
Abstract
Gastric cancer is regarded as the fifth most common cancer globally but the third most common cancer death. Although systemic chemotherapy is the primary treatment for advanced gastric cancer patients, the outcome of chemotherapy is unsatisfactory. Novel therapeutic strategies and potential alternative treatments are therefore needed to overcome the impact of this disease. At a cellular level, mitochondria play an important role in cell survival and apoptosis. A growing body of studies have shown that mitochondria play a central role in the regulation of cellular function, metabolism, and cell death during carcinogenesis. Interestingly, the impact of mitochondrial dynamics, including fission/fusion and mitophagy, on carcinogenesis and cancer progression has also been reported, suggesting the potential targeting of mitochondrial dynamics for the treatment of cancer. This review not only comprehensively summarizes the homeostasis of gastric cancer cells, but the potential therapeutic interventions for the targeting of mitochondria for gastric cancer therapy are also highlighted and discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer is the fifth most common cancer worldwide with a mortality rate of between 8 and 13% [1,2,3,4,5,6]. However, the etiology of gastric cancer cannot be specified with any high degree of certainty due to the involvement of multiple factors, including tumor suppressor genes, deoxyribonucleic acid (DNA) repair genes, and cell cycle signaling [7, 8]. A previous study has shown that infection with Helicobacter pylori (HP) is a risk factor of non-cardia gastric cancer, whereas cardia gastric cancer may be associated with HP infection or reflux [1]. Besides HP infection, dietary factors including dietary salt, a low fruit diet, grilled meat, alcohol drinking and smoking may increase the risk of gastric cancer [1, 9].
Gastric cancer is classified using various pathohistological classifications, including the World Health Organization (WHO) classification, Lauren’s classification, and the modified WHO classification [10, 11]. The WHO classification is divided into five categories: papillary, tubular, mucinous, mixed, and poorly cohesive [10, 11]. The Lauren’s classification consists of intestinal, diffuse, and indeterminate types [10, 11]. The modified WHO classification includes grouping into differentiated and undifferentiated types [10, 11]. Although there are several widely recognized classifications for gastric cancer, there is no definite classification for the correlation of prediction outcome between the type of gastric cancer and the treatment [10]. This limitation is due to the heterogeneity of disease.
As regards diagnosis the WHO and Lauren classifications are the most used systems regarding clinical practice guidelines [8, 10, 11]. The optimal treatment for gastric cancer depends on the stage of disease, resection being the standard curative treatment [12, 13]. The primary therapy for locally advanced gastric cancer is neoadjuvant chemotherapy followed by surgery [3, 5, 14, 15]. However, the overall response rate to systemic treatment is between 20 and 40% as the anticancer therapy is not effective [3, 16,17,18]. The 5-year survival rate is 70–95% in early cancer and only 5–25% in advanced cancer [5, 6, 14, 19]. In addition, half of the resectable gastric cancer cases require adjuvant treatment in which drug resistance may occur [3, 10, 17, 19]. Metastatic gastric cancer patients are predicted as a poor overall survival group [2, 19]. The median overall survival time is 6–13 months [20].
Unfortunately, the outcome of the current treatment is not satisfactory, therefore novel alternative strategies to improve gastric cancer treatment outcomes are urgently needed. A growing body of research has shown that targeting mitochondria is a potential alternative treatment because gastric cancer homeostasis and cell death depend mainly on mitochondria and oxidative stress (OS). Several steps have been identified as potential targets for intervention, including mitophagy, autophagy, mitochondrial fission and fusion, ROS production and elimination, apoptosis, ATP production, and cell cycle arrest. In this review, we not only comprehensively summarize the homeostasis of gastric cancer cells, but the potential therapeutic interventions for the targeting of mitochondria for gastric cancer therapy are also highlighted and discussed.
Gastric cancer homeostasis
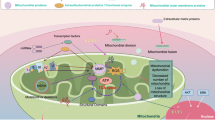
There are low levels of certainty as regards the risk factors or mechanisms involved in gastric cancer development. However, OS plays an important role in tumor proliferation and progression [7, 21, 22]. At a cellular level, mitochondria are essential organelles that regulate the homeostasis of cancer cells and programmed cell death [7, 23]. One of the key regulatory processes is mitophagy which decreases mitochondrial OS, inhibits mitochondrial pro-apoptotic factor leakage, and increases intracellular adenosine triphosphate (ATP) generation [2]. In addition to mitophagy, mitochondrial dynamics, calcium buffering by the endoplasmic reticulum (ER), and autophagy of the damaged organelles are also involved in the homeostasis of gastric cancer [23]. Noticeably, OS is a typical influencer in all previously mentioned mechanisms, and mitochondria are the main source of production of cellular reactive oxygen species (ROS) [7, 23]. OS is an important factor that induces cell apoptosis by causing DNA damage through caspase-dependent and caspase-independent pathways [24, 25]. However, the role of OS in mitochondrial dynamics is uncertain. One report showed that mitochondrial ROS induced mitochondrial fragmentation, and ATP is generated by mitochondrial respiration using oxidative phosphorylation [26]. For survival under low oxygen conditions, the cancer cell can produce ATP by increasing glucose uptake and carrying out aerobic glycolysis, a process known as the Warburg effect [26, 27]. Although there is homeostatic regulation in cancer cells, overproduction of ROS above specific levels could result in decreased cell viability due to apoptosis [2, 23, 28], as shown in Fig. 1.

