
Abstract
Tissue homeostasis in metazoa requires the rapid and efficient clearance of dying cells by professional or semi-professional phagocytes. Impairment of this finely regulated, fundamental process has been implicated in the development of autoimmune diseases, such as systemic lupus erythematosus. Various studies have provided us a detailed understanding of the interaction between dying cells and phagocytes as well as the current concept that apoptotic cell removal leads to a non- or anti-inflammatory response, whereas necrotic cell removal stimulates a pro-inflammatory reaction. In contrast, our knowledge about the soluble factors released from dying cells is rather limited, although meanwhile it is generally accepted that not only the dying cell itself but also the substances liberated during cell death contribute to the process of corpse clearance and the subsequent immune response. This review article is intended as an up-to-date survey over attraction and danger signals of apoptotic, primary and secondary necrotic cells, their function as chemoattractants in phagocyte recruitment, additional effects on the immune system, and the receptors, which are engaged in this scenario.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Controlled cell death comprises a vital element for the maintenance of tissue homeostasis in metazoa. Cells die by apoptosis and the body has to deal with billions of corpses during daily cell turnover and tissue renewal. The disposal of the apoptotic remains is fundamentally important, since otherwise apoptotic cells tend to become secondary necrotic, release intracellular contents, and instigate inflammation and autoimmunity [1, 2]. Although considered non-physiological, necrosis produces dead cells as well. The elimination of necrotic cells represents an even greater challenge for the phagocytic apparatus, since due to their leaky plasmamembrane not only the corpses but also the accidentally liberated intracellular constituents have to be removed. To this end, in multicellular organisms apoptotic and necrotic cells are rapidly engulfed either by phagocytosis-competent neighboring cells or by professional phagocytes [3]. But how do phagocytes reach their prey in time, since usually they are not located directly next to the dying cell? One solution would be that the dying cell secretes soluble mediators, which attract the scavengers. Particularly in the supernatants of apoptotic cells diverse attraction or ‘find-me’ signals have been identified. The chemoattractants produced by apoptotic cells do not only recruit macrophages to the site of cell death, they also modulate their activation and differentiation and thereby can influence the consecutive immune response. The effects on phagocyte activation and the subsequent immune response are especially well studied for soluble factors released by primary necrotic cells. However, the process of phagocyte recruitment here is poorly understood. Even less is known about the factors released by secondary necrotic cells. So far it remains elusive if the milieu created around a secondary necrotic cell is an additive mixture of factors released during the apoptotic and the post-apoptotic state or if additional factors are liberated and non-additive effects can be observed.
This review article addresses the soluble attraction and/or danger signals secreted by dying (apoptotic) and dead (primary and secondary necrotic) cells as well as the phagocyte’s sensing and response mechanisms.
‘Find-me’ signals of apoptotic cells
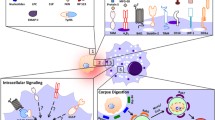
‘Find-me’ signals released from apoptotic cells play a critical role in their timely removal. In the following we will discuss the different attraction signals, which have been described in the context of apoptotic cell removal (Fig. 1).

Attraction signal synapse between an apoptotic cell and a target cell. Casp Caspase, EMAP II endothelial monocyte-activating polypeptide II, FKN fractalkine, G2A G-protein-coupled receptor G2A, HBD heparin-binding domain, iPLA 2 β Calcium-independent phospholipase A2β, LPC lysophosphatidylcholine, LTF lactoferrin, μBleb micro Bleb, NTP nucleoside triphosphate, P2X purinergic receptor X, P2Y purinergic receptor Y, PC phosphatidylcholine, RP S19 ribosomal protein S19, S1P sphingosine-1-phosphate, Sph sphingosine, TGase 2 transglutaminase 2, TSP-1 thrombospondin-1, TyrRS tyrosyl tRNA synthetase
Dimer of ribosomal protein S19 (dRP S19)
The covalent dimer of ribosomal protein S19 (dRP S19) was the first attraction signal of apoptotic cells to be identified [4]. Originally characterized as a part of the small subunit of ribosomes, RP S19 was later described as the essential monocyte chemoattractant in synovial extracts of patients with rheumatoid arthritis [5]. When apoptotic HL-60 cells, which release a cross-linked dimer of RP S19 (dRP S19), were injected intradermally into guinea pigs, a strong monocyte/macrophage infiltration was observed resulting in a rapid phagocytosis of the apoptotic cells [4]. The authors hypothesized that transglutaminase 2 was responsible for covalent dimerization of RP S19. Intriguingly, only the dimer (dRP S19) exerted the specific chemoattractant effect on monocytes and macrophages [6, 7]. The use of neutralizing antibodies and receptor antagonists identified the G-protein coupled receptor (GPCR) CD88 as the crucial receptor in dRP S19 stimulated chemotaxis [8].
Additionally, dRP S19 exerted mitigating effects on macrophages and neutrophils [9], thus, contributing to the anti-inflammatory character of apoptotic cell clearance. However, further studies have to clarify, how dRP S19 is secreted and whether the secretion is independent of the cell type as well as the apoptotic stimulus employed.
Endothelial monocyte-activating polypeptide II (EMAP II)
Another apoptotic cell-derived attraction signal is the endothelial monocyte-activating polypeptide II (EMAP II) [10]. EMAP II was purified from the supernatant of murine fibrosarcoma cells on the basis of its ability to induce tissue factor expression on endothelial cells [11]. Later it became obvious that it acts as an in vitro chemoattractant for neutrophils and monocytes [12]. In their initial study Knies et al. observed a profound abundance of EMAP II mRNA at sites of active tissue remodeling during mouse embryogenesis. The authors spotted an accumulation of F4/80-positive macrophages adjacent to the EMAP II mRNA signal in apoptotic cells and hypothesized that EMAP II was responsible for phagocyte accumulation in vivo [10].
Translation of the EMAP II mRNA results in a precursor protein (43 kDa), which comparable to pro-IL-1β lacks a conventional secretion signal and has to be proteolytically processed in order to mature and release the active 23 kDa protein. This cleavage occurs at an aspartate residue (ASTD↓) during apoptosis but not necrosis [10] and is mediated by the executioner caspases-3 and -7 [13]. Pro-EMAP II p43 today is known to be identical to the p43 subunit of the mammalian tRNA multi-synthetase complex [14]. Yet, it has to be mentioned, that pro-EMAP II processing and release of mature EMAP II p23 have been observed to be rather late events in the course of apoptosis occurring several hours after poly (ADP-ribose) polymerase (PARP) cleavage. Unfortunately, it was not examined, whether mature EMAP II p23 was released from apoptotic cells with an intact plasmamembrane or rather by secondary necrotic cells with compromised membrane integrity. Since in vivo apoptotic cells are phagocytosed at an early stage, Behrensdorf et al. [13] suggested that mature EMAP II p23 serves as a chemoattractant back-up signal at sites of excessive cell death where the additional recruitment of mononuclear phagocytes would contribute to the clearance process. This model also takes into account the formerly described pro-inflammatory effects of EMAP II p23 (i.e. myeloperoxidase release in neutrophils and stimulation of TNF-α production in monocytes [15]), and of pro-EMAP II p43 (i.e. induction of TNF-α synthesis, ICAM-1 adhesion molecule and several other cytokines and chemokines (IL-8, MCP-1, MIP-1α, MIP-1β, MIP-2α, RANTES, and IL-1β) in THP-1 cells [16]). Taken together, these findings suggest a function of EMAP II as an attraction and danger signal of secondary necrosis rather than of early apoptosis.
The only receptor, which has been reported in the context of EMAP II p23-stimulated lymphocyte and endothelial cell migration, is CXCR3 [17]. But it remains to be clarified, whether CXCR3 is the crucial sensor for EMAP II p23 in monocyte and granulocyte chemotaxis, since so far it is mainly known to be engaged in lymphocyte recruitment [18].
Fragments of human tyrosyl tRNA synthetase (TyrRS)
An additional ‘unusual’ chemokine, which similar to EMAP II is released during apoptosis and which in healthy cells also plays a role in aminoacylation of tRNAs, is human tyrosyl tRNA synthetase (TyrRS) [19]. Under apoptotic conditions TyrRS has been observed to be released into the culture supernatant of U937 cells that still have retained an intact plasmamembrane. Once secreted, TyrRS has to be proteolytically processed in order to function as a chemokine. Wakasugi and Schimmel [19] identified human neutrophil elastase as one putative extracellular protease capable of cleaving TyrRS in vitro into an N-terminal (mini TyrRS) and a C-terminal fragment. This cleavage was an absolute prerequisite for the function as attraction signal, since only the fragments but not the full length TyrRS exhibited the chemokine activity. Mini TyrRS comprising a conserved ELR motif, which precedes the first cysteine residue at the N-terminus, acts as chemoattractant only for granulocytes, whereas the C-terminal domain sharing 51% of sequence identity to mature EMAP II stimulates migration of granulocytes and mononuclear phagocytes. By in vitro competition binding assays CXCR1 was identified as the receptor mediating mini TyrRS-induced chemotaxis. The phagocyte sensor responsible for monocyte and neutrophil migration to the EMAP II-like C-terminal domain so far remains elusive.
