
Abstract
Taura syndrome in shrimp, caused by Taura syndrome virus (TSV), is a disease listed by the World Organisation for Animal Health (OIE) that has been identified in five continents, causing serious economic losses in years. Due to the lack of specific drugs available to treat Taura syndrome, an effective TSV detection method is urgently needed to ensure pathogen prediction and disease control. In this study, we developed and evaluated an isothermal enzymatic recombinase amplification–based assay (TSV-ERA) that was able to diagnose TSV within 20 min at an optimal temperature of 42 °C. In the reaction system, the optimal concentrations of the primer and the probe were 500 nM and 120 nM, respectively. When the plasmid standards were used as a detection template, the check-out time of TSV-ERA was 12 min (21.76 ± 0.60 Ct), and the limit of detection (LOD) was 3.6 × 101 copies/µL. Compared to the sensitivity of the TSV detection method recommended by the OIE, TSV-ERA was equal to real-time PCR (qPCR) and higher than one-step PCR. This assay was highly specific for TSV and had no cross-reactivity with Enterocytozoon hepatopenaei (EHP), white spot syndrome virus (WSSV), Vibrio parahaemolyticus strain causing acute hepatopancreatic necrosis disease (VpAHPND), infectious hypodermal and hematopoietic necrosis virus (IHHNV), and the genomic DNA of specific pathogen-free (SPF) shrimp. Compared with the common detection methods, the TSV-ERA assay was able to shorten the detection time and decrease the detection temperature while sustaining accurate sensitivity. Based on these results, our TSV-ERA detection method is simple, rapid, and effective, and has great potential in the detection of TSV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Shrimp is one of the most valuable marine culture species globally, constituting approximately 61% of total global crustacean aquaculture production and contributing to economic growth in many developing countries (FAO 2020; Prochaska et al. 2022; Rob 2020). In recent years, increased shrimp farming has also been accompanied by outbreaks of various diseases, resulting in huge economic losses (Arulmoorthy et al. 2020; Thitamadee et al. 2016). Among these diseases, various viruses can cause more serious damage than bacteria- and fungi-based diseases (Lee et al. 2022). Taura syndrome virus (TSV) is an RNA virus; along with white spot syndrome virus (WSSV) and yellow head virus (YHV), it is one of the most dangerous shrimp viruses, as it has severely disrupted shrimp aquaculture in the past 20 years. TSV infects ectoderm- and mesoderm-derived tissues, especially hematopoietic tissue, subepidermal connective tissue, epidermal epithelium, lymphoid organs, and antennal glands. Shrimp infected with TSV are characterized by irregular black spots and reddish body color, in addition to anorexia, lethargy, soft cuticle, unstable swimming behavior, flaccid body, and opaque musculature, with a mortality rate of 60–90% (Dhar et al. 2004; Poulos et al. 2006; Tang et al. 2004). TSV was first reported in Ecuador in June 1992, and it was spread across Asia by transboundary shrimp transportation, while new strains continued to emerge. In 2000, TSV was listed as a designated disease by the World Organisation for Animal Health (OIE), especially in America and Asia, where it is widespread and difficult to control (George et al. 2011; Thitamadee et al. 2016). However, there is a lack of specific drugs for the treatment of TSV. Therefore, it is critical to establish fast and sensitive TSV detection technology to ensure pathogen prediction and disease control.
Currently, detection methods of TSV mainly include immunological detection and molecular-based detection, such as reverse transcriptase polymerase chain reaction (RT-PCR) (Nunan et al. 1998), nested RT-PCR, real-time PCR (qPCR), and so on. It had been reported that immunological assays with complex procedures failed to detect several isolates of TSV (Erickson et al. 2002, 2005; Robles-Sikisaka et al. 2002), and were usually less sensitive than in situ hybridization (ISH) (Poulos et al. 2008). Although PCR-based methods for pathogen diagnosis are associated with higher sensitivity and specificity, the reaction temperature must be precisely controlled by expensive equipment. Although isothermal nucleic acid–based amplification (NASBA) does not require a thermal circulator, it was found to be impractical for pond-side TSV detection due to the long reaction time (Teng et al. 2006). While the reverse transcription loop-mediated isothermal amplification (RT-LAMP) reaction was able to complete detection at 60–65 ℃ for 30 min through a relatively simple operation, non-specific amplification occasionally occurred (Kiatpathomchai et al. 2007, 2008; Phromjai et al. 2015; Sappat et al. 2011). Moreover, the LAMP reaction system requires multiple primers, the design of which is complex and prone to the risk of mismatch. In brief, it is particularly vital to establish a practical and effective diagnostic method for the pre-detection and management control of TSV in remote and underdeveloped areas.