Gastric cancer homeostasis. Targeting homeostasis in gastric cancer cells has the potential to improve the efficacy of systemic therapy with the purposes of decreasing cell viability and invasion by inhibiting mitophagy and autophagy, disturbing mitochondrial dynamics, decreasing calcium buffering and cellular ATP, activating the pro-apoptosis pathway, and promoting OS. In addition, the cell cycle arrest may be interrupted to knock out tumor proliferation and progression. OS oxidative stress, X inhibition
Current status of chemotherapy for gastric cancer
Chemotherapy is the major treatment of gastric cancer, especially in advanced gastric cancer patients. However, its efficacy is limited by drug resistance and poor response. For better treatment outcomes, a combination of anticancer agents is the preferred approach [29]. Chemotoxicity is a major issue therefore a two-drug regimen is frequently a better option in comparison to a three-drug regimen [29]. However, triple agents are an option in the case of medically fit selected patients [29]. The National Comprehensive Cancer Network (NCCN) guidelines version 2.2021 suggest fluoropyrimidine (fluorouracil or capecitabine) plus oxaliplatin for postoperative regimens [29]. For unresectable patients, fluorouracil plus oxaliplatin or fluorouracil plus cisplatin are recommended [29]. Fluoropyrimidine (fluorouracil or capecitabine) plus paclitaxel are the other regimens for unresectable cases [29]. The Asia guidelines, including Japanese and Korean guidelines, recommend S-1 or capecitabine plus oxaliplatin or cisplatin for adjuvant chemotherapy [20, 30]. Mechanistically, these anticancer drugs affect cancer cells by inducing DNA damage, angiogenesis inhibition, and apoptosis [31].
Fluoropyrimidine is recommended by several regimens [32]. There are oral and intravenous forms, capecitabine and S-1 are the oral forms, and the intravenous form is 5-fluorouracil (5-FU) [32]. Capecitabine is a prodrug converted to fluorouracil by three enzymes specifically carboxylesterase, cytidine deaminase, and thymidine phosphorylase [32, 33]. In gastric cancer tissue, cytidine deaminase and thymidine phosphorylase are highly specific therefore, there is higher amount of fluorouracil in cancer tissue than normal tissue [32]. S-1 is tegafur in combination with gimeracil: a dihydropyridine dehydrogenase inhibitor which prevents fluorouracil degradation, and oteracil: a pyrimidine phosphoribosyltransferase inhibitor which inhibits fluorouracil phosphorylation in the gastrointestinal tract [32]. Hence, there are lower gastrointestinal side effects in comparison with 5-FU [32]. 5-FU is an antimetabolite agent which is used in gastrointestinal cancer therapy [28, 34]. However, the monotherapy of 5-FU is ineffective [12, 28]. The anticancer effect depends on fluorodeoxyuridine monophosphate, the active metabolite that inhibits thymidine synthase which is associated with DNA replication [28, 34]. Fluorouridine triphosphate and fluorodeoxyuridine triphosphate inhibit ribonucleic acid (RNA) mutation and induce DNA disruption [28]. Previous studies reported that apoptosis induced by 5-FU was associated with p53 phosphorylation and the p53 increased OS by activating mitochondrial ferredoxin reductase [28]. The increased OS damaged DNA a finding verified by 8-OH-dG expression [28]. Although the excessive OS induced cell death, a lack of antioxidant enzymes could have the same effect on cell viability [28]. The authors of the study concluded that the response rate of 5-FU therapy depended on OS induced by p53 expression [28].
Platinum-based chemotherapy consists of oxaliplatin, cisplatin and carboplatin. Both oxaliplatin and cisplatin are used in gastric cancer treatment [29]. Oxaliplatin is a diaminocyclohexane carrier ligand that induces apoptosis by inhibiting DNA replication and repair [35]. Cisplatin was shown to cause cancer cell apoptosis by damaging the DNA damage [36]. Although both oxaliplatin and cisplatin provide therapeutic effects in gastric cancer treatment, the toxicity of cisplatin is higher than oxaliplatin regimens [29, 32]. Paclitaxel is a taxane which induces tubulin polymerization, DNA fragmentation and apoptosis however the exact mechanism of its ability to induce cell death is unknown [37].
Some anticancer agents are not recommended in the current guidelines. However, previous studies have shown that they have therapeutic potential for treating gastric cancer [38,39,40]. Doxorubicin (DOX) is an example of a gastric cancer chemotherapy agent which is not recommended recently because there are other agents which give a better response rate and have a lower incidence of side effects. DOX is an anthracyclin which inhibits DNA and RNA synthesis [38]. Additionally, anticancer effects and side effects of DOX are associated with the mitochondrial apoptotic pathway which is induced by releasing cytochrome c (Cyto c) from mitochondria [39, 40]. Previous studies have shown that DOX increased ROS production and decreased extracellular signal-related kinase (ERK) 1/2 phosphorylation which signaled via the caspase-dependent pathway and caused internal programmed cell death resulting in apoptosis [40, 41]. ERK protein is one of the mitogen-activated protein kinases (MAPKs) which are involved in cell survival and cell death, other MAPKs being c-Jun N-terminal kinase (JNK) and p38 [41]. Although chemotherapy agents are often viewed as a strategy that mainly affects cancer cells, accumulating evidence indicates that these agents also affect normal cell function resulting in various side effects. Therefore, highly effective strategies and novel alternative treatments for gastric cancer are required.
Mitochondrial targeting therapy as an alternative treatment for gastric cancer
Mitochondria play an essential role in the regulation of cancer cell homeostasis and programmed cell death [7, 23]. Therefore, mitochondrial targeting therapy may be a potential alternative strategy for treating gastric cancer. There are several steps during cellular stress responses that have been identified as potential targets for intervention including mitophagy, autophagy, mitochondrial fission and fusion, ROS production and elimination, apoptosis, ATP production and cell cycle arrest.
Targeting mitophagy and autophagy
Mitophagy is selective mitochondrial degradation by autophagy, whereas autophagy is general organelle degradation to prevent persistent cell damage and maintain cellular health [23]. These processes are essential in cancer cell homeostasis, the cells responding to excessive OS by reducing mitochondrial injury, inhibiting pro-apoptotic factor leakage, and increasing ATP synthesis [2]. There are several pathways involved in mitophagy and autophagy. The inhibition of mitophagy or autophagy enhances anticancer effects by increasing ROS and inducing apoptosis. The agents that have been designed to target mitophagy and autophagy are indomethacin, transient receptor potential melastatin-2 (TRPM2), and Yes-associated protein (YAP) knockdown [2, 3].