Beyond its chemoattractant function the C-terminal fragment of TyrRS has been shown to induce the production of TNF-α and tissue factor in mononuclear phagocytes and to instigate myeloperoxidase release by granulocytes [19]. Thus, it can be concluded that the chemokine properties of cleaved TyrRS appropriately fit into the hypothesis that apoptotic cells release ‘find-me’ signals in order to trigger their timely and efficient removal; yet the pro-inflammatory effects of the EMAP II-like C-terminal domain are not in accordance with the concept that apoptotic cell clearance generally is anti-inflammatory. Future studies should address this conflict and clarify which phagocyte receptor is the sensor for the EMAP II-like C-terminal domain of TyrRS.
Thrombospondin 1 (TSP-1) and its heparin-binding domain (HBD)
The homotrimetric matricellular glycoprotein Thrombospondin 1 (TSP-1) and its 26 kDa fragment comprising the N-terminal heparin-binding domain (HBD) are further proteinaceous factors, whose production and release during apoptosis have been reported [20]. TSP-1 previously had been shown to modulate a number of cellular processes, including migration, proliferation and angiogenesis [21].Yet, Krispin et al. [20] were the first to report that mRNA as well as protein synthesis of TSP-1 and its subsequent release are actively induced in apoptosing peripheral monocytes and neutrophils, although a role of TSP-1 as bridging protein in the phagocytic synapse of apoptotic cell removal had already been acknowledged [22]. Apart from the full length TSP-1 the free N-terminus comprising the heparin-binding domain (HBD) was detected in the culture supernatants of apoptotic peripheral monocytes [20]. Unfortunately, the authors did neither examine the destiny of the C-terminal fragment nor the chemotactic potential of the culture supernatants. This would have been highly interesting, since the moiety responsible for the induction of monocyte chemotaxis had previously been mapped to the 140 kDa C-terminal region [23], whereas neutrophils had been reported to migrate only in response to the full length protein or a combination of the N- and the C-terminal fragments [24]. However, the final proof that TSP-1 or its fragments released during apoptosis act as ‘find-me’ signals for professional phagocytes is still missing and the phagocyte sensor(s) involved remain obscure. Yet, seminal migration data collected with neutralizing antibodies suggest that the vitronectin receptor (αvβ3 integrin) might be involved [25].
In addition to its putative role as apoptotic ‘find-me’ signal TSP-1 has been shown to reduce lipopolysaccharide (LPS)-induced upregulation of MHC class II and CD86 surface expression in iDCs and their T cell activating capacities [20]. Unfortunately the authors failed to identify a single phagocyte receptor mediating this tolerizing effect but rather observed a simultaneous participation of CD29, CD36, CD47, CD51, and CD91, respectively [20].
Soluble IL-6 receptor (sIL-6R)
IL-6 trans-signalling, a process known to be an important regulator of immune responses in the context of innate as well as acquired immunity, has been reported to play a role in monocyte recruitment by apoptosing neutrophils. The basis of IL-6 trans-signalling is the release of soluble IL-6 receptor (sIL-6R), which in turn binds IL-6 to create an agonistic IL 6/sIL-6R complex. This complex subsequently interacts with ubiquitously expressed CD130 on the target cell and, thus, is capable of activating cell types, which due to a lack of IL-6R expression would otherwise not respond to IL-6 itself [26]. Chalaris et al. [27] were the first to observe IL-6R shedding from the surface of apoptosing neutrophils under participation of the metalloproteinase ADAM 17.
Importantly, the authors provided evidence for a contribution of IL-6R shedding to the resolution of inflammation. Utilizing a murine carrageenan air pouch model of acute inflammation they could demonstrate that neutrophil depletion resulted in reduced local sIL-6R levels and a concomitant decrease in the number of recruited mononuclear cells [27]. Thus, it was concluded that apoptosis-induced IL-6R shedding from neutrophils promotes IL-6 trans-signalling and regulates the transition from the initial neutrophil infiltration to a more sustained population of mononuclear cells, which finally are engaged in the clearance of apoptotic neutrophils and hence the resolution of inflammation. Intriguingly, this cascade has been shown to be nonfunctional in IL6−/− mice and transgenic mice overexpressing a soluble form of CD130 [28, 29].
Fractalkine (FKN)
Another facette of monocyte attraction by apoptotic cells, which closely resembles that of ADAM 17-mediated IL-6R shedding, has been described by Truman et al. [30]. In this study the authors portrayed the release of membrane-bound fractalkine by a so far unknown protease in apoptotic Burkitt lymphoma cells leading to the CX3CR1-dependent recruitment of macrophages [30]. Intriguingly, fractalkine so far is the only classical chemokine, which has been reported to play a ‘find-me’-signal role in the context of apoptotic cell removal. The CX3CR1-fractalkine receptor–ligand system seems to be of special importance in the germinal centers, since significantly fewer macrophages were observed in the germinal centers of CX3CR1−/− mice when compared to the wildtype controls. However, despite the lower macrophage numbers no accumulation of apoptotic or secondary necrotic corpses was described suggesting that the clearance process was not detectably compromised. Hence, additional studies are required to further validate the attraction signal function of fractalkine in this context- in particular because fractalkine expression is rather limited to certain cell types, such as B cells and neurons.
Apart from macrophage recruitment fractalkine can exert different other (mainly anti-inflammatory) effects. Thus, CX3CR1 signalling has been reported to suppress inflammatory neurotoxicity in microglia [31]. Furthermore, there is evidence for fractalkine modulating the macrophages’ capacity to engulf apoptotic prey cells by inducing the expression of the bridging protein MFG-E8 [32, 33].
Lysophosphatidylcholine (LPC)
In 2003 we identified lysophosphatidylcholine (LPC) as the first apoptotic cell-derived lipid ‘find-me’ signal being generated due to the caspase-3 mediated activation of the calcium-independent, cytosolic phospholipase A2β (iPLA2β) [34]. LPC had already been described as surface bound target for natural IgM antibodies [35] and may, therefore, fulfill a dual role as membrane bound ‘eat-me’ and as soluble ‘find-me’ signal.
Our recent analyses revealed that only LPC but none of its metabolic derivates or related lysophospholipids exhibits a chemotaxis-stimulating effect on monocytic cell lines and that the GPCR G2A is crucially engaged in this scenario [36]. G2A knock-out mice previously had been reported to develop the typical autoimmune phenotype of a late-onset, multi-organ inflammatory syndrome closely resembling human systemic lupus erythematosus (SLE) [37], which was already known from mice with other deficiencies in apoptotic cell recognition [38–40]. This observation strongly supports the current notion that defects in apoptotic cell clearance contribute to chronic inflammation and the onset of autoimmune diseases [1].
Lysophosphatidylcholine has various other immunomodulatory effects. Exemplarily, administration of LPC induced the expression of the classical monocyte attracting chemokines MCP-1 and RANTES in human vascular endothelial cells, thus acting as an amplification loop of phagocyte recruitment [41]. Treatment with LPC also enhanced Fc-receptor-mediated phagocytosis [42, 43]. If this also holds true for Fc-receptor-independent phagocytic removal of apoptotic cells, remains to be clarified. Therapeutic in vivo administration of LPC in a mouse sepsis model markedly enhanced the clearance of intraperitoneal bacteria and the bactericidal activity of neutrophils [44]. LPC also significantly suppressed the endotoxin-induced release of high-mobility group box 1 (HMGB-1) protein from monocytes and macrophages in endotoxemia and sepsis [45]. From this it was concluded that LPC confers protection against lethal sepsis by facilitating the elimination of the invading pathogens and by inhibiting the endotoxin-induced release of the pro-inflammatory cytokine HMGB-1.
However, pro-inflammatory effects of LPC have also been described. Amongst them were stimulation of phospholipase D activity in murine peritoneal macrophages [46], augmentation of IL-1β-stimulated inducible NO synthase (iNOS) expression [47], promotion of dendritic cell (DC) maturation [48], and increase in anti-CD3-stimulated interferon-γ (IFN-γ) production and CD40 ligand expression in CD4+ T cells [49]. Future studies have to clarify how these observations fit into the current concept that the timely clearance of apoptotic cells basically constitutes an anti-inflammatory process.
Sphingosine-1-phosphate (S1P)
Sphingosine-1-phosphate (S1P) has been described as a second apoptotic lipid attraction signal [50]. This study revealed a profound increase in sphingosine kinase 1 (SphK1) protein expression with a concomitant enhancement of SphK1 activity and subsequent S1P release into the culture supernatants of apoptosing Jurkat cells. The latter (S1P release during apoptosis) had already been observed by Weigert et al. [51, 52]; however, these authors identified SphK2 as the crucial S1P generating enzyme. As shown by Gude et al. [50], purified S1P exhibits a chemotactic effect on monocytes and macrophages. Unfortunately, the authors did not demonstrate that S1P derived from apoptotic Jurkat cells is of crucial importance for the chemotactic activity of the corresponding culture supernatants. This question could be resolved by analyses employing S1P degrading enzymes or other means of S1P depletion. Moreover, it should be clarified, if S1P release is a general apoptotic phenomenon or if it is restricted to certain cell types. The receptors engaged in S1P-stimulated chemotaxis are 5 related GPCRs termed S1P-R1 to S1P-R5. However, so far no data is available if one of the S1P-Rs is of special importance, since mononuclear phagocytes express all of the known family members [53].