Recombinase polymerase amplification (RPA) assay is a new type of nucleic acid isothermal amplification technology which has been increasingly applied in scientific research because of its outstanding advantages such as fast reaction speed, low reaction temperature, and accurate detection results. Enzymatic recombination amplification (ERA) is an improved RPA technology (Piepenburg et al. 2006; Zhao et al. 2015) which modifies and optimizes tool enzymes such as DNA polymerase and endonuclease derived from bacteria, viruses, and phages. Without the use of any thermal circulators, ERA can be completed in 20 min at 25–45 ℃ (Bergeron et al. 2016; Boonbanjong et al. 2022; Mayboroda et al. 2018). ERA has been proven to be more accurate, simpler to operate, and less time-consuming than RT-PCR, nested RT-PCR, RT-LAMP, and other commonly used detection methods. Recently, ERA has been applied to the detection of multi-pathogens, such as WSSV (Zhang et al. 2023a), Vibrio parahaemolyticus strain causing acute hepatopancreatic necrosis disease (VpAHPND) (Zhou et al. 2023), Mycobacterium tuberculosis, porcine epidemic diarrhea virus (PEDV), Porcine circovirus type 3 (PCV3), beer spoilage bacteria, and even SARS-CoV-2 (Liu et al. 2021a, b; Yang et al. 2021; Zhang et al. 2021; Meng et al. 2021; Xia and Chen 2020), suggesting that ERA has potential research value and application prospects in the field of detection. In this study, we developed an easy, rapid, and effective ERA-based assay (TSV-ERA) for TSV detection. Through the screening of primers, the optimization of reaction conditions, and the investigation of sensitivity and specificity, TSV-ERA was finally established. It has the advantages of reduced time consumption and high specificity, provides results comparable to those of conventional one-step PCR and qPCR prescribed by the OIE, and is suitable for field detection to prevent disease outbreaks.
Materials and methods
Pathogen samples and genomic DNA extraction
The common pathogens used in this study, including Enterocytozoon hepatopenaei (EHP), WSSV, VpAHPND, and infectious hypodermal and hematopoietic necrosis virus (IHHNV), are preserved in the Key Laboratory of Tropical Aquatic Germplasm of Hainan Province of Sanya Oceanographic Institution, Ocean University of China, China. Specific pathogen-free (SPF) shrimp of Penaeus vannamei were provided by a farm in Wenchang, Hainan, China, and confirmed with either our previously established pathogen detection method or commercial kits (Li et al. 2023; Xu et al. 2022, 2023; Zhang et al. 2023a, b, 2024; Zhou et al. 2023). According to the manufacturer’s instructions, genomic DNA from 30 mg hepatopancreases of shrimp was extracted using the nucleic acid release agent (DNA) kit (NR201, GenDx, China). The tissue to be tested was added to a 1.5-mL centrifuge tube with 150 µl of nucleic acid releasing agent, mixed via shaking, and added to a 95 °C water bath for 3 min. The mixed lysate was directly used for amplification. The DNA concentrations were measured using a NanoDrop One spectrophotometer (Thermo Fisher Scientific, USA). All extracted DNA samples were diluted to 10 ng/µL and then frozen at −20 ℃.
Construction of plasmid standard
A 802-bp TSV gene fragment named TSV-1 (GenBank accession no. AF277675, nt 6810–7611) and a 272-bp TSV gene fragment named TSV-2 (GenBank accession no. AF277675, nt 904–1175) were synthesized and ligated into the pUC57-kan vector, which were used as plasmid standards. The gene fragment was synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The plasmids were transferred into Escherichia coli Trans1-T1 competent cells (CB501, TransGen, China), and then three independent clones were selected for confirmation by sequencing. Positive recombinants were extracted with an EasyPure HiPure Plasmid MiniPrep Kit (EM121, Tiangen, China), and the concentration and purity of the plasmid were measured with a NanoDrop One spectrophotometer (Thermo Fisher Scientific, USA). The copy number of the recombinant plasmid was calculated using the following equation: plasmid DNA copy number (copies/µL) = [concentration (ng/µL) × 10−9 × 6.02 × 1023 (copies/mol)] / [fragment size (bp) × 660 (g/mol/bp)]. The concentration of the TSV-1 plasmid DNA used for the ERA assay and one-step PCR was 155.3 ng/µL and equal to 3.6 × 1010 copies/µL. The concentration of the TSV-2 plasmid DNA used for qPCR was 285.2 ng/µL and equal to 7.8 × 1010 copies/µL.
Primer and probe design
The primers and probe of TSV-ERA were designed using Primer Premier 5.0 according to the ERA assay guide, based on the TSV capsid protein 2 gene (VP2) (GenBank accession no. AF277675, nt 6953–7611). The primer length of ERA was 28–35 bases. The Tm value was between 50 and 70 ℃. GC content should be between 30 and 70% to avoid the formation of primer dimers and a hairpin structure. The probe was designed based on the sequences between primers. The probe contained 46–52 nucleotides, with no less than 30 nucleotides at the 5′ end of THF and no less than 15 nucleotides at the 3′ end. The optimal primer set for the reaction was determined using comparison tests and used to validate subsequent tests. The primers and probes in this study are listed in Table 1.