Indomethacin, a nonsteroidal anti-inflammatory drug or NSAID, was found to induce lysosomal dysfunction and inhibit autophagy which induced mammalian target of rapamycin (mTOR)-independent apoptosis in gastric cancer cells [42]. Indomethacin increased oxaliplatin chemosensitivity [42]. Together with a TRPM2 channel blocker, such as clotrimazole, they have been shown to inhibit autophagy and mitophagy in gastric cancer [3, 43]. Previous studies have shown that TRPM2 knockdown inhibited autophagy through downregulation of the mTOR-independent but JNK-dependent pathway which interfered with mitochondrial metabolism, increasing ROS, and leading to cell damage [3, 43]. Moreover, they suggested that TRPM2 knockdown inhibited mitophagy by lowering Bcl-2/adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) expression (Table 3) [3]. Outer mitochondrial membrane fusion mediator mitofusin 2 (MFN2), which is located at the mitochondrial outer membrane, plays an important role in the mitochondrial fusion process [44]. Additionally, a previous study has shown that the Hippo-YAP pathway was associated with cancer cell progression [2]. Specifically, the YAP-knockdown inhibited mitophagy-SIRT1/MFN2 pathway which increased ROS and apoptosis (Table 3) [2].
Targeting mitochondrial dynamics
Mitochondrial dynamics regulate mitochondrial size, shape, and distribution [26, 45]. This process consists of fission and fusion, which protects the cell from mitochondrial DNA mutations [26]. Mitochondrial fission is the process by which mitochondria divide, which is mediated by the constricting action of GTPase Dynamin-related protein 1 (DRP1) [45]. Mitochondrial fusion has two separate processes, one which is mediated by MFN1 and MFN2 and occurs in the outer mitochondrial membrane, and one in the inner membrane, which is mediated by optic atrophy 1 (OPA1) [45]. Interestingly, a previous study has shown that indomethacin disrupts mitochondrial dynamics by increasing mitochondrial fission through protein kinase-C (PKC) activation followed by p38 phosphorylation and DRP1 activation, leading to apoptosis of both gastric cancer and normal gastric cells (Table 3) [23].
In addition to the inhibition of mitophagy, YAP-knockdown was found to inhibit mitochondrial fusion mediated by MFN2, resulting in cancer cell apoptosis (Table 3) [2]. In vitro and in vivo studies have shown that MFN2 expression was lower in gastric cancer tissue compared with normal gastric tissue, and lower MFN2 expression was directly correlated to small tumor size [44]. MFN2 inhibited cell proliferation, decreased cell invasion, and induced apoptosis suggesting that MFN2 suppression may be used as an anticancer agent [44]. These findings suggest that activation of mitochondrial fission or inhibition of mitochondrial fusion could promote apoptosis and cell death in gastric cancer cells.
Targeting antioxidant enzymes
There is a higher level of OS in cancer cells in comparison to normal cells [46]. ROS are generated in ER, cytoplasm, the cell membrane, and especially in the mitochondria [24]. The common forms of ROS are superoxide anions, hydroxyl radicals, and hydrogen peroxide (H2O2) [7]. The mitochondrial ROS are produced by the electron transport chain (ETC) on the inner mitochondrial membrane during oxidative phosphorylation [26]. Mitochondrial ROS are also induced by the production of pro-inflammatory cytokines [26].
Mitochondrial antioxidant enzymes are transferred into the mitochondria and attenuate mitochondrial ROS and toxicity [26]. There are several antioxidant enzymes including superoxide dismutase (SODs), glutathione peroxidase (GPx), catalase, peroxiredoxins and thioredoxins [26]. In mammals, there are three isoforms of SOD: SOD1/copper-zinc SOD (CuZnSOD) which is found in the nucleus and mitochondria, SOD2/MnSOD which is a scavenger of the superoxide in mitochondria, and SOD3 which is a metalloenzyme predominantly located in the extracellular space. [22, 26, 46, 47].
MnSOD plays an important role during cancer cell proliferation and invasion; however, the role of mitochondrial ROS in cancer cell invasion is controversial. Tamaru et al. reported that MnSOD decreased mitochondrial ROS levels leading to the inhibition of tumor cell invasion [22]. In contrast, a previous study has shown that MnSOD promoted interaction of actin, S100A4 and Talin, and enhanced rat gastric tumor cell invasion [46]. The effects of MnSOD overexpression on cell viability and invasion of rat gastric cancer cells were shown in Table 1. In a clinical study, patients with early stage gastric cancer had lower MnSOD expression in comparison with advanced gastric cancer patients [47]. Moreover, under certain conditions, non-mitochondrial generated ROS were found to augment mitochondrial ROS production, a process known as “ROS-induced ROS” [26]. p47phox cytosolic subunit translocation activated phagosomal NADPH oxidase resulting in increased ROS and cell apoptosis (Table 3) [25]. Tumor-associated NADH oxidase (tNOX) has been shown to exert anti OS effects [4].