In addition to its effects on monocyte migration S1P liberated during apoptosis might have other functions. Thus, it was suggested that S1P derived from apoptotic tumor cells stimulates macrophage polarization towards a tumor-associated macrophage phenotype with reduced capacity to produce pro-inflammatory cytokines, such as TNF-α and IL-12, and instead enhanced release of immuno-suppressive mediators, including IL-10 and PGE2 [52, 54]. These studies imply that the liberation of S1P during apoptosis might contribute to the anti-inflammatory character of apoptotic cell clearance.
Apoptotic micro-blebs
In 1999, Segundo et al. [55] described an apoptotic cell-derived attraction signal with undefined molecular character: apoptotic micro-blebs. The authors observed that membraneous particles (0.2 μm in size), which are actively released by apoptotic human germinal center B cells, exhibit chemoattractive activity on monocytes. After depletion of the micro-blebs by ultrafiltration or ultracentrifugation this chemoattractant activity was almost abrogated. Yet, the responsible molecular entity, the range of action and the phagocyte sensor(s) involved in this context remain to be resolved.
Nucleotides
In a very recent report Elliot et al. [56] identified the nucleotides ATP and UTP as a new class of apoptotic cell-derived attraction signals. The authors reported the caspase-dependent release of ATP and UTP in an early phase of apoptosis across an intact plasma membrane resulting in a selective attraction of monocytes and macrophages. Utilizing two murine in vivo models they observed a crucial role of the purinergic P2Y2 receptor in nucleotide-mediated monocyte/macrophage attraction. However, further studies are needed to clarify the molecular mechanisms underlying apoptosis-associated nucleotide secretion and to analyze the phenotype of the P2Y2 knockout-mouse in terms of chronic inflammation and autoimmunity. Most importantly, the apparent discrepancy between ATP’s function in non-inflammatory apoptotic cell removal and its role as a classical danger signal in the context of tissue damage, allergen-induced asthmatic airway inflammation and the T cell response against tumor cells stimulated to undergo an immunogenic form of cell death (see the paragraph on ATP below and [57, 58]) has to be resolved.
Lactoferrin (LTF)
Amongst the soluble factors being liberated during apoptosis, lactoferrin (LTF), a 75–80 kDa transferrin-related, iron-binding protein constituent of colostrums, saliva, lacrimal fluid and neutrophil secondary granules, plays a special role. Its de novo expression and release during apoptosis have been observed by Bournazou et al. [59], but in contrast to the ‘find-me’ signals described above, LTF exerts the function of a neutrophilic anti-attraction or ‘keep-out’ signal. LTF was reported to profoundly inhibit neutrophil migration towards different classical chemoattractants, including fMLP, C5a, IL-8 or LTB4, in vitro and in vivo. Importantly, this inhibition was only to be observed with polymorphonuclear but not with mononuclear phagocytes [59]. In vivo the lack of granulocyte recruitment to apoptotic zones represents a key feature of the non-inflammatory apoptosis program, clearly discriminating areas of physiological cell death from sites of pathological tissue destruction or infection—a finding, which now can be explained at least in part by the presence of LTF. The identity of the LTF receptor on neutrophils remains unknown.
Apart from its neutrophilic ‘keep-out’ signal function, other LTF-mediated anti-inflammatory effects, including inhibition of neutrophil activation [59], inhibition of TNF-α, IL-1, and IL-6 production [60, 61] and stimulation of IL-10, IL-4 and TGF-β production [62, 63] have been reported. In conclusion, LTF seems to be a central anti-inflammatory component of the apoptotic milieu and is the first neutrophilic ‘keep-out’ signal, which has been identified. Future studies are required in order to identify other ‘keep-out’ signals and to validate their putative involvement in pathologic disorders, like SLE or rheumatoid arthritis.
Danger signals of primary necrotic cells
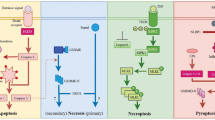
Necrosis is considered as an accidental, uncontrolled type of cell death. However, recent data describe forms of necrosis displaying features of a well controlled cell death occurring not only under pathophysiological conditions but also physiologically during development (e.g. the death of chondrocytes controlling the longitudinal growth of bones) and tissue homeostasis (e.g. the death of intestinal epithelial cells) [64–67]. In contrast to apoptosis necrosis generally is considered to be associated with inflammation, although there are scattered reports describing conflicting observations—particularly when the dying cells are separated from their soluble releases [68, 69]. Potentially cytotoxic and autoantigenic intracellular components get exposed and act as endogenous danger signals or damage associated molecular patterns (DAMPs) that ‘inform’ the immune system about the tissue damage [70, 71]. During necrosis most of the intracellular content is released, however, only a handful of molecules have been identified to elicit necrosis-induced immune signalling (Fig. 2).

Danger signal synapse between a primary necrotic cell and a target cell. COX-2 cyclooxygenase-2, GP glycoprotein, HDGF hepatoma-derived growth factor, HMGB-1 high mobility group box 1 protein, HSP heat-shock protein, ICAM intercellular adhesion molecule, iNOS inducible NO-synthase, MSU monosodium urate, RAGE receptor for advanced glycation end products, snRNP small nuclear ribonucleoprotein, SR scavenger receptor, TLR toll-like receptor, VCAM vascular cell adhesion molecule
High mobility group box 1 protein (HMGB-1)
HMGB-1 is a 30 kDa ubiquitous, nuclear, non-histone protein loosely bound to chromatin [72] with cytokine-like activity in the context of tissue injury and inflammation [73–75].
The detailed mechanisms of nuclear export and subsequent HMGB-1 release during necrosis induced by various stimuli, including ionomycin, carbonyl cyanide 3-chlorophenylhydrazone, deoxyglucose or azide, are not fully understood, but importantly HMGB-1 secretion does not occur during apoptosis—presumably due to freezing of HMGB-1 to chromatin or to hypoacetylated proteins within the apoptotic nucleus. This clear difference between apoptotic and necrotic cells in terms of HMGB-1 release might contribute to the vital distinction in the activation of an anti- or pro-inflammatory response to dying and dead cells. Furthermore, apoptotic cells have been shown to oxidatively modify HMGB-1, thereby switching its pro-inflammatory to a rather anti-inflammatory character [76].
Apart from its passive liberation during necrosis HMGB-1 has been reported to be expressed on the cell surface and to be actively secreted by monocytes, macrophages, and DCs in response to inflammatory stimuli, such as IL-1, TNF-α, IFN-γ, or LPS [77, 78]. Here, it exhibits the characteristics of a leaderless cytokine not being translocated from the Golgi apparatus to the cell membrane directly after synthesis (like IL-2), but rather (similar to cytokines of the IL-1 family) requiring alternative means of secretion by organelles that belong to the endolysosomal compartment. In this context hyperacetylation of HMGB-1 at several of its 43 lysine residues prevented nuclear-import and consequently led to cytosolic HMGB-1 accumulation in cytoplasmatic secretory vesicles ready to be released into the extracellular space [77]. Notably, pre-treatment with LPC inhibited activation-induced HMGB-1 release, suggesting a cross-talk between apoptotic ‘find-me’ and necrotic danger signals [45]. Although the detailed signalling mechanisms regulating activation-induced HMGB-1 secretion remain elusive, a positive feedback loop may operate at sites of necrosis.
Extracellular HMGB-1 has been shown to incite various immune cell reactions. In vitro HMGB-1 exerted a direct chemotactic effect on neutrophils [79] and accompanied by activation of endothelial cells with enhanced expression of vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1) and endothelial cell selectin (E-selectin) this accounted for transendothelial migration of neutrophils in vivo [80]. Transendothelial migration of monocytes under autocrine participation of surface bound HMGB-1 was described by Rouhiainen et al. [81], but in this study no direct chemotactic effect of soluble HMGB-1 could be detected. In concert with IL-1 and IL-2 family members HMGB-1 has been reported to promote the interaction between natural killer (NK) cells and monocytes or iDCs leading to enhanced DC maturation [75, 82, 83].
Furthermore, HMGB-1 has been described to directly modulate cytokine expression. In vitro culture of monocytes with purified HMGB-1 resulted in secretion of the pro-inflammatory cytokines TNF-α, IL-1α, IL-1β, IL-6, IL-8, MIP-1α, and MIP-1β [84]. Increased TNF-α production could also be observed in monocytes that had been incubated with necrotic HMGB-1+/+ but not with HMGB-1−/− fibroblasts [73], and in vivo injection of purified HMGB-1 led to rising TNF-α serum levels. Yet, the direct effect of HMGB-1 on cytokine production still is uncertain, since stimulation with highly purified recombinant HMGB-1 induced only a very weak pro-inflammatory cytokine response in mononuclear cells [85]. Consequently, the authors proposed that HMGB-1 might need a cofactor, like lipopolysaccharide or single stranded DNA, in order to exert its immune modulatory effects. This hypothesis has to be carefully validated in future studies.
At least 3 different multi-ligand receptors have been described to be engaged in HMGB-1 dependent immune signalling: The receptor for advanced glycation end products (RAGE) and the toll-like receptors (TLR) 2 and 4 with the common downstream pathway engaging myeloid differentiation primary response protein 88 (MyD88) and NF-κB [86–88].