Establishment and optimization of ERA assay
Primers were screened with an ERA basic-type nucleic acid amplification kit (KS101, GenDx, China). The reaction mixture (50 µL) included 20 µL of lysate, 2.5 µL of 10 µM forward primer, 2.5 µL of 10 µM reverse primer, 2 µL of DNA template, 21 µL of ddH2O, and 2 µL of ERA activator (MgOAc). This reaction procedure was performed using a Veriti 96-Well Thermal Cycler (Thermo Fisher Scientific, USA) at 40 ℃ for 20 min, and then 7.5 µL of loading buffer was added to the system and incubated at 56 ℃ for 5 min. ERA amplification products were electrophoresed on 1.5% agarose gel to screen primers, and ImageJ was employed for detailed gel analysis to ensure accurate interpretation of the results. An ERA fluorescent-type nucleic acid amplification kit (KS103, GenDx, China) was used to carry out the TSV-ERA reaction. The reaction system comprised 20 µL of lysate, 2 µL of DNA template (TSV-1 plasmid), 2.5 µL of 10 µM forward primer, 2.5 µL of 10 µM reverse primer, 0.6 µL of 10 µM probe, 21.2 µL of ddH2O, and 2 µL of ERA activator (MgOAc). The reaction was carried out in a CFX96 real-time PCR instrument (Bio-Rad, USA) at 40 ℃ for 20 min. To determine the optimum temperature, TSV-ERA was conducted at 30 ℃, 33 ℃, 36 ℃, 39 ℃, 42 ℃, and 45 ℃ for 20 min. To determine the optimum final concentration of primers and probes in the system, TSV-ERA was carried out at the most suitable temperature with four amounts of primer added (1.5, 2.0, 2.5, and 3.0 µL, respectively) and three amounts of probe added (0.6, 0.8, and 1.0 µL, respectively), respectively. Each reaction was repeated three times.
Analytic sensitivity and specificity of TSV-ERA
The TSV-1 plasmid with a 10-fold gradient dilution (3.6 × 101 − 3.6 × 100 copies/µL) was used to evaluate the sensitivity of TSV-ERA. The genomic DNA of EHP, WSSV, IHHNV, VpAHPND, and SPF shrimp were used to validate the specificity of the TSV-ERA. The TSV-1 recombinant plasmid was used as the positive control. Reactions with no template served as the negative control (NTC).
Sensitivity of one-step PCR and qPCR
The LOD of one-step PCR and qPCR prescribed by the OIE for TSV detection was compared with the established TSV-ERA. The one-step PCR volume was 25 µL, including 12.5 µL of 2 × Taq Master Mix, 1 µL of DNA template (TSV-1 plasmid), 1 µL of forward 9992 F primer (10 µM), 1 µL of reverse 9195R primer (10 µM), and 9.5 µL of ddH2O. The program was as follows: denaturation for 1 min 30 s at 94 ℃, followed by 40 cycles of 20 s at 94 ℃, 20 s at 60 ℃ and 25 s at 72 ℃, and an extension step of 5 min at 72 ℃. The obtained products were electrophoresed on 1.5% agarose gel. The qPCR was carried out in a 25-µL system, containing 12.5 µL of 2 × Premix Ex Taq, 1 µL of DNA template (TSV-2 plasmid), 1 µL of forward 1004 F primer (10 µM), 1 µL of reverse 1075R primer (10 µM), 0.5 µL of TSV TM-probe (10 µM), and 9 µL of ddH2O. The reaction protocol was 95 ℃ for 30 s, followed by 40 cycles of 5 s at 95 ℃ and 30 s at 60 ℃.
Statistical analysis
Values were given as the mean ± standard deviation (n = 3). GraphPad Prism 8.4.0 was used for statistical analysis and illustration.
Results
Screening of primer pairs
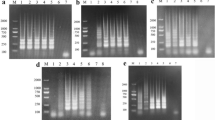
Two different concentrations of the TSV-1 plasmid were used as a template to evaluate and compare the performance of the five primer sets. Based on the results of agarose gel electrophoresis and ImageJ analysis, the TSV-F1/R1 primers were observed to have non-specific amplification, although the band was brightest at a concentration of 3.6 × 108 copies/µL (Fig. 1a). When the concentration was 3.6 × 105 copies/µL, only the TSV-F4/R4 primers were observed at specific amplifications and at the brightest band, which were selected to verify all further assays (Fig. 1b).

Screening of primer pairs. a Agarose gel electrophoresis of the five primer sets’ amplified products was performed using an ERA basic kit at 3.6 × 108 copies/µL template concentration. M, DL2000 DNA marker; 1–5: TSV-F1/R1, TSV-F2/R2, TSV-F3/R3, TSV-F4/R4, TSV-F5/R5. b Agarose gel electrophoresis of the five primer sets’ amplified products was performed using an ERA basic kit at 3.6 × 105 copies/µL template concentration. M, DL2000 DNA marker; 1–5: TSV-F1/R1, TSV-F2/R2, TSV-F3/R3, TSV-F4/R4, TSV-F5/R5
Optimization of the ERA assay
The TSV-ERA reaction system was optimized with the TSV-1 plasmid at a concentration of 3.6 × 105 copies/µL. In six different temperature tests, it was observed that the reaction was successfully amplified at 30–45 ℃, and the fluorescence signals began to decline above 42 ℃ (Fig. 2a). Based on the minimum Ct and the maximum fluorescence value, 42 ℃ was selected as the optimum temperature for TSV-ERA. As illustrated in Fig. 2b and c, in the 50-µL reaction system, the final concentrations of the primer and probe were 500 nM (2.5µL) and 120 nM (0.6 µL), respectively. The optimal reaction system was comprised of 20 µL of lysate, 2.0 µL of DNA template, 2.5 µL of 10 µM forward primer, 2.5 µL of 10 µM reverse primer, 0.6 µL of 10 µM probe, 2 µL of ERA activator, and 20.4 µL of ddH2O, and the reaction procedure took place at 42 ℃ for 20 min.