Several agents targeted antioxidant enzymes, which played a significant role in decreasing ROS levels and increasing apoptosis [4, 22]. TPT, a Topo I inhibitor could inhibit glutamine uptake by reducing alanine-serine-cysteine transporter (ASCT2) glutamine transporter activities [48]. ASCT2 knockdown markedly decreased GSH and increased ROS in gastric cancer cell lines, resulting in induced caspase-dependent apoptosis, reduced cell proliferation, and invasion (Tables 1 and 3) [48]. A previous study has shown that capsaicin suppressed the activity of tNOX, leading to excessive ROS levels and activation of the caspase-dependent apoptotic pathway (Table 2) [4]. In addition, the inhibition of SOD and GSH-Px activities by a novel nitric oxide prodrug (NG) resulted in increased ROS from lipid peroxidation products, which verified by malondialdehyde (MDA: a reactive aldehyde) levels (Table 2) [13]. The ethanol extraction of Vitex has been shown to exert anti-tumor effects in human cell lines including cells from breast cancer, lung cancer, gastric cancer, colon cancer, ovarian cancer, uterine cervical carcinoma, and uterine cervical fibroblast [49]. Additionally, a previous study reported that Vitex increased OS, measured by mRNA levels of tumor necrosis factor-alpha (TNF-alpha), heme oxygenase-1 (HO-1), CU/ZnSOD, and thioredoxin (TXN) [49]. An increase in ROS production resulted in early apoptosis (Table 3) [49]. SL3 from Artemisia argyi, a Chinese herb, significantly increased ROS production by activating NADPH oxidase levels in the p47phox cytomembrane resulting in cell apoptosis (Table 3) [25]. Decreased expression of MnSOD by 17-DMAG, a heat shock protein 90 (HSP90) inhibitor, promoted gastric cancer cell apoptosis and decreased cell proliferation (Table 3) [15]. Furthermore, several natural or novel agents could increase ROS synthesis without directly involving antioxidant enzymes, which induced apoptosis via both caspase-dependent and caspase-independent pathways. These findings suggest that OS are an important factor in the induction of cell apoptosis through caspase-dependent, caspase-independent pathways and DNA damage [24, 25].
Targeting pro-apoptotic factors
Apoptosis is an essential mechanism in the maintenance of cancer cell homeostasis [24]. Thus, apoptosis is the target of cancer treatment bases on the evidence showing a lack in apoptosis increases carcinogenesis [50]. This internal programming cell death is regulated by pro-apoptotic proteins such as Bax and Bak, and anti-apoptotic proteins including Bcl-2 and Bcl-XL [24, 50]. There are several apoptotic pathways including mitochondrial, death receptor, and ER pathways [48]. The caspase-dependent apoptotic pathway is related to mitochondrial pathways [48]. The caspases are classified into three groups by peptide analysis, including: (1) caspase-1,4, and 5 (2) caspase-2,3, and 7 (3) caspase-6, 8, and 9 [50]. In addition to apoptosis, group 1 is involved in cytokine production, whereas the role of group 2 is apoptotic activation, and group 3 is a cell death signal magnifier [50]. The most common trigger of the mitochondrial-mediated apoptotic pathway is OS, which increases mitochondrial membrane permeability, and causes a decrease in mitochondrial membrane potential (MMP) [7, 13, 48]. Following this apoptotic proteins are released into the cytoplasm and subsequently apoptosis occurs [13]. Several studies pointed out that the mitochondrial-dependent apoptotic pathway could be detected by a decrease in mitochondrial membrane potential (MMP), including 5F, topotecan, capsaicin, farrerol, indomethacin, melittin, PDOX, tomentosin, TRPM2 knockdown and purified polysaccharide (WATP) [2, 4, 5, 14, 18, 23, 24, 40, 48, 51]. A previous study suggested that excessive H2O2 induced p53 phosphorylation, upregulated pro-apoptotic Bax, and downregulated anti-apoptotic Bcl-2 (Table 2) [7].
ROS production induced apoptosis via both caspase-dependent and caspase-independent pathways. It has been found that many agents revealed are involved in the multitude of steps leading to apoptosis. Several agents increased ROS production and were involved with apoptotic protein, controlling internal programming cell death, including capsaicin, TPT. Capsaicin, present in chilies, induced Bcl-2 related apoptosis in a AGS cell line and decreased cell viability (Table 3) [9]. Tomentosin induced apoptosis by increasing the expression of Bax and decreasing that of Bcl-2 (Table 3) [14]. Moreover, the downregulation of ASCT2 by TPT increased Bax and decreased Bcl-2 levels, which resulted in apoptosis (Tables 1 and 3) [48]. In addition, Mito-FF was selectively taken up into the mitochondria, which resulted in mitochondrial membrane disruption and the leakage of mitochondrial contents, and cell apoptosis (Table 3) [17].
Cyto c release into the cytoplasm was the first trigger point in the case of various targeting agents which induced gastric cancer cell apoptosis. Anemarrhena asphodelodies induced apoptosis via Cyto c release into the cytoplasm followed by the stimulation of the caspase-3 dependent but p53-independent pathway and inhibited cancer cell growth [50]. PDOX, a doxorubicin prodrug, increased ROS production and decreased ERK1 phosphorylation which initiated by the release of Cyto c from the mitochondria resulting in caspase-dependent apoptosis (Table 2) [40]. The mechanism underlying its anticancer effect was not different from DOX but its side effects were lower [40]. Isothiocyanate (ITC), a compound found in cruciferous plant, could inhibit cell proliferation and increase cell apoptosis in various cancer cells by acting on thiol groups [6]. The binding of PITC with GSH reduced cellular antioxidants, followed by an increase in ROS, DNA fragmentation, mitochondrial damage, and the release of Cyto c into the cytoplasm [6]. In addition, initiated p53 phosphorylation and inhibited Bcl-2, could also induce apoptosis [6] (Tables 2 and 3). PsL, a Chinese herb with anti-tumor effects, has been shown to induced ROS production in gastric cancer cell lines via activating p53 [24]. Bax decreased MMP and induced cell membrane disruption followed by the release of apoptosis-inducing factor (AIF) and Cyto c, causing DNA fragmentation and the caspase-dependent pathway [24]. However, inhibition of caspase-3 by Z-DEVE-FMK did not alter cell apoptosis by 5F (Tables 2 and 3) [24]. Excessive ROS production following NG-induced caspase 3,9 mediated apoptosis through Bax-Bcl2 regulation and Cyto c and AIF released into the cytoplasm (Table 2) [13]. Farrerol induced Cyto c releasing and caspase-mediated apoptosis pathway (Table 2) [51]. Melittin is the component in bee venom that induces apoptosis in cancer cell lines, including melanoma and ovarian cancer [5]. In gastric cancer cells, melittin increased ROS production followed caspase-3 mediated apoptotic [5]. Melittin increased mitochondrial permeability followed by the release of Cyto c, the mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (Smac/Diablo), AIF, and EndoG proteins, suggesting that melittin could induce human gastric cancer cell apoptosis via activation of mitochondrial pathway (Table 2) [5]. Vitex initiated APAF1 and TNF-alpha activation leading to apoptosis [49]. In addition to the binding of APAF1, Cyto c was released into the cytoplasm, and both mechanisms activated the caspase-dependent pathway [49]. Following the binding of TNF-alpha, Fas-associated death receptor and caspase-8 were activated [49]. These results suggested that Vitex induced apoptosis through mitochondrial and death ligand receptor pathways. A previous study showed the association between GSH reduction and early apoptosis, suggesting that the increase of OS induced early apoptosis (Table 3) [49]. YAP-knockdown inhibited Bcl-xL anti-apoptotic factor and induced the caspase-9 apoptotic pathway through the Hippo-YAP pathway (Table 3) [2].