Increased extracellular HMGB-1 expression in serum and synovial fluid has been reported to be involved in pathologic conditions, such as sepsis [89] or arthritis [90, 91], suggesting that HMGB-1 might be a future target for therapeutic interventions of acute or chronic inflammatory disorders.
Hepatoma-derived growth factor (HDGF)
Hepatoma-derived growth factor (HDGF) is another candidate for a necrotic cell-derived danger signal that is related to HMGB-1 (32% amino acid sequence homology). HDGF is a ubiquitously expressed protein with a bipartite nuclear localization signal lacking a classical signal peptide for secretion [92]. Besides its mainly nuclear localization HDGF is passively released during ionomycin- and carbonyl cyanide 3-chlorophenylhydrazone-mediated necrosis and (in very little amounts) actively secreted by healthy cells [93]. In the course of apoptosis HDGF has been reported to be retained in the nucleus and consequently trapped inside apoptotic cells. Further studies are necessary to explore the immunological consequences of HDGF secretion during necrosis and its putative role as danger signal.
The calgranulin family of proteins (S100 proteins)
The members of the calgranulin family of proteins, also referred to as S100 proteins (Soluble in 100% ammonium sulfate at neutral pH), comprise more than 20 related, small (10–20 kDa), acidic, homo- and hetero-dimerizing, calcium-binding proteins of the helix-loop-helix (‘EF-hand type’) conformation. S100 proteins are tissue specifically expressed and serve a variety of intracellular functions, including signal transduction and regulation of proliferation [94]. Extracellular danger signal functions have been reported for the family members S100A8, S100A9, S100A12 predominantly expressed in granulocytes, monocytes and immature macrophages, and S100B mainly found in the brain [95–97]. A potent chemoattraction of neutrophils was exerted by purified S100A8 in vitro and in vivo [98, 99]. Treatment with S100A9 and the S100A8-S100A9 heterodimer, but not with S100A8 alone, led to an increased CD11b expression strengthening the interaction between monocytes and endothelial cells. S100A8 and S100A9 have been found in high levels in the endothelium of inflamed tissues close to transmigrating leukocytes. Neutralizing antibodies against S100A9 or the S100A8-S100A9 heterodimer strongly inhibited transendothelial migration of monocytes supporting the notion that S100A8 and A9 are involved in monocyte recruitment [100]. A role of the S100A8-S100A9 heterodimer in the propagation of inflammation has been suggested [101], but there are also conflicting reports showing a rather anti-inflammatory effect on macrophage and lymphocyte activation [102, 103].
Another pro-inflammatory S100 family member is S100A12. Endothelial cells have been described to respond to S100A12 with a strong upregulation of VCAM-1 and ICAM-1 expression accompanied by enhanced adhesion of mononuclear cells and granulocytes—a prerequisite for extravasation [104]. Moreover, S100A12 stimulated a strong chemotactic response in monocytes and induced IL-1β, TNF-α and IL-2 secretion in murine macrophages and human PBMC, respectively. In vivo systemic infusion of S100A12 led to an increase in VCAM-1 expression in the lung and injection of S100A12 into mouse footpads stimulated a profound inflammatory response characterized by leukocyte infiltration and subcutaneous edema. Due to competition studies the effects of S100A12 were at least in part mediated by RAGE, the only known receptor for S100 proteins so far [104].
Apart from myeloid S100A8, S100A9, and S100A12, the brain protein S100B has been described to exert various pro-inflammatory effects, like NF-κB-dependent induction of IL-6 mRNA in neurons, iNOS activation in astrocytes, and increased cyclooxygenase-2 (COX-2) expression in microglia [105–107]. Yet, the RAGE-dependent induction of iNOS mRNA expression as well as NO secretion by microglia was only to be observed in combined treatment with S100B and IFN-γ [108, 109]. Since at least some of the effects mentioned above were only dependent on the presence of the extracellular but not the signal transducing domain of RAGE, the involvement of RAGE in S100B-mediated microglia activation remains obscure.
One of the open questions concerning the role of S100 proteins as necrotic cell-derived danger signals is if and how they are released during necrosis, since so far only for S100B a damage-associated release has been described in an in vitro stretch injury model [110, 111]. However, due to their high expression levels and small molecular weight, it is realistic that also other S100 proteins might be released during necrosis.
In addition to the passive release an activation-induced secretion of S100A8, S100A9, and S100A12 by human monocytes has been observed after interaction with TNF-α-activated endothelial cells [112]. Similar to HMGB-1 S100 proteins lack a conventional leader signal and they are supposed to be secreted via a nonclassical pathway involving a functional tubulin network [113]. S100B is constitutively secreted by astroglia, which can be further augmented by a number of agents [114–116].
The S100A8-S100A9 complex and S100A12 are useful diagnostic markers of inflammatory diseases, like arthritis and chronic inflammatory lung or bowel disease. During inflammation high concentrations of S100 proteins have been found in synovial fluid, sputum, stool and blood [117]. They are useful biomarkers of disease activity and response to treatment, since they sensitively indicate phagocyte activation and exhibit a strong correlation to the status of inflammation [112]. Future studies have to clarify, whether S100 proteins also represent therapeutic targets.
Heat-shock proteins (HSPs)
The highly conserved heat-shock proteins (HSPs) comprise a set of proteins abundantly present in prokaryotes as well as eukaryotes and act as molecular chaperones assisting the correct folding of nascent and stress-accumulated misfolded proteins to prevent their aggregation [118]. Additionally, they exert various extracellular and immunomodulatory functions. Under conditions of stress, including inflammation and infections, HSPs have been found in human sera [119, 120].
In mammals they have been grouped into five families, according to their molecular weight: HSP100, HSP90, HSP70, HSP60, and the small HSPs. The families are composed of several members expressed either constitutively (like HSC70) or inductively (like HSP70) exhibiting various subcellular localizations. The cellular stimuli that induce HSP expression include heat-shock, oxidative stress, and other types of cellular insult.
HSP70, HSP90, GP98 and calreticulin have been shown to be passively released during heat-induced necrosis, but similar to HMGB-1 not during apoptosis [121, 122]. Yet, again similar to HMGB-1, HSPs have also been reported to be actively secreted by various cell types in response to a variety of stress and activation signals [123]. In this scenario the mechanisms of HSP transport to the plasma membrane and export remain enigmatic, since cytosolic HSPs do not contain leader peptides for secretion via the classic pathway. Therefore, HSPs like several other ‘leaderless’ proteins are supposed to be secreted via noncanonical pathways, including exosomes (HSP27, HSP70, HSC70, and HSP90), secretory lysosomal endosomes (HSP70), and a pathway involving masking of the KDEL ER-retention signal (GP96) [124–127]. Yet, the knowledge about the secretion of ‘leaderless’ proteins is still incomplete and needs further studies for clarification.
In the extracellular environment HSPs can exert potent effects on immune cells and modulate the immune response in a multitude of ways [128]. HSP70, HSP90, and GP96 have been shown to stimulate the production of the pro-inflammatory cytokines TNF-α, IL-1β, IL-6, and IL-12 in human monocytes and DCs [121, 129, 130] and treatment with exogenous GP96 induced maturation of mouse and human DCs [131]. But HSPs can also exert indirect effects as peptide carriers. Since HSPs are molecular chaperons, they bind peptides, including antigenic ones, and piggyback them outside the cell. Such HSP-peptide complexes have been observed to strongly promote T cell activation by DCs. HSP-chaperoned peptides were swiftly internalized by DCs and consecutively processed via the endosomal pathway [132]. The former HSP-bound peptide was then cross-presented in the context of MHC class I molecules on the surface of the DC [133]. In vitro and in vivo in tumor mouse models cross-presentation initiated robust antigen-specific CD8+-T cell responses [134]. Thus, HSPs might represent putatively powerful vaccination agents for tumor immunotherapy, since they strongly foster the cross-presentation of tumor-derived, antigenic peptides.
Heat-shock proteins also display anti-inflammatory effects, which can be particularly noted in the context of chronic inflammatory diseases. As an example, rheumatoid arthritis was postulated to be triggered by cross reactive T cells recognizing common epitopes of mammalian and highly immunogenic prokaryotic HSPs [135]. Administration of the corresponding mammalian HSP could ameliorate the pro-inflammatory responses to bacterial HSP epitopes leading to remission of inflammation [136]. Additionally, treatment with HSP72 attenuated the LPS- or TNF-α-induced HMGB-1 release from activated macrophages in vitro [137]. In conclusion, depending on the context, HSPs may be key players of pro- as well as anti-inflammatory responses.
The heterogeneous group of HSPs is mirrored by the confusingly large group of putative HSP receptors, including CD91, CD40, the heterotrimeric TLR2/TLR4/CD14 cluster, LOX-1, and the scavenger receptors SCREC-1, CLEVER-1, and SR-A [138–145], stimulating diverse cellular signalling pathways, like for example the MyD88/IRAK pathway for NFκB-activation [130, 146]. Conceivably, different HSPs recognize different receptors on different cell types and the multiplicity of receptors indicates a certain specialization: TLR2, TLR4, and CD40 might be adapted for transmembrane signalling, while, in the context of cross-presentation, CD91 and the scavenger receptors might be more important for the internalization of HSPs.