Optimization of TSV-ERA. a Optimization of temperature. Curves amplified at 30 ℃, 33 ℃, 36 ℃, 39 ℃, 42 ℃, and 45 ℃, respectively. a, b Optimization of primer and probe concentration. Curves amplified with different primer amounts (1.5, 2.0, 2.5, and 3.0 µL, respectively) (a) and probe amounts (0.6, 0.8, and 1.0 µL, respectively) (b). Vertical bars represent mean ± standard deviation (n = 3) of three repeats
Specificity of the TSV-ERA assay
To evaluate the specificity of the assay, four different pathogens were tested separately using TSV-ERA. The genomic DNA samples from EHP, WSSV, IHHNV, VpAHPND, and SPF shrimps were amplified individually using TSV-ERA. The positive template was the TSV-1 plasmid. As shown in Fig. 3, no fluorescence signal was observed in other aquatic pathogens, demonstrating good specificity for TSV-ERA.

Specificity of TSV-ERA. Templates for the TSV-ERA were the DNA extracted from EHP, WSSV, IHHNV, VpAHPND, and SPF shrimp. The TSV-1 plasmid was used as a positive control. NTC represents negative control
Sensitivity of TSV-ERA, one-step PCR, and qPCR
The 10-fold dilution of the TSV-1 and TSV-2 recombinant plasmids was used as a template for the sensitivity analysis and comparison of TSV-ERA, one-step PCR, and qPCR. As illustrated in Fig. 4a, TSV-ERA was subjected to analysis for 12 min (21.76 ± 0.60 Ct), and the limit of detection (LOD) was 3.6 × 101 copies/µL. The standard curve in the range of 3.6 × 105 − 3.6 × 101 DNA copies/µL suggested that the TSV-ERA had a high correlation coefficient (R2 = 0.9779) (Fig. 4b). The LOD of the one-step PCR was 3.6 × 102, but only a very faint band was observed at the concentration of 3.6 × 101 copies/µL (Fig. 5). The qPCR could detect up to 7.8 × 101 copies/µL (Fig. 6a) and had a high correlation coefficient (R2 = 0.9928, Fig. 6b). When using standard plasmids as templates, the sensitivity of TSV-ERA was consistent with that of qPCR, but ten times higher than that of one-step PCR.

Sensitivity of TSV-ERA. a Curve of TSV-ERA amplified using TSV-1 plasmid (3.6 × 105 − 3.6 × 100 copies/µL). b Standard curve of TSV-ERA

Sensitivity of one-step PCR. Agarose gel electrophoresis of one-step PCR using TSV-1 plasmid standard templates. M, DL2000 DNA marker; 1–8: 3.6 × 107 − 3.6 × 100 copies/µL; N, negative control

Sensitivity of qPCR assay. a Curve of qPCR amplified using TSV-2 plasmid (7.8 × 105 − 7.8 × 100 copies/µL). b Standard curve of qPCR
Discussion
Taura syndrome, caused by TSV, is a disease listed by the OIE and has been identified in five continents, leading to significant economic losses (Wertheim et al. 2009). It has been proven that TSV can infect juvenile and adult shrimp, as well as seedlings or later larval stages, and surviving individuals can become lifelong carriers of the virus (Cao et al. 2010; Stentiford et al. 2009). TSV is highly resistant, remains pathogenic for up to 3 weeks in dead shrimp, and remains infectious even after prolonged freezing (Dhar et al. 2004; Stentiford et al. 2009). Because there is no specific drug to treat TSV, early diagnosis of infectious pathogens is essential for the prediction, prevention, and control of potential outbreaks. Therefore, a rapid, easy-to-operate, and practical TSV detection tool is desperately needed. In this study, we successfully established an effective TSV-ERA assay which was suitable for field detection.
The VP2 gene has been widely used as a target gene for TSV detection, including by the OIE (Fadilah and Fasya 2021; Ochoa et al. 2020; Phromjai et al. 2015). It has been confirmed that amplicons originating from the VP2 gene in the TSV genome provided the most successful amplification (Ochoa et al. 2020). Therefore, we designed five primer sets based on the VP2 gene, and TSV-F4/F4 was selected as the best primer combination to validate all further tests (Fig. 1). In the temperature optimization experiment, it was found that the ERA reaction was successfully amplified in a wide temperature range of 30–45 ℃ (Fig. 2a), which was consistent with previous research results (Lobato and O’Sullivan 2018). Among the six different temperatures tested (30, 33, 36, 39, 42, and 45 ℃), 42 ℃ was selected as the optimum reaction temperature for TSV-ERA considering the lowest Ct value and maximum fluorescence signal. Compared with conventional diagnostic methods, TSV-ERA required no accurate temperature control and could be performed at a comparatively low temperature. This implies that TSV-ERA can be carried out at ambient temperature, decreasing the demand for expensive thermal cycling equipment, eliminating dependence on laboratories, and increasing its potential for widespread application in the disease diagnosis industry (Li et al. 2018).