Thus, there are several potential target pathways for the induction of apoptosis from both internal and external stimuli; however, OS seems to be a key player in the induction of apoptosis. The initial process involves the release or activation of apoptotic proteins, including those in the Bcl-2 family and Cyto c, both caspase-dependent, and caspase-independent. The interventions targeting the apoptotic pathway are illustrated in Fig. 2.

Apoptosis as a targeting therapy. There are extrinsic and intrinsic pathways inducing apoptosis, including those involving mitochondrial dynamics, death receptors, and ER [48]. Potential interventions targeting apoptosis pathways are illustrated
Targeting calcium buffering by ER and ATP generation
WATP extracted from Aster tataricus, a Chinese herb, increases intracellular calcium and decreases MMP, followed by an increase in cancer cell apoptosis (Table 3) [18]. Mechanistically, the initial apoptotic pathway could possibly involve ER, which is one of the key players that controls the homeostasis of intracellular calcium. At a cellular level, ATP is generated by mitochondrial respiration mainly via oxidative phosphorylation, which occurs in the ETC [26]. Thereby, the inhibition of the ETC could reduce cellular ATP and ROS production. However, the Warburg effect enables the cancer cells to escape cell death due to cell energy depletion. NG inhibits the ETC at complexes I, II, and IV, decreasing ATP generation (Table 2) [13]. In addition, 17-DMAG and TRPM2 knockdown significantly decreased gastric cancer cell survival mainly through the inhibition of autophagy, mitochondrial function, and ATP production [15, 43]. Specifically, 17-DMAG competes with ATP to bind with HSP90, which decreases cancer cell viability [15]. It couples with TRPM2, maintaining cancer cell viability by its involvement in the ETC. TRPM2 knockdown was accompanied by decreasing ATP levels, and ROS levels were reduced, resulting in the inhibition of autophagy (Table 3).
Targeting cell cycle arrest
Excessive OS damages DNA, which the cell compensates by arresting the cell cycle for DNA repair [7]. The prolonged excessive OS induces irreversible DNA damage, which leads to cell apoptosis. A few studies showed that anticancer interventions inhibited the cell cycle at different phases [4, 6, 40]. PDOX caused cell cycle arrest at the G2/S phase (Table 2) [40]. PITC induced cell cycle arrest by increasing the S-phase and decreasing Cyclin A1 (Tables 2 and 3) [6]. Capsaicin induced cell cycle arrest at the G0/G1 phase, in association with decreasing Rb phosphorylation, Cyclin D1 and increasing p53 phosphorylation (Tables 2 and 3) [4].
Others
Yang et al. reported that the pro-inflammatory cytokine levels including interleukin-1 (IL-1), interleukin-6 (IL-6), interleukin-8 (IL-8), and TNF-alpha were decreased in human gastric cancer cell lines treated with Tomentosin 20 µM/ml at 24 h which correlated with a reduction in cell proliferation and an increase in cell apoptosis (Table 3) [14]. An ex vivo study in primary cell culture and gastric cancer cell lines found that increasing IL-8 levels predicted chemoresistance to platinum-based chemotherapy [52]. A decrease in pro-inflammatory cytokine levels might increase chemosensitivity by decreasing cell proliferation and increasing cell apoptosis.
In brief, the effects of interventions on gastric cancer cell lines mainly involve three parts, oxidative stress, apoptosis, and mitochondrial function. The final endpoints evaluated are cancer cell proliferation and progression, reported as changes in cell morphology, proliferation rate, cell growth rate, percentage of viable cells, rate of apoptosis, number of cell deaths, percentage of cell migration, and cell cycle distribution. The effects of interventions can be evaluated by the differentiation of gastric cancer cell lines coupled with the WHO classification used in clinical practice guidelines. Tables 1, 2, 3 and 4 summarize the results from in vitro studies. Table 1 shows the effects of MnSOD overexpression and topotecan on cell viability and invasion of rat and xenograft in nude mice gastric cancer cells. Table 2 shows the results found from studies on differentiated human gastric cancer cell lines including MGC-803, MKN-28, TMC-1, and SGC-7901. Table 3 demonstrates the results of undifferentiated cell lines KATO-III, HGC-27, MKN-45, AGS, and SNU-1. A summary of the targets of potential interventions on cancer cell proliferation and invasion classified by cancer cell differentiation are shown in Fig. 3. From the empirical evidence, the chemical and genetic interventions involve multiple mechanisms associated with decreasing cell viability and cell invasion. The combination of an anticancer agent with a mitochondrial targeting agent provides the synergistic effects by increasing apoptosis, as shown in Table 4. In undifferentiated gastric cancer cell lines, Mito-FF induced chemosensitivity of 5-FU and TRPM2-knockdown increased the anticancer effects of paclitaxel and DOX [3, 17]. Additionally, indomethacin increased oxaliplatin chemosensitivity by causing cell death [42].

