacol 175:3190–3199. https://doi.org/10.1111/bph.14116
Acknowledgements
AKV gratefully acknowledges the Council of Scientific and Industrial Research (CSIR), Government of India for providing financial support in the form of a Senior Research Fellowship (09/001(0426)/2018-EMR-I). The Department of Biotechnology, Government of India, provided financial support for this work through a project of “Research Resources, Service Facilities, and Platforms”. The FIST Grant of DST, New Delhi, and SAP DRS I from the UGC, India, both provide funding for the Department of Biochemistry.
Author information
Authors and Affiliations
Contributions
AKV prepared the first draft of the manuscript. The figures were prepared by AKV and SS. SIR assisted with subsequent drafts of the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Verma, A.K., Singh, S. & Rizvi, S.I. Therapeutic potential of melatonin and its derivatives in aging and neurodegenerative diseases. Biogerontology 24, 183–206 (2023). https://doi.org/10.1007/s10522-022-10006-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-022-10006-x