Histological or molecular methods have been used to diagnose TSV infection in the acute, transitional, and early chronic stages. However, it is difficult to histologically detect low viral levels in the chronic phase (Poulos et al. 2006). Molecular biology methods with high sensitivity have been widely used in virus detection. As previously reported, TSV testing was carried out using RT-PCR (Koiwai et al. 2018; Navarro et al. 2009; Tang et al. 2017), nested RT-PCR, qPCR (Tang et al. 2004), and RT-LAMP (Phromjai et al. 2015; Sappat et al. 2011). ERA is an isothermal amplification technique, which has the characteristics of higher sensitivity and lower detection temperature. In this study, TSV-ERA was subjected to 12 min of analysis (21.76 ± 0.60 Ct) with the LOD of 3.6 × 101 copies/µL (Fig. 2). Using the plasmid as a template, compared with the TSV detection method recommended by the OIE, the sensitivity of TSV-ERA was equal to qPCR, but higher than one-step PCR (Figs. 4, 5, and 6). Moreover, TSV-ERA was outstanding in simplicity and rapidity in addition to possessing higher sensitivity. As shown in Fig. 4, TSV-ERA was able to complete the amplification reaction within 20 min, while the product amplification was observed at 2 min (3.64 ± 0.20 Ct). In contrast, TSV-ERA was much faster than one-step PCR (60–90 min for PCR amplification, and 30 min for electrophoresis) and two times faster than qPCR when achieving the same detection sensitivity (Table 2). RT-LAMP assays to detect TSV at 63–65 ℃ have been successfully established, but these methods showed shortcomings such as time-consuming gel electrophoresis and difficulty in visually measuring turbidity, especially for weakly positive samples (Kiatpathomchai et al. 2007, 2008; Sappat et al. 2011). It was reported that a visual colorimetric method based on RT-LAMP combined with nanogold-oligo probes could detect 1 pg total RNA from TSV-infected shrimp in only 15 min at 65 ℃, but this method was not quantitative (Phromjai et al. 2015), while LAMP still involved the use of a temperature controller and two or three pairs of primers with complex designs, which was prone to false positive results (Notomi et al. 2015). Through experiments and comparisons, we found that TSV-ERA has obvious potential in scientific research and practical applications.
ERA is a modified version of RPA and may become a potential alternative method of RPA (Li et al. 2020; Lobato and O’Sullivan 2018). Compared to other common methods, ERA provides better device compatibility. As long as this detector can excite and then accept and analyze the chosen fluorophore while maintaining a temperature between 30 and 45 ℃, it is fit for use in the TSV-ERA assay (Li et al. 2018; Zhao et al. 2015). Remarkably, ERA is increasingly being applied to samples of different species, such as pig, goose, and human (Zhang et al. 2021; Yang et al. 2022; Liu et al. 2021a, b; Zhong et al. 2022). Moreover, in the specificity test, there was no cross-reaction with the genomic DNA of the other four aquatic pathogens (EHP, WSSV, IHHNV, and VpAHPND) and SPF shrimp, revealing the high specificity for TSV-ERA (Fig. 3). Compared to other methods, TSV-ERA costs about USD 6 per test, eliminating the need to invest in expensive equipment or specialized laboratories. Therefore, ERA may hold promise as a practical TSV detection technique.
In conclusion, the TSV-ERA method has been successfully established, which can be used to notably shorten reaction time consumption and decrease detection temperature while sustaining accurate sensitivity. Most importantly, TSV-ERA had the distinct advantages of strong specificity, simple operation, and requiring neither laboratory facilities nor professional operators. If used in conjunction with a fast and easy nucleic acid extraction method, TSV-ERA has the potential to be used in the field detection of TSV and possibly significantly reduce its diagnostic cost. All these results suggest that TSV-ERA could be used for the diagnosis of TSV infection in shrimp aquaculture, and it has potentially broad prospects in laboratory and practical applications.
Data availability
No datasets were generated or analyzed during the current study.