Uric acid (UA)
In 2003 uric acid (UA) was described as endogenous danger signal released from dying cells displaying a strong adjuvant activity by priming CD8+-T cell responses when coinjected with antigen into mice [147]. Being the crucial intracellular antioxidant in humans UA is constitutively present in high intracellular concentrations (up to 4 mg/ml in liver cells) and its concentration still increases during cell injury, most likely due to an augmented metabolization of nucleic acids [147]. Apart from CD8+-T cell responses extracellular UA has also been reported to enhance CD4+-T cell responses towards particulate antigens [148, 149]. The authors concluded that UA affects a step required for the generation of helper and cytotoxic T cell responses and, consequently, examined the reaction of antigen presenting cells (APCs) towards stimulation with UA. In vitro murine bone marrow-derived DCs responded to UA treatment with maturation [147]. Furthermore, the application of UA as adjuvant in an in vivo immunotherapy model with dying tumor cells enhanced antibody production and improved inhibition of tumor growth [150].
Uric acid readily forms monosodium urate (MSU) microcrystals in extracellular body fluids suggesting that MSU crystals are the biologically active extracellular form. In vitro treatment with MSU crystals stimulated DC maturation and in vivo reducing UA levels via administration of allopurinol or metabolization by uricase inhibited the immune response to antigens associated with transplanted syngeneic cells and the proliferation of autoreactive T cells in a transgenic model of diabetes [151].
When MSU crystals precipitate and accumulate in joints and periarticular tissues they cause serious inflammatory attacks, referred to as gout [152]. The presence of MSU crystals has been reported to augment the respiratory burst capacity in polymorphonuclear granulocytes and macrophages in this disease [153, 154] and to induce the production of the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 in human monocytes and synovial cells [155–157]. Furthermore, MSU crystals induced the production of neutrophil chemotactic factors, such as IL-8 (in humans), KC, MIP-2 (in rodents), and consequently neutrophil infiltration [158–161]. An additional role in neutrophil attraction was attributed to the MSU-dependent release of S100A8/A9 and S100A12, respectively [162, 163].
In order to unravel the responsible receptor, intraperitoneal injection of MSU crystals in mice deficient for various TLRs and adaptor proteins with a Toll/IL-1 receptor (TIR) domain and subsequent analyses of neutrophil infiltration were employed [164]. None of the TLR deficient mice (TLR-1, -2, -4, -2/-4, -6, -7, -9, or -11) showed a reduction in MSU crystal-induced neutrophil recruitment, although TLR2 and TLR4 had been shown to be required for this activity in a murine air-pouch model [165]. Chen and coworkers only observed a reduced neutrophil recruitment in mice lacking MyD88. The authors identified the IL-1 receptor as upstream activator of MyD88 and concluded that MSU crystals initially stimulate the inflammasome-dependent IL-1β production in macrophages, leading to IL-1R- and MyD88-dependent neutrophil attraction. This is substantiated by the fact that MSU crystals have been described to activate the NALP3 inflammasome resulting in the production of IL-1β and IL-18 [166]. Moreover, macrophages from mice deficient in various components of the inflammasome have been shown to be defective in MSU crystal-induced IL-1β activation and mice deficient in the inflammasome or the IL-1R exhibited impaired peritoneal neutrophil infiltration after injection of MSU crystals [166]. These results enable first insights into the molecular processes underlying the inflammatory conditions of gout, and further support a pivotal role of the endogenous danger signal UA in this scenario.
Adenosine-5′-triphosphate (ATP)
The intracellular function in energy metabolism and the extracellular function in neurotransmission of ATP are well established since decades. Additionally, extracellular ATP can act as danger signal released by necrotic cells [167]. In healthy tissues the intracellular ATP levels are high (5–10 mM), whereas its concentration in the extracellular compartment is only in the nanomolar range. However, transient or permanent ruptures of cell membranes by stretch injury can lead to a substantial increase in the extracellular ATP concentration [168, 169]. Moreover, ATP can be actively secreted by various cell types [170]. The molecular mechanisms of this release are far from being understood, but they are supposed to involve exocytosis with complete or transient fusion between vesicle and cell membrane (‘kiss and run’ release [171]) or the liberation through connexin hemichannels [172].
Once in the extracellular space, ATP exerts a plethora of effects on various cell types. Thus, it can act as chemoattractant for microglia and immature DCs in vitro and in vivo [173–176]. Besides its chemokine function extracellular ATP has been described to modulate cytokine production and immune cell activation. Stimulation with ATP activated the NALP3 inflammasome and triggered IL-1β release by human macrophages and DCs [57, 177, 178]. ATP-treatment of iDCs also promoted maturation as mirrored by upregulated expression of CXCR4 and CCR7 and downregulated expression of CCR5 [179]. Functionally this was reflected in DC migration towards SDF-1α and ELC and decreased migration towards MIP-1β, thus, favoring lymphnode localization. Additionally, stimulation with ATP simultaneously augmented the constitutive production of MDC and inhibited the LPS-induced IP-10 and RANTES production. This led to a strong Th2 but not Th1 cell chemotaxis towards DC supernatants. Since DCs matured by LPS or CD40L in the presence of ATP also failed to induce the production of IL-6, IL-1-α, IL-1β, TNF-α, and IL-12, whereas the up-regulation of CD80 and CD86 and the release of the anti-inflammatory cytokines IL-10 and IL-1 receptor antagonist remained unaffected [180, 181], la Sala and colleagues concluded that ATP shifts DC maturation towards a Th2-polarized immune response. This is further substantiated by the observation that bone marrow-derived mast cells release the Th2-like cytokines IL-4, IL-6, IL-13, and TNF-α when stimulated with ATP [182].
Extracellular nucleotides are recognized by P2 purinergic receptors (P2Rs), which are ubiquitously expressed on the surface of a variety of cells and are divided into the P2XR and the P2YR subfamilies. P2XR and P2YR have been identified as multimeric ligand-gated plasma membrane ion channels and as GPCRs, respectively [169]. The different subfamilies seem to transduce different physiological effects in response to ATP ligation. Whereas the molecular sensors mediating migratory responses towards extracellular ATP belong to the P2YR subfamily [173–175], the modulatory effects of ATP on cytokine production and DC activation/maturation have been linked to the P2XR subfamily [57, 178, 180]. Activation of P2XRs by extracellular ATP has been described to lead to an increase in plasma membrane permeability for several ions, like Na+, K+, and Ca2+ [169]. P2X7 (formerly referred to as P2Z) differs from the other P2XR family members due to its ability to form a non-selective, reversible membrane pore, which is permeable for low molecular weight hydrophilic solutes. As downstream targets of P2XR signalling the transcription factors NF-κB and NFAT have been identified [183, 184]. Conversely, ligation of P2YR induced phospholipase C-mediated inositol trisphosphate generation, Ca2+ release from intracellular stores, and adenylate cyclase stimulation or inhibition by utilization of different G proteins (Gi/o or Gq/11) [169]. Intriguingly, P2R-mediated responses are differentially regulated by the local ATP concentration. The EC50-values of P2XRs have been determined to be in the μM range, whereas P2YRs already respond to nM concentrations of ATP [169]. These observations are in concordance with the different biological functions of the two subfamilies. For intermediate and long distance chemoattraction receptors with higher affinity (P2YRs) are required. In contrast, immune cell activation and maturation should only take place close to the site of cellular injury, where the ATP-concentration is high. Hence, receptors with lower affinity (P2XRs) are suitable. The extracellular nucleotide metabolism also modulates P2R-mediated responses, since ATP can be hydrolyzed to adenosine-5′-diphosphate (ADP) by ecto-ATP/ADPase (CD39) and NTPDase 2 (CD73) [185, 186]. CD39 can also hydrolyze ADP to adenosine-5′-monophosphate (AMP) and AMP can be further cleaved to form adenosine by the ubiquitous 5′-ectonucleotidase CD73. Adenosine itself has been reported to exert a variety of effects on different immune cells and it remains to be clarified in how far metabolization of ATP might contribute to its function as danger signal.