The summary of the targets of potential interventions on cancer cell proliferation and invasion classified by cancer cell differentiation. From the empirical evidence, the chemical and genetic interventions involve multiple mechanisms for decreasing cell viability and cell invasion. a Differentiated gastric cancer cell line. b Undifferentiated gastric cancer cell line. KD knockdown
Table 5 shows the effects of chemical interventions on tumor size of gastric cancer cells in in vivo studies. Both Mito-FF and 5-FU were proved to have an effect on antioxidant enzyme downregulation and induce cancer cell apoptosis which resulted in a decrease in tumor size [17]. Mito-FF plus 5-FU enhanced the inhibition of tumor growth by increasing apoptosis and mitochondrial ROS synthesis [17]. Topotecan exerted its anti-cancer effect through a reduction in ASCT2 expression in a BALC/c nude mice model [14, 48]. Similarly, 17-DMAG induced a reduction in cancer cell proliferation, tumor weight and volume by decreasing antioxidant enzymes and increasing apoptosis [15].
Potential markers for gastric cancer treatment
In addition to the histological expression of gastric cancer cell lines, the phenotype and genotype could influence the treatment outcomes. The example gastric cancer cell lines were used to show p53 expression status. These, included the MKN-28 cell line: p53 mutation, MKN-45 and MKN-74 cell lines: wild-type p53, and KATO-III cell line: p53 deletion [53]. The efficacy of several agents was dependent on p53 expression. p53 is a tumor suppressor protein that regulates cell apoptosis, cell cycle arrest, DNA repair, and glycolysis [27, 50]. A previous study reported that wild-type p53 inhibited glycolysis and induced oxidative phosphorylation, and wild-type p53 mutation increased cancer cell proliferation and invasion [27]. Accordingly, gastric cancer patients with a wild-type p53 mutation were associated with poor prognosis [27]. The chemosensitivity of 5-FU was found to be related to p53 expression [12, 28]. p53 increased OS, which induced cell apoptosis [28]. The Bcl-2 family has been proposed as a potential apoptotic activator of targeting agents such as 5F and PITC, which induce p53-dependent apoptosis [6, 24]. Capsaicin also increased p53 expression [4]. However, several agents, including Anemarrhena asphodelodies, have been shown to induce apoptosis through the p53-independent pathway [50]. A previous study demonstrated that treatment with Anemarrhena asphodelodies increased apoptosis in both MKN-45 and KATO-III cells [50].
From in vivo study, MnSOD was found to be involved in cancer cell proliferation and invasion; however, the role of mitochondrial ROS in cancer cell invasion was controversial. In the clinical study, MnSOD expression was increased in gastric cancer patients, and the early gastric cancer patients had lower levels of MnSOD expression in comparison with advanced gastric cancer patients [47]. Malafa et al. reported that MnSOD expression was increased in gastric cancer patients with lymph node metastasis; in contrast, increased MnSOD expression was not associated with increased tumor depth invasion [47]. High MnSOD expression has been found to predict the advance of the disease in terms of lymph node metastasis. The analysis of gastrectomy specimens showed that 17-DMAG downregulated the antioxidant enzymes in both normal and cancerous gastric tissue [15]. TRPM2 expression was reported as being associated with a decrease in overall survival of gastric cancer patients and inhibition of TRPM2 increased the chemosensitivity of paclitaxel and doxorubicin [43]. TRPM2 expression may be used as a prognostic factor, particularly in stage 3 and 4 gastric cancer patients, high TRPM2 expression being associated with poor overall survival [3]. Thus, these findings suggested that p53, MnSOD, and TRPM2 expression may be used as predictive markers. A summary of the potential prediction markers for gastric cancer are shown in Table 6.
Conclusion and perspective
To date, the outcomes of chemotherapy in gastric cancer cases is unsatisfactory. Thus, to improve the outcomes of gastric cancer treatment, novel alternative interventions are needed. At a cellular level, mitochondria play an essential role in cancer cell homeostasis suggesting that therapies to target mitochondria may be useful in treatment of gastric cancer. Multiple potential targets have been reported including mitophagy, autophagy, mitochondrial fission and fusion, ROS production and elimination, apoptosis, ATP production, and cell cycle arrest. A growing body of basic research has shown that several natural, chemical, and genetic interventions can exert anticancer effects. However, based on the clinical findings to date, there is insufficient evidence to demonstrate their beneficial effects against gastric cancer in the affected patients. Therefore, to proceed with the clinical application of any of these approaches with any degree of certainty additional information is required around the mechanisms of action, appropriate dosage, and side effects before any of these alternative interventions in gastric cancer patients can be used with confidence in the near future.
Data availability
None.
Code availability
None.