References
Arulmoorthy MP, Anandajothi E, Vasudevan S, Suresh E (2020) Major viral diseases in culturable penaeid shrimps: a review. Aquacult Int 28(5):1939–1967. https://doi.org/10.1007/s10499-020-00568-3
Bergeron MG, Boissinot M, Stewart G, Daher RK (2016) Recombinase polymerase amplification for diagnostic applications. Clin Chem 62(7):947–958. https://doi.org/10.1373/clinchem.2015.245829
Boonbanjong P, Treerattrakoon K, Waiwinya W, Pitikultham P, Japrung D (2022) Isothermal amplification technology for disease diagnosis. Biosensors 12(9):677. https://doi.org/10.3390/bios12090677
Cao Z, Wang SY, Breeland V, Moore AM, Lotz JM (2010) Taura syndrome virus loads in Litopenaeus vannamei hemolymph following infection are related to differential mortality. Dis Aquat Organ 91(2):97–103. https://doi.org/10.3354/dao02258
Dhar AK, Cowley JA, Hasson KW, Walker PJ (2004) Genomic organization, biology, and diagnosis of Taura syndrome virus and yellowhead virus of penaeid shrimp. Adv Virus Res 63:353–421. https://doi.org/10.1016/S0065-3527(04)63006-5
Erickson HS, Zarain-Herzberg M, Lightner DV (2002) Detection of Taura syndrome virus (TSV) strain differences using selected diagnostic methods: diagnostic implications in penaeid shrimp. Dis Aquat Organ 52(1):1–10. https://doi.org/10.3354/dao052001
Erickson HS, Poulos BT, Tang KF, Bradley-Dunlop D, Lightner DV (2005) Taura syndrome virus from Belize represents a unique variant. Dis Aquat Organ 64(2):91–98. https://doi.org/10.3354/dao064091
Fadilah AN, Fasya AH (2021) Examination of Taura syndrome virus (TSV) in white shrimp (Litopenaeus vannamei) and tiger prawn (Penaeus monodon) with polymerase chain reaction (PCR) method. IOP Conference Series: Earth and Environmental Science, 679(1). https://doi.org/10.1088/1755-1315/679/1/012069
FAO (2020) The state of world fisheries and aquaculture. Food and Agriculture Organization of the United Nations
George SK, Kaizer KN, Betz YM, Dhar AK (2011) Multiplication of Taura syndrome virus in primary hemocyte culture of shrimp (Penaeus vannamei). J Virol Methods 172(1–2):54–59. https://doi.org/10.1016/j.jviromet.2010.12.020
Kiatpathomchai W, Jareonram W, Jitrapakdee S, Flegel TW (2007) Rapid and sensitive detection of Taura syndrome virus by reverse transcription loop-mediated isothermal amplification. J Virol Methods 146(1–2):125–128. https://doi.org/10.1016/j.jviromet.2007.06.007
Kiatpathomchai W, Jaroenram W, Arunrut N, Jitrapakdee S, Flegel TW (2008) Shrimp Taura syndrome virus detection by reverse transcription loop-mediated isothermal amplification combined with a lateral flow dipstick. J Virol Methods 153(2):214–217. https://doi.org/10.1016/j.jviromet.2008.06.025
Koiwai K, Kodera T, Thawonsuwan J, Riani S, Kawase M, Kondo H, Hirono I (2018) Rapid diagnosis of three shrimp RNA viruses using RT-PCR-DNA chromatography. J Fish Dis. https://doi.org/10.1111/jfd.12821
Lee D, Yu YB, Choi JH, Jo AH, Hong SM, Kang JC, Kim JH (2022) Viral shrimp diseases listed by the OIE: a review. Viruses 14(3):585. https://doi.org/10.3390/v14030585
Li J, Macdonald J, von Stetten F (2018) Review: a comprehensive summary of a decade development of the recombinase polymerase amplification. Royal Soc Chem 144(1):31–67. https://doi.org/10.1039/c8an01621f
Li J, Macdonald J, von Stetten F (2020) Correction: review: a comprehensive summary of a decade development of the recombinase polymerase amplification. Royal Soc Chem 145(5):1950–1960. https://doi.org/10.1039/c9an90127b
Li J, Wang Y, Hu J, Bao Z, Wang M (2023) An isothermal enzymatic recombinase amplification (ERA) assay for rapid and accurate detection of Enterocytozoon hepatopenaei infection in shrimp[J]. J Invertebr Pathol 197:107895. https://doi.org/10.1016/j.jip.2023.107895
Liu P, Wang X, Liang J, Dong Q, Zhang J, Liu D, Wang S, Bi J, Liu W, Wang Z, Chen L, Liu L, Huang X, Zhang G (2021a) A recombinase polymerase amplification-coupled Cas12a mutant-based module for efficient detection of streptomycin-resistant mutations in Mycobacterium tuberculosis. Front Microbiol 12:796916. https://doi.org/10.3389/fmicb.2021.796916
Liu Y, Chen Y, Dang L, Liu Y, Huang S, Wu S, Ma P, Jiang H, Li Y, Pan Y, Wei Y, Ma X, Liu M, Ji Q, Chi T, Huang X, Wang X, Zhou F (2021b) EasyCatch, a convenient, sensitive and specific CRISPR detection system for cancer gene mutations. Mol Cancer 20(1):157. https://doi.org/10.1186/s12943-021-01456-x
Lobato IM, O’Sullivan CK (2018) Recombinase polymerase amplification: basics, applications and recent advances. Trends Anal Chem 98:19–35. https://doi.org/10.1016/j.trac.2017.10.015