A crucial role for extracellular ATP in allergic reactions was described by Idzko et al. [58]. Utilizing an experimentally induced mouse asthma model they have shown that intrapulmonal allergen challenge causes an acute accumulation of ATP in the airways—a finding, which could also be observed in asthmatic humans. Importantly, when lung ATP levels were neutralized using apyrase or when mice were treated with P2R antagonists, all cardinal features of asthma were abrogated. Furthermore, Th2 sensitization to inhaled antigen was enhanced by endogenous or exogenous ATP. These adjuvant effects of ATP were due to the recruitment and activation of lung myeloid DCs, which induced Th2 responses in the mediastinal lymph nodes. In conclusion, these results suggest that the danger signal ATP and subsequent purinergic signalling events exert a key role in the allergen-driven lung inflammation rendering ATP an attractive target for future therapies.
mRNA, small nuclear ribonucleoproteins (snRNPs) and genomic DNA
It is well accepted that non-host dsRNA (e.g. viral RNA) and non-host DNA (e.g. non-methylated CpG DNA) exert immunostimulatory effects [187]. But there is also evidence that pro-inflammatory reactions can be stimulated by self-derived nucleic acids. Necrotic cells generated by freeze-thawing have been reported to release cytosolic mRNA and incubation with extracellular mRNA led to an activation and in vitro maturation of DCs [188]. Thus, extracellular mRNA exhibited a DC stimulating potency similar to LPS, dsRNA and CD40L, and induced production of the pro-inflammatory cytokines IL-12, IFN-α, IL-8, and TNF-α. Furthermore, on the surfaces of mRNA-treated DCs the chemokine receptors CCR5 and CCR6 were down- and CXCR4 was upregulated, preparing the DCs for lymph node homing. In addition to mRNA prepared in vitro RNA released from or associated with necrotic cells stimulated DCs leading to induction of IFN-α secretion [189]. Ni et al. [188] suggested the involvement of the P2YR subfamily in extracellular mRNA-signalling, since Poly(A) RNA mimicked at least some of the described effects, which furthermore could be strongly reduced by pre-incubation with pertussis toxin or ATP-mediated desensitization. However, in vitro transcribed RNA lacking a Poly(A)-tail induced DC maturation as well—although to a lesser extent. Thus, other sensors apart from the P2YR family have to be involved. In a follow-up study TLR3 has been implicated to serve as a further mRNA receptor [189]. Heterologous RNA released from necrotic cells or generated by in vitro transcription stimulated TLR3- and NF-κB-dependent DC maturation and pro-inflammatory cytokine (IL-8, IL-12 and IFN-α) expression [189]. Yet, the ubiquitous presence of RNases in the extracellular space and the inherent instability of mRNA limit its danger signal function both temporally and spatially. In addition, TLR3 is known to be located in the endosomal compartment. Therefore, further studies are required to analyze if mRNA released by necrotic cells is complexed with stabilizing compounds in order to increase its longevity and to target it to endosomes, a prerequisite for its function as damaged cell-derived trigger of TLR3.
In addition to encoding mRNA also structural RNA was reported to be released by necrotic cells and to stimulate a proinflammatory response, particularly when complexed in small nuclear ribonucleoproteins, like U1 snRNP [190]. U1 snRNP stimulated TLR7 signalling-dependent IFN-α secretion from plasmacytoid DCs and TLR8 signalling-dependent TNF-α secretion from ectopically TLR8-expressing cells, respectively. In both cases the RNA component of the particle was mandatory for the stimulation, since it was sensitive to RNase but not proteinase treatment. Moreover, the activity could be mimicked by short synthetic oligoribonucleotides. Notably, U1 snRNPs were only stimulatory for plasmacytoid DCs if transfected or taken up via Fcγ-receptor II as RNP immune complexes. Hence, snRNPs have to be taken up and directed to the endosomal compartment in order to activate endosomally located TLR7 or TLR8—a conclusion, which is further strengthened by the observation that small ‘antimalarial’ molecules inhibiting the endosomal pathway can block the immunostimulatory effects of U1 snRNPs [190].
Recently, while searching a ligand for the orphan macrophage-inducible C-type lectin (Mincle) Yamasaki et al. [191] identified the spliceosome-associated protein 130 (SAP130), a component of the U2 small nuclear ribonucleoprotein (U2 snRNP), as another danger signal, which is released during primary as well as secondary necrosis. Ligation of Mincle by SAP130 stimulated FcRγ- and CARD-9-dependent cytokine (TNF-α, IL-6) and chemokine (MIP-2, KC) production in vitro. Administration of neutralizing anti-Mincle antibodies resulted in reduced neutrophil recruitment into the thymus after whole-body irradiation-induced thymocyte necrosis and co-injection of theses antibodies with dead cells into the peritoneal cavity led to a strongly reduced MIP-2 production with concomitantly decreased neutrophil invasion. Yet, in how far the SAP130-Mincle ligand–receptor-system contributes to the pathogenesis of chronic inflammation and autoimmunity requires further examination, but it is obvious that SAP130 is a component of the spliceosome, which is known to be a major nuclear antigen associated with some of these diseases, including SLE [192].
In addition to mRNA and snRNPs, double stranded genomic dsDNA released from sheared or osmotically shocked necrotic cells has been shown to exert immunostimulatory effects on murine macrophages and bone marrow-derived murine DCs as reflected by upregulated surface expression of MHC I/II and costimulatory molecules (CD40, CD54, CD69) [193]. Subsequent reports could also show an induction of pro-inflammatory cytokines, such as IFN-α, TNF-α, and IL-6, by DCs in response to extracellular DNA [194–196]. Notably, transfection of dsDNA strongly augmented its DC maturating capacity in comparison to mere incubation with naked dsDNA and inhibitors of endocytosis and endosomal acidification substantially reduced DC maturation and cytokine production [194–196]. This suggests that the endosomal pathway is crucially involved. Functionally, dsDNA enhanced APC function in vitro as analyzed by the generation of antigen-specific T cells. This finding could also be observed in vivo, since significantly higher OVA-specific antibody titers were induced by immunization of mice with the combination of OVA antigen plus dsDNA in comparison to administration of OVA alone. Coadministering OVA plus dsDNA also led to a profoundly increased OVA-specific cytotoxic T cell activity. Consecutive studies by different groups identified the endosomally located TLR9 and subsequent MyD88 signalling to be vital elements in the context of genomic dsDNA-stimulated DC activation [194–197].
IL-6
Cells induced to undergo necrosis by various agents, including TNF stimulation, have also been reported to autonomously produce and release the pro-inflammatory cytokine IL-6 [198]. The authors observed that necrotic but not apoptotic cell death coincided with NF-κB-and p38 MAPK-mediated upregulation and secretion of IL-6. Thus, necrotically dying cells themselves are involved in the expression and secretion of inflammatory cytokines besides their capacity to induce inflammatory responses caused by the leakage of intracellular danger signals.
Danger signals of post-apoptotic, secondary necrotic cells
The clearance of apoptotic cells is a complex process and must be considered being an integral part of the apoptotic program. However, if the process of apoptotic cell clearance is delayed or even fails, primary apoptotic cells become secondary necrotic, as characterized by a loss of plasmamembrane integrity and subsequent release of intracellular contents. It could be argued that the repertoire of soluble mediators released by secondary necrotic cells should be a sum of the factors released by apoptotic and necrotic cells. However, one has to pay regard to the fact that LPC, dRP S19, EMAP-II or TyRS, are already released during bona fide apoptosis and that due to the ubiquitous presence of degrading enzymes in the extracellular space the stability of these compounds is supposedly rather limited, rendering it uncertain if these mediators are still present at the onset of secondary necrosis. Furthermore, the factors liberated during primary and secondary necrosis should differ fundamentally, since the crucial danger signals described in the context of primary necrosis (discussed above) are processed during apoptosis: ATP is consumed, intracellular proteins are proteolytically processed, RNA is degraded and DNA is internucleosomally cleaved [199]. However, the data on this issue are scarce. The secondary necrotic cell-derived danger signals convincingly described so far will be discussed in the following (Fig. 3).

Danger signal synapse between a secondary necrotic cell and a target cell
Uric acid (UA)
The effects of UA and its monosodium salt MSU have already been described above. All this also holds true for secondary necrosis, where the amount of available and releasable UA has been reported to be even higher than in the case of primary necrosis, most likely due to the degradation of chromatin, which is enhanced in the late phases of apoptosis [147].
Nucleosomes
Genomic DNA fragments in form of oligonucleosomes are crucial danger signals of post-apoptotic cells. Whereas during necrosis unspecific DNA cleavage leads to large-scale fragmentation and cleavage products of many kbp in length, apoptosis-associated internucleosomal DNA yields free nucleosomes containing small DNA fragments of 180 bp or multiples of 180 bp in length. The small oligonucleosomal DNA fragments can readily leave the nucleus via the nuclear pore complexes, accumulate in the cytosol and are finally released during secondary necrosis. At very late stages of apoptosis the core histones and low molecular weight DNA (approx 180 bp) could be detected in cell-free supernatants of spontaneously apoptozing lymphocytes [200]. Importantly, nucleosomes released from dying human lymphocytes exerted a significant proliferative and stimulatory effect on lymphocytes [200]. In vitro stimulation with purified nucleosomes of mouse bone marrow-derived DCs, human monocyte-derived DCs, and purified human myeloid DCs induced their maturation and their capability to stimulate allogeneic T cells [197]. Recent reports have shown that HMGB-1 associated with those secondary necrotic cell-derived nucleosomes is of crucial importance [201, 202]. HMGB-1–nucleosome complexes from secondary necrotic cells induced cytokine expression in macrophages and DC maturation. In contrast, nucleosomes from viable cells devoid of HMGB-1 induced no marked cytokine release. One of the receptors engaged in HMGB-1-nucleosome-induced immune activation was identified to be TLR2 with consecutive activation of MyD88 [197, 202].
Patients with SLE have been reported to exhibit defects in apoptotic cell removal and increased appearance of secondary necrosis [203]. Circulating nucleosomes could be detected in the plasma of nearly 50% of all patients but in less than 5% of normal healthy donors [202, 204]. There was a significant correlation between disease activity score (SLEDAI) and serum concentration of nucleosomes. Most importantly, immunization with HMGB-1-nucleosomes resulted in significantly increased anti-dsDNA and anti-histone antibody titers in nonautoimmune mice [202]. Hence, nucleosomes with tightly bound HMGB-1 may play a crucial role in breaking the immunological tolerance against dsDNA, which represents a key autoantigen in SLE (discussed further in this issue by Kruse et al.).