References
Sung H, Ferlay J, Siegel RL et al (2020) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. https://doi.org/10.3322/caac.21660
Yan H, Qiu C, Sun W et al (2018) Yap regulates gastric cancer survival and migration via SIRT1/Mfn2/mitophagy. Oncol Rep. https://doi.org/10.3892/or.2018.6252
Almasi S, Kennedy BE, El-Aghil M et al (2018) TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J Biol Chem. https://doi.org/10.1074/jbc.M117.817635
Wang HM, Chuang SM, Su YC et al (2011) Down-regulation of tumor-associated NADH oxidase, tNOX (ENOX2), enhances capsaicin-induced inhibition of gastric cancer cell growth. Cell Biochem Biophys. https://doi.org/10.1007/s12013-011-9218-0
Kong GM, Tao WH, Diao YL et al (2016) Melittin induces human gastric cancer cell apoptosis via activation of mitochondrial pathway. World J Gastroenterol. https://doi.org/10.3748/wjg.v22.i11.3186
Huang L, Cai C, Dang W et al (2019) Propyl isothiocyanate induces apoptosis in gastric cancer cells by oxidative stress via glutathione depletion. Oncol Lett. https://doi.org/10.3892/ol.2019.10875
Mao Y, Song G, Cai Q et al (2006) Hydrogen peroxide-induced apoptosis in human gastric carcinoma MGC803 cells. Cell Biol Int. https://doi.org/10.1016/j.cellbi.2005.12.008
Barati N, Momtazi-Borojeni AA, Majeed M et al (2019) Potential therapeutic effects of curcumin in gastric cancer. J Cell Physiol. https://doi.org/10.1002/jcp.27229
Lo Y-C, Yang Y-C, Wu IC et al (2005) Capsaicin-induced cell death in a human gastric adenocarcinoma cell line. World J Gastroenterol. https://doi.org/10.3748/wjg.v11.i40.6254
Wang Q, Liu G, Hu C (2019) Molecular classification of gastric adenocarcinoma. Gastroenterol Res. https://doi.org/10.14740/gr1187
Berlth F, Bollschweiler E, Drebber U et al (2014) Pathohistological classification systems in gastric cancer: diagnostic relevance and prognostic value. World J Gastroenterol. https://doi.org/10.3748/wjg.v20.i19.5679
Matsuhashi N, Saio M, Matsuo A et al (2005) The evaluation of gastric cancer sensitivity to 5-FU/CDDP in terms of induction of apoptosis: time- and p53 expression-dependency of anti-cancer drugs. Oncol Rep 14:609–615
Liu L, Li T, Tan J et al (2014) NG as a novel nitric oxide donor induces apoptosis by increasing reactive oxygen species and inhibiting mitochondrial function in MGC803 cells. Int Immunopharmacol. https://doi.org/10.1016/j.intimp.2014.08.005
Yang H, Zhao H, Dong X et al (2020) Tomentosin induces apoptotic pathway by blocking inflammatory mediators via modulation of cell proteins in AGS gastric cancer cell line. J Biochem Mol Toxicol. https://doi.org/10.1002/jbt.22501
Kim JG, Lee SC, Kim O-H et al (2017) HSP90 inhibitor 17-DMAG exerts anticancer effects against gastric cancer cells principally by altering oxidant-antioxidant balance. Oncotarget. https://doi.org/10.18632/oncotarget.17007
Cao W, Yang W, Lou G et al (2009) Phase II trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) as first-line treatment for advanced gastric cancer. Anticancer Drugs. https://doi.org/10.1097/CAD.0b013e3283273509
Kim DJ, Jeena MT, Kim OH et al (2020) Novel therapeutic application of self-assembly peptides targeting the mitochondria in in vitro and in vivo experimental models of gastric cancer. Int J Mol Sci. https://doi.org/10.3390/ijms21176126
Zhang Y, Wang Q, Wang T et al (2012) Inhibition of human gastric carcinoma cell growth in vitro by a polysaccharide from Aster tataricus. Int J Biol Macromol. https://doi.org/10.1016/j.ijbiomac.2012.06.019
Marin JJ, Al-Abdulla R, Lozano E et al (2016) Mechanisms of resistance to chemotherapy in gastric cancer. Anticancer Agents Med Chem. https://doi.org/10.2174/1871520615666150803125121
Guideline Committee of the Korean Gastric Cancer Association DWG, Review P (2018) Korean practice guideline for gastric cancer 2018: an evidence-based, multi-disciplinary approach. J Gastric Cancer. https://doi.org/10.5230/jgc.2019.19.e8
Wu Z, Wang L, Wen Z et al (2021) Integrated analysis identifies oxidative stress genes associated with progression and prognosis in gastric cancer. Sci Rep. https://doi.org/10.1038/s41598-021-82976-w
Tamura M, Matsui H, Tomita T et al (2014) Mitochondrial reactive oxygen species accelerate gastric cancer cell invasion. J Clin Biochem Nutr. https://doi.org/10.3164/jcbn.13-36
Mazumder S, De R, Debsharma S et al (2019) Indomethacin impairs mitochondrial dynamics by activating the PKCζ-p38-DRP1 pathway and inducing apoptosis in gastric cancer and normal mucosal cells. J Biol Chem. https://doi.org/10.1074/jbc.RA118.004415
Liu Z, Ng EK, Liang NC et al (2005) Cell death induced by Pteris semipinnata L. is associated with p53 and oxidant stress in gastric cancer cells. FEBS Lett. https://doi.org/10.1016/j.febslet.2005.01.050
Zhang XW, Wang S, Tu PF et al (2018) Sesquiterpene lactone from Artemisia argyi induces gastric carcinoma cell apoptosis via activating NADPH oxidase/reactive oxygen species/mitochondrial pathway. Eur J Pharmacol. https://doi.org/10.1016/j.ejphar.2018.07.053
Li X, Fang P, Mai J et al (2013) Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. https://doi.org/10.1186/1756-8722-6-19
Liu Y, Zhang Z, Wang J et al (2019) Metabolic reprogramming results in abnormal glycolysis in gastric cancer: a review. Onco Targets Ther. https://doi.org/10.2147/OTT.S189687
Matsunaga T, Tsuji Y, Kaai K et al (2010) Toxicity against gastric cancer cells by combined treatment with 5-fluorouracil and mitomycin c: implication in oxidative stress. Cancer Chemother Pharmacol. https://doi.org/10.1007/s00280-009-1192-5
(NCCN) NCCN. Gastric Cancer. 2021 [updated March 9, 2021]; https://www.nccn.org/professionals/physician_gls/pdf/gastric.pdf.