Mayboroda O, Katakis I, O’Sullivan CK (2018) Multiplexed isothermal nucleic acid amplification. Anal Biochem 545:20–30. https://doi.org/10.1016/j.ab.2018.01.005
Meng Q, Yang H, Zhang G, Sun W, Ma P, Liu X, Dang L, Li G, Huang X, Wang X, Liu J, Leng Q (2021) CRISPR/Cas12a-assisted rapid identification of key beer spoilage bacteria. Innovative Food Sci Emerg Technol 74:102854. https://doi.org/10.1016/j.ifset.2021.102854
Navarro SA, Tang KFJ, Lightner DV (2009) An improved Taura syndrome virus (TSV) RT-PCR using newly designed primers. Aquaculture 293(3–4):290–292. https://doi.org/10.1016/j.aquaculture.2009.05.003
Notomi T, Mori Y, Tomita N, Kanda H (2015) Loop-mediated isothermal amplification (LAMP): principle, features, and future prospects. J Microbiol 53(1):1–5. https://doi.org/10.1007/s12275-015-4656-9
Nunan LM, Poulos BT, Lightner DV (1998) Reverse transcription polymerase chain reaction (RT-PCR) used for the detection of Taura syndrome virus (TSV) in experimentally infected shrimp. Dis Aquat Organ 34(2):87–91. https://doi.org/10.3354/dao034087
Ochoa LM, Cruz-Flores R, Dhar AK (2020) Detection and phylogenetic analyses of Taura syndrome virus from archived Davidson’s-fixed paraffin-embedded shrimp tissue. Viruses 12(9):1030. https://doi.org/10.3390/v12091030
Phromjai J, Mathuros T, Phokharatkul D, Prombun P, Suebsing R, Tuantranont A, Kiatpathomchai W (2015) RT-LAMP detection of shrimp Taura syndrome virus (TSV) by combination with a nanogold-oligo probe. Aquac Res 46(8):1902–1913. https://doi.org/10.1111/are.12345
Piepenburg O, Williams CH, Stemple DL, Armes NA (2006) DNA detection using recombination proteins. PLoS Biol 4(7):e204. https://doi.org/10.1371/journal.pbio.0040204
Poulos BT, Tang KFJ, Pantoja CR, Bonami JR, Lightner DV (2006) Purification and characterization of infectious myonecrosis virus of penaeid shrimp. J Gen Virol 87(Pt 4):987–996. https://doi.org/10.1099/vir.0.81127-0
Poulos BT, Noble BW, Lightner DV (2008) Comparison of Taura syndrome virus (TSV) detection methods during chronic-phase infection in Penaeus vannamei. Dis Aquat Organ 82(3):179–185. https://doi.org/10.3354/dao01996
Prochaska J, Poompuang S, Koonawootrittriron S, Sukhavachana S, Na-Nakorn U (2022) Evaluation of a commercial SPF Litopenaeus vannamei shrimp breeding program: resistance to infectious myonecrosis virus (IMNV), Taura syndrome virus (TSV), and white spot syndrome virus (WSSV) from laboratory challenges. Aquaculture 554:738145. https://doi.org/10.1016/j.aquaculture.2022.738145
Rob F (2020) Fresh insights into the $26.7 billion shrimp sector. the Fish Site
Robles-Sikisaka R, Hasson KW, Garcia DK, Brovont KE, Cleveland KD, Klimpel KR, Dhar AK (2002) Genetic variation and immunohistochemical differences among geographic isolates of Taura syndrome virus of penaeid shrimp. J Gen Virol 83(Pt 12):3123–3130. https://doi.org/10.1099/0022-1317-83-12-3123
Sappat A, Jaroenram W, Puthawibool T, Lomas T, Tuantranont A, Kiatpathomchai W (2011) Detection of shrimp Taura syndrome virus by loop-mediated isothermal amplification using a designed portable multi-channel turbidimeter. J Virol Methods 175(2):141–148. https://doi.org/10.1016/j.jviromet.2011.05.013
Stentiford GD, Bonami JR, Alday-Sanz V (2009) A critical review of susceptibility of crustaceans to Taura syndrome, yellowhead disease and white spot disease and implications of inclusion of these diseases in European legislation. Aquaculture 291(1–2):1–17. https://doi.org/10.1016/j.aquaculture.2009.02.042
Tang KFJ, Wang J, Lightner DV (2004) Quantitation of Taura syndrome virus by real-time RT-PCR with a TaqMan assay. J Virol Methods 115(1):109–114. https://doi.org/10.1016/j.jviromet.2003.09.021
Tang KFJ, Aranguren LF, Piamsomboon P, Han JE, Maskaykina IY, Schmidt MM (2017) Detection of the microsporidian Enterocytozoon hepatopenaei (EHP) and Taura syndrome virus in Penaeus vannamei cultured in Venezuela. Aquaculture 480:17–21. https://doi.org/10.1016/j.aquaculture.2017.07.043
Teng PH, Chen CL, Wu CN, Wu SY, Ou BR, Lee PY (2006) Rapid and sensitive detection of Taura syndrome virus using nucleic acid-based amplification. Dis Aquat Organ 73(1):13–22. https://doi.org/10.3354/dao073013
Thitamadee S, Prachumwat A, Srisala J, Jaroenlak P, Salachan PV, Sritunyalucksana K, Flegel TW, Itsathitphaisarn O (2016) Review of current disease threats for cultivated penaeid shrimp in Asia. Aquaculture 452:69–87. https://doi.org/10.1016/j.aquaculture.2015.10.028