Caspase-/granzyme B-generated autoantigens
Casciola-Rosen and colleagues [205–207] have shown that secondary necrotic cells can act as autoantigen reservoirs potentially initiating and driving systemic autoimmunity in susceptible hosts. During apoptosis many autoantigens targeted in systemic autoimmunity undergo posttranslational modifications, such as caspase- or in particular granzyme B-mediated cleavage or oxidation potentially enhancing their immunogenicity [208]. Immunoblot analyses with lysates of apoptotic and secondary necrotic cells using highly specific human autoantibodies revealed that several autoantigens, including poly (ADP-ribose) polymerase, topoisomerase I, SSB/La, U1-70 kd, or the pyruvate dehydrogenase complex, were cleaved into their signature apoptotic fragments, which were recognized by human autoantibodies [207, 209]. However, some autoantigens (e.g. ribosomal RNP, Ku, and SSA/Ro) appeared to be resistant to proteolysis during cell death and, therefore, other posttranslational modifications might have initiated the responses against these autoantigens [206]. The authors suggested that the release of autoantigenic neoepitopes in the context of the pro-inflammatory milieu induced by dying cell-derived danger signals might instigate a robust autoimmune response in the patients. It remains to be clarified, if autoantigens liberated from secondary necrotic cells can trigger this autoimmune response in their free form or if complexing by HSPs and consecutive antigen cross-presentation might contribute to this scenario. Furthermore, since classical danger signals of necrotic cells, like HSP90, have also been reported to be caspase substrates and, thus, to be proteolytically processed during apoptosis [210], it is intriguing to find out, if these modifications increase, decrease or abrogate their functions as dead cell-derived danger signals. Although the generation of autoantibodies against a plethora of intracellular antigens in different autoimmune diseases supports the concept of a correlation between the release of intracellular antigens during secondary necrosis and autoimmunity, a causative relationship still has to be finally proven.
Conclusions
The clearance of dying and dead cells has been examined during the last years in numerous studies. In addition to surface interactions, where receptors of the phagocyte get into contact with the dying cell, soluble factors have been identified to be released from the dying cell and to operate over intermediate or even long distances. Multiple attraction and danger signals have been characterized to be orchestrating the process of dead cell removal, including the subsequent response of the innate as well as the acquired immune system in terms of pro- or anti-inflammation, immune activation, and tolerance induction. As summarized here, the repertoire of released substances is dependent on the form of cell death. Consequently, apoptotic, primary, and secondary necrotic cells obviously ‘smell’ and ‘feel’ different for the immune system. Until now we have only gained first insights into the plethora of effects exerted by dRP S19, TyRS, TSP-1, EMAP-II, LPC, HMGB-1, HPSs, S100 proteins, uric acid, ATP, genomic DNA and others. Future studies will help to resolve the issue why and especially how the immune system responds differently to the ‘interstitial fingerprint’ of apoptotic, primary and secondary necrotic cells. This is of particular interest, since chronic inflammation, autoimmunity, and missing tumor surveillance have been linked to a deregulation of the appearance and the function of some of these factors. Thus, endogenous danger molecules and attraction signals might represent promising biomarkers for various conditions of disease and may also provide potential targets for future therapeutic intervention.
Abbreviations
- ADAM 17:
-
A disintegrin and metalloproteinase 17
- APC:
-
Antigen presenting cell
- C5a:
-
Complement protein 5a
- CARD:
-
Caspase recruitment domain
- CLEVER-1:
-
Common lymphatic endothelial and vascular endothelial receptor 1
- COX-2:
-
Cyclooxygenase-2
- DAMP:
-
Damage associated molecular pattern
- DC:
-
Dendritic cell
- dRP S19:
-
Dimer of ribosomal protein S19
- ELC:
-
EBV-induced molecule 1 ligand chemokine
- EMAP II:
-
Endothelial monocyte-activating polypeptide II
- FcRγ:
-
Receptor for Fc region of IgG
- fMLP:
-
N-Formylmethionyl-leucyl-phenylalanine
- GP96:
-
Glycoprotein 96
- GPCR:
-
G protein coupled receptor
- HBD:
-
Heparin binding domain
- HDGF:
-
Hepatocyte derived growth factor
- HMGB-1:
-
High mobility group box 1 protein
- HSP:
-
Heat shock protein
- ICAM:
-
Intercellular adhesion molecule
- iDC:
-
Immature dendritic cell
- IFN:
-
Interferon
- IL:
-
Interleukin
- IP-10:
-
Inducible protein of 10 kDa
- iPLA2 :
-
Calcium-independent phospholipse A2
- IRAK:
-
Interleukin-1 receptor-associated kinase
- KC:
-
Keratinocyte chemoattractant
- Ku:
-
Ku autoantigen
- LOX-1:
-
Lectin-like oxidized low-density lipoprotein receptor
- LPC:
-
Lysophosphatidylcholine
- LPS:
-
Lipopolysaccharide
- LTB4 :
-
Leukotrien B4
- LTF:
-
Lactoferrin
- MAPK:
-
Mitogen-activated protein kinase
- MCP:
-
Monocyte chemotactic protein
- MDC:
-
Macrophage-derived chemokine
- MFG-E8:
-
Milk-fat globule EGF-factor 8
- Mincle:
-
Macrophage-inducible C-type lectin
- MIP:
-
Macrophage inflammatory protein
- MSU:
-
Monosodium urate
- MyD88:
-
Myeloid differentiation primary response gene 88
- NALP3:
-
NACHT domain- leucine-rich repeat- and PYD-containing protein 3
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NK cell:
-
Natural killer cell
- NOS:
-
NO synthase
- OVA:
-
Ovalbumin
- P2X:
-
Purinergic receptor X
- P2Y:
-
Purinergic receptor Y
- PARP poly:
-
Poly (ADP-ribose) polymerase
- PBMC:
-
Peripheral blood mononuclear cell
- PGE2 :
-
Prostaglandin E2
- RAGE:
-
Receptor for advanced glycation end products
- RANTES:
-
Regulated upon activation, normal T-cell expressed and secreted
- RNP:
-
Ribonucleoprotein
- S1P:
-
Sphingosin-1-phosphate
- SAP130:
-
Spliceosome-associated polypeptide of 130 kDa
- SDF-1:
-
Stromal cell-derived factor-1
- SLE:
-
Systemic lupus erythematosus
- SphK:
-
Sphingosin kinase
- SR-A:
-
Scavenger receptor A
- SREC-1:
-
Scavenger receptor class F member 1
- SSA/Ro:
-
Sjoegren syndrome antigen A/autoantigen Ro
- SSB/La:
-
Sjoegren syndrome antigen B/autoantigen La
- TGase 2:
-
Transglutaminase 2
- TGF-β:
-
Transforming growth factor β
- TIR:
-
Toll/IL-1 receptor domain
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- TSP-1:
-
Thrombospondin-1
- TyrRS:
-
Tyrosyl tRNA synthetase
- U1-70 kd:
-
70 kDa polypeptide of U1 small nuclear RNP
- UA:
-
Uric acid
- VCAM:
-
Vascular cell adhesion molecule
References
Gaipl US, Franz S, Voll RE, Sheriff A, Kalden JR, Herrmann M (2004) Defects in the disposal of dying cells lead to autoimmunity. Curr Rheumatol Rep 6:401–407
Lauber K, Blumenthal SG, Waibel M, Wesselborg S (2004) Clearance of apoptotic cells; getting rid of the corpses. Mol Cell 14:277–287
Krysko DV, Denecker G, Festjens N, Gabriels S, Parthoens E, D’Herde K, Vandenabeele P (2006) Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell Death Differ 13:2011–2022
Horino K, Nishiura H, Ohsako T, Shibuya Y, Hiraoka T, Kitamura N, Yamamoto T (1998) A monocyte chemotactic factor, S19 ribosomal protein dimer, in phagocytic clearance of apoptotic cells. Lab Invest 78:603–617
Nishiura H, Shibuya Y, Matsubara S, Tanase S, Kambara T, Yamamoto T (1996) Monocyte chemotactic factor in rheumatoid arthritis synovial tissue. Probably a cross-linked derivative of S19 ribosomal protein. J Biol Chem 271:878–882
Nishimura T, Horino K, Nishiura H, Shibuya Y, Hiraoka T, Tanase S, Yamamoto T (2001) Apoptotic cells of an epithelial cell line, AsPC-1, release monocyte chemotactic S19 ribosomal protein dimer. J Biochem 129:445–454
Umeda Y, Shibuya Y, Semba U, Tokita K, Nishino N, Yamamoto T (2004) Guinea pig S19 ribosomal protein as precursor of C5a receptor-directed monocyte-selective leukocyte chemotactic factor. Inflamm Res 53:623–630
Nishiura H, Shibuya Y, Yamamoto T (1998) S19 ribosomal protein cross-linked dimer causes monocyte-predominant infiltration by means of molecular mimicry to complement C5a. Lab Invest 78:1615–1623
Yamamoto T (2007) Roles of the ribosomal protein S19 dimer and the C5a receptor in pathophysiological functions of phagocytic leukocytes. Pathol Int 57:1–11
Knies UE, Behrensdorf HA, Mitchell CA, Deutsch U, Risau W, Drexler HC, Clauss M (1998) Regulation of endothelial monocyte-activating polypeptide II release by apoptosis. Proc Natl Acad Sci USA 95:12322–12327