Arai H, Iwasa S, Boku N et al (2019) Fluoropyrimidine with or without platinum as first-line chemotherapy in patients with advanced gastric cancer and severe peritoneal metastasis: a multicenter retrospective study. BMC Cancer. https://doi.org/10.1186/s12885-019-5720-3
Matsuhashi N, Saio M, Matsuo A et al (2004) Expression of p53 protein as a predictor of the response to 5-fluorouracil and cisplatin chemotherapy in human gastrointestinal cancer cell lines evaluated with apoptosis by use of thin layer collagen gel. Int J Oncol 24:807–813
Wagner AD, Wedding U (2009) Advances in the pharmacological treatment of gastro-oesophageal cancer. Drugs Aging. https://doi.org/10.2165/11315740-000000000-00000
Desmoulin F, Gilard V, Malet-Martino M et al (2002) Metabolism of capecitabine, an oral fluorouracil prodrug: (19)F NMR studies in animal models and human urine. Drug Metab Dispos. https://doi.org/10.1124/dmd.30.11.1221
Holohan C, Van Schaeybroeck S, Longley DB et al (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. https://doi.org/10.1038/nrc3599
Cunningham D (2006) Is oxaliplatin the optimal platinum agent in gastric cancer? Eur J Cancer Suppl. https://doi.org/10.1016/S1359-6349(06)70003-9
Huang D, Duan H, Huang H et al (2016) Cisplatin resistance in gastric cancer cells is associated with HER2 upregulation-induced epithelial-mesenchymal transition. Sci Rep. https://doi.org/10.1038/srep20502
Sakamoto J, Matsui T, Kodera Y (2009) Paclitaxel chemotherapy for the treatment of gastric cancer. Gastric Cancer. https://doi.org/10.1007/s10120-009-0505-z
Doxorubicin AK (2007). In: Enna SJ, Bylund DB (eds) xPharm: the comprehensive pharmacology reference. Elsevier, New York, pp 1–5
Florou D, Patsis C, Ardavanis A et al (2013) Effect of doxorubicin, oxaliplatin, and methotrexate administration on the transcriptional activity of BCL-2 family gene members in stomach cancer cells. Cancer Biol Ther. https://doi.org/10.4161/cbt.24591
Zhong YJ, Liu SP, Firestone RA et al (2013) Anticancer effects of Ac-Phe-Lys-PABC-doxorubicin via mitochondria-centered apoptosis involving reactive oxidative stress and the ERK1/2 signaling pathway in MGC-803 cells. Oncol Rep. https://doi.org/10.3892/or.2013.2629
Tangchirakhaphan S, Innajak S, Nilwarangkoon S et al (2018) Mechanism of apoptosis induction associated with ERK1/2 upregulation via goniothalamin in melanoma cells. Exp Ther Med. https://doi.org/10.3892/etm.2018.5762
Vallecillo-Hernández J, Barrachina MD, Ortiz-Masiá D et al (2018) Indomethacin disrupts autophagic flux by inducing lysosomal dysfunction in gastric cancer cells and increases their sensitivity to cytotoxic drugs. Sci Rep. https://doi.org/10.1038/s41598-018-21455-1
Miller BA (2019) TRPM2 in cancer. Cell Calcium. https://doi.org/10.1016/j.ceca.2019.03.002
Zhang G-E, Jin H-L, Lin X-K et al (2013) Anti-tumor effects of Mfn2 in gastric cancer. Int J Mol Sci. https://doi.org/10.3390/ijms140713005
Tilokani L, Nagashima S, Paupe V et al (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. https://doi.org/10.1042/ebc20170104
Indo HP, Matsui H, Chen J et al (2015) Manganese superoxide dismutase promotes interaction of actin, S100A4 and Talin, and enhances rat gastric tumor cell invasion. J Clin Biochem Nutr. https://doi.org/10.3164/jcbn.14-146
Malafa M, Margenthaler J, Webb B et al (2000) MnSOD expression is increased in metastatic gastric cancer. J Surg Res. https://doi.org/10.1006/jsre.1999.5773
Wang L, Liu Y, Zhao TL et al (2019) Topotecan induces apoptosis via ASCT2 mediated oxidative stress in gastric cancer. Phytomedicine. https://doi.org/10.1016/j.phymed.2018.12.011
Ohyama K, Akaike T, Imai M et al (2005) Human gastric signet ring carcinoma (KATO-III) cell apoptosis induced by Vitex agnus-castus fruit extract through intracellular oxidative stress. Int J Biochem Cell Biol. https://doi.org/10.1016/j.biocel.2005.02.016
Takeda Y, Togashi H, Matsuo T et al (2001) Growth inhibition and apoptosis of gastric cancer cell lines by Anemarrhena asphodeloides Bunge. J Gastroenterol. https://doi.org/10.1007/s005350170135
Liu E, Liang T, Wang X et al (2015) Apoptosis induced by farrerol in human gastric cancer SGC-7901 cells through the mitochondrial-mediated pathway. Eur J Cancer Prev. https://doi.org/10.1097/cej.0000000000000104
Limpakan Yamada S, Wongsirisin P, Yodkeeree S et al (2019) Interleukin-8 associated with chemosensitivity and poor chemotherapeutic response to gastric cancer. J Gastrointest Oncol. https://doi.org/10.21037/jgo.2019.09.02
Nagamine M, Okumura T, Tanno S et al (2003) PPAR gamma ligand-induced apoptosis through a p53-dependent mechanism in human gastric cancer cells. Cancer Sci. https://doi.org/10.1111/j.1349-7006.2003.tb01443.x
Funding
This work was supported by the NSTDA Research Chair grant from the National Science and Technology Development Agency Thailand (NC), The National Research Council of Thailand 2563NRCT321511 (KS), the Chiang Mai University Fundamental Fund (KS), the Senior Research Scholar grant from the National Research Council of Thailand (SCC), and the Chiang Mai University Center of Excellence Award (NC).
Author information
Authors and Affiliations
Contributions
All authors contributed to the conception. Literature search and data collection were performed by PT. The first draft of the manuscript was written by PT and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tanprasert, P., Limpakan (Yamada), S., Chattipakorn, S.C. et al. Targeting mitochondria as a therapeutic anti-gastric cancer approach. Apoptosis 27, 163–183 (2022). https://doi.org/10.1007/s10495-022-01709-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-022-01709-0