Wertheim JO, Tang KF, Navarro SA, Lightner DV (2009) A quick fuse and the emergence of Taura syndrome virus. Virology 390(2):324–329. https://doi.org/10.1016/j.virol.2009.05.010
Xia S, Chen X (2020) Single-copy sensitive, field-deployable, and simultaneous dual-gene detection of SARS-CoV-2 RNA via modified RT-RPA. Cell Discovery 6(1):37. https://doi.org/10.1038/s41421-020-0175-x
Xu Y, Wang Y, Hu J, Bao Z, Wang M (2022) Development and visualization improvement for the rapid detection of Decapod Iridescent Virus 1 (DIV1) in Penaeus vannamei based on an Isothermal recombinase polymerase amplification assay. Viruses 14(12):2752. https://doi.org/10.3390/v14122752
Xu Y, Wang Y, Hu J, Bao Z, Wang M (2023) The development of a novel quantitative assay for the detection of convert mortality nodavirus (CMNV) in Litopenaeus vannamei. Aquaculture 577:739923. https://doi.org/10.1016/j.aquaculture.2023.739923
Yang K, Liang Y, Li Y, Liu Q, Zhang W, Yin D, Song X, Shao Y, Tu J, Qi K (2021) Reverse transcription-enzymatic recombinase amplification coupled with CRISPR-Cas12a for rapid detection and differentiation of PEDV wild-type strains and attenuated vaccine strains. Anal Bioanal Chem 413(30):7521–7529. https://doi.org/10.1007/s00216-021-03716-7
Yang K, Zhang W, Xu L, Liu Q, Song X, Shao Y, Tu J, Qi K (2022) Facile, ultrasensitive, and highly specific diagnosis of goose astrovirus via reverse transcription-enzymatic recombinase amplification coupled with a CRISPR-Cas12a system detection. Poult Sci 101(12):102208. https://doi.org/10.1016/j.psj.2022.102208
Zhang W, Xu L, Liu Q, Cao Y, Yang K, Song X, Shao Y, Tu J, Qi K (2021) Enzymatic recombinase amplification coupled with CRISPR-Cas12a for ultrasensitive, rapid, and specific porcine circovirus 3 detection. Mol Cell Probes 59:101763. https://doi.org/10.1016/j.mcp.2021.101763
Zhang L, Wang Y, Hu J, Bao Z, Wang M (2023a) Rapid detection of white spot syndrome virus in Penaeus vannamei based on real-time enzymatic recombinase amplification. Aquaculture 566. https://doi.org/10.1016/j.aquaculture.2022.739196
Zhang L, Liu K, Liu M, Bao Z, Wang M (2023b) Development of a real-time enzymatic recombinase amplification assay for rapid detection of infectious hypodermal and hematopoietic necrosis virus (IHHNV) in shrimp Penaeus vannamei. J Invertebr Pathol 201:108024. https://doi.org/10.1016/j.jip.2023.108024
Zhang L, Wang Y, Liu M, Hu J, Bao Z, Wang M (2024) Development of a multiplex real-time enzymatic recombinase amplification assay for differentiation of yellow head virus genotype 1 and 2 in Penaeus vannamei. Aquaculture 582:740564. https://doi.org/10.1016/j.aquaculture.2024.740564
Zhao Y, Chen F, Li Q, Wang L, Fan C (2015) Isothermal amplification of nucleic acids. Chem Rev 115(22):12491–12545. https://doi.org/10.1021/acs.chemrev.5b00428
Zhong M, Chen K, Sun W, Li X, Huang S, Meng Q, Sun B, Huang X, Wang X, Ma X, Ma P (2022) PCDetection: PolyA-CRISPR/Cas12a-based miRNA detection without PAM restriction. Biosens Bioelectron 214:114497. https://doi.org/10.1016/j.bios.2022.114497
Zhou Q, Wang Y, Hu J, Bao Z, Wang M (2023) Development of a real-time enzymatic recombinase amplification assay (RT-ERA) and an ERA combined with lateral flow dipsticks (LFD) assay (ERA-LFD) for rapid detection of acute hepatopancreatic necrosis disease (AHPND) in shrimp Penaeus vannamei. Aquaculture 566:739205. https://doi.org/10.1016/j.aquaculture.2022.739205
Funding
This work was supported by the PI Project of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML20220018), the Project of Sanya Yazhou Bay Science and Technology City Management Foundation (SKJC-2023-01-004), and the High-end Talent Research Project of Hebei Province (2021HBQZYCSB001).
Author information
Authors and Affiliations
Contributions
Jiaobing Li: writing—original draft, conducted the experiments, and wrote the manuscript. Jingjie Hu: conceived the study and designed the experiments, checked, and modified the manuscript. Zhenmin Bao: conceived the study and designed the experiments, checked, and modified the manuscript. Mengqiang Wang: conceived the study and designed the experiments, checked, and modified the manuscript. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Handling Editor: Brian Austin
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, J., Hu, J., Bao, Z. et al. Rapid and sensitive detection of Taura syndrome virus (TSV) in shrimp based on an isothermal enzymatic recombinase amplification (ERA) assay. Aquacult Int 32, 6211–6225 (2024). https://doi.org/10.1007/s10499-024-01462-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10499-024-01462-y