Kao J, Ryan J, Brett G, Chen J, Shen H, Fan YG, Godman G, Familletti PC, Wang F, Pan YC et al (1992) Endothelial monocyte-activating polypeptide II. A novel tumor-derived polypeptide that activates host-response mechanisms. J Biol Chem 267:20239–20247
Kao J, Houck K, Fan Y, Haehnel I, Libutti SK, Kayton ML, Grikscheit T, Chabot J, Nowygrod R, Greenberg S et al (1994) Characterization of a novel tumor-derived cytokine. Endothelial-monocyte activating polypeptide II. J Biol Chem 269:25106–25119
Behrensdorf HA, van de Craen M, Knies UE, Vandenabeele P, Clauss M (2000) The endothelial monocyte-activating polypeptide II (EMAP II) is a substrate for caspase-7. FEBS Lett 466:143–147
Quevillon S, Agou F, Robinson JC, Mirande M (1997) The p43 component of the mammalian multi-synthetase complex is likely to be the precursor of the endothelial monocyte-activating polypeptide II cytokine. J Biol Chem 272:32573–32579
Kao J, Fan YG, Haehnel I, Brett J, Greenberg S, Clauss M, Kayton M, Houck K, Kisiel W, Seljelid R et al (1994) A peptide derived from the amino terminus of endothelial-monocyte-activating polypeptide II modulates mononuclear and polymorphonuclear leukocyte functions, defines an apparently novel cellular interaction site, and induces an acute inflammatory response. J Biol Chem 269:9774–9782
Ko YG, Park H, Kim T, Lee JW, Park SG, Seol W, Kim JE, Lee WH, Kim SH, Park JE, Kim S (2001) A cofactor of tRNA synthetase, p43, is secreted to up-regulate proinflammatory genes. J Biol Chem 276:23028–23033
Hou Y, Plett PA, Ingram DA, Rajashekhar G, Orschell CM, Yoder MC, March KL, Clauss M (2006) Endothelial-monocyte-activating polypeptide II induces migration of endothelial progenitor cells via the chemokine receptor CXCR3. Exp Hematol 34:1125–1132
Cascieri MA, Springer MS (2000) The chemokine/chemokine-receptor family: potential and progress for therapeutic intervention. Curr Opin Chem Biol 4:420–427
Wakasugi K, Schimmel P (1999) Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science 284:147–151
Krispin A, Bledi Y, Atallah M, Trahtemberg U, Verbovetski I, Nahari E, Zelig O, Linial M, Mevorach D (2006) Apoptotic cell thrombospondin-1 and heparin-binding domain lead to dendritic-cell phagocytic and tolerizing states. Blood 108:3580–3589
Adams JC (2001) Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol 17:25–51
Savill J, Hogg N, Ren Y, Haslett C (1992) Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest 90:1513–1522
Mansfield PJ, Suchard SJ (1994) Thrombospondin promotes chemotaxis and haptotaxis of human peripheral blood monocytes. J Immunol 153:4219–4229
Mansfield PJ, Boxer LA, Suchard SJ (1990) Thrombospondin stimulates motility of human neutrophils. J Cell Biol 111:3077–3086
Lymn JS, Patel MK, Clunn GF, Rao SJ, Gallagher KL, Hughes AD (2002) Thrombospondin-1 differentially induces chemotaxis and DNA synthesis of human venous smooth muscle cells at the receptor-binding level. J Cell Sci 115:4353–4360
Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seegert D (2007) The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets 11:613–624
Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, Jones SA, Rose-John S, Scheller J (2007) Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood 110:1748–1755
Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, Rose-John S, Fuller GM, Topley N, Jones SA (2001) Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 14:705–714
Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS, Jones SA, Rose-John S, Scheller J (2008) Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood 111:1021–1028
Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, Graham G, Combadiere C, Gregory CD (2008) CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112:5026–5036
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924
Leonardi-Essmann F, Emig M, Kitamura Y, Spanagel R, Gebicke-Haerter PJ (2005) Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia. J Neuroimmunol 160:92–101
Miksa M, Amin D, Wu R, Ravikumar TS, Wang P (2007) Fractalkine-induced MFG-E8 leads to enhanced apoptotic cell clearance by macrophages. Mol Med 13:553–560
Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S (2003) Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113:717–730
Kim SJ, Gershov D, Ma X, Brot N, Elkon KB (2002) I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med 196:655–665
Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, Wesselborg S, Lauber K (2007) Migration to apoptotic ‘find-me’ signals is mediated via the phagocyte receptor G2A. J Biol Chem
Le LQ, Kabarowski JH, Weng Z, Satterthwaite AB, Harvill ET, Jensen ER, Miller JF, Witte ON (2001) Mice lacking the orphan G protein-coupled receptor G2A develop a late-onset autoimmune syndrome. Immunity 14:561–571
Botto M (1998) C1q knock-out mice for the study of complement deficiency in autoimmune disease. Exp Clin Immunogenet 15:231–234
Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK (2001) Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411:207–211
Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S (2004) Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304:1147–1150
Murugesan G, Sandhya Rani MR, Gerber CE, Mukhopadhyay C, Ransohoff RM, Chisolm GM, Kottke-Marchant K (2003) Lysophosphatidylcholine regulates human microvascular endothelial cell expression of chemokines. J Mol Cell Cardiol 35:1375–1384
Ngwenya BZ, Yamamoto N (1985) Activation of peritoneal macrophages by lysophosphatidylcholine. Biochim Biophys Acta 839:9–15
Homma S, Yamamoto M, Yamamoto N (1993) Vitamin D-binding protein (group-specific component) is the sole serum protein required for macrophage activation after treatment of peritoneal cells with lysophosphatidylcholine. Immunol Cell Biol 71(Pt 4):249–257
Yan JJ, Jung JS, Lee JE, Lee J, Huh SO, Kim HS, Jung KC, Cho JY, Nam JS, Suh HW, Kim YH, Song DK (2004) Therapeutic effects of lysophosphatidylcholine in experimental sepsis. Nat Med 10:161–167
Chen G, Li J, Qiang X, Czura CJ, Ochani M, Ochani K, Ulloa L, Yang H, Tracey KJ, Wang P, Sama AE, Wang H (2005) Suppression of HMGB1 release by stearoyl lysophosphatidylcholine:an additional mechanism for its therapeutic effects in experimental sepsis. J Lipid Res 46:623–627
Gomez-Munoz A, O’Brien L, Hundal R, Steinbrecher UP (1999) Lysophosphatidylcholine stimulates phospholipase D activity in mouse peritoneal macrophages. J Lipid Res 40:988–993
Taniuchi M, Otani H, Kodama N, Tone Y, Sakagashira M, Yamada Y, Mune M, Yukawa S (1999) Lysophosphatidylcholine up-regulates IL-1 beta-induced iNOS expression in rat mesangial cells. Kidney Int Suppl 71:S156–S158
Coutant F, Perrin-Cocon L, Agaugue S, Delair T, Andre P, Lotteau V (2002) Mature dendritic cell generation promoted by lysophosphatidylcholine. J Immunol 169:1688–1695
Sakata-Kaneko S, Wakatsuki Y, Usui T, Matsunaga Y, Itoh T, Nishi E, Kume N, Kita T (1998) Lysophosphatidylcholine upregulates CD40 ligand expression in newly activated human CD4 + T cells. FEBS Lett 433:161–165
Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, Barbour SE, Milstien S, Spiegel S (2008) Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J 22:2629–2638
Weigert A, Johann AM, von Knethen A, Schmidt H, Geisslinger G, Brune B (2006) Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood 108:1635–1642
Weigert A, Tzieply N, von Knethen A, Johann AM, Schmidt H, Geisslinger G, Brune B (2007) Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol Biol Cell 18:3810–3819
Rosen H, Goetzl EJ (2005) Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol 5:560–570
Johann AM, Weigert A, Eberhardt W, Kuhn AM, Barra V, von Knethen A, Pfeilschifter JM, Brune B (2008) Apoptotic cell-derived sphingosine-1-phosphate promotes HuR-dependent cyclooxygenase-2 mRNA stabilization and protein expression. J Immunol 180:1239–1248
Segundo C, Medina F, Rodriguez C, Martinez-Palencia R, Leyva-Cobian F, Brieva JA (1999) Surface molecule loss and bleb formation by human germinal center B cells undergoing apoptosis: role of apoptotic blebs in monocyte chemotaxis. Blood 94:1012–1020
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS (2009) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461:282–286
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15:1170–1178
Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, Hoogsteden HC, Luttmann W, Ferrari D, Di Virgilio F, Virchow JC Jr, Lambrecht BN (2007) Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med 13:913–919
Bournazou I, Pound JD, Duffin R, Bournazos S, Melville LA, Brown SB, Rossi AG, Gregory CD (2009) Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J Clin Invest 119:20–32
Crouch SP, Slater KJ, Fletcher J (1992) Regulation of cytokine release from mononuclear cells by the iron-binding protein lactoferrin. Blood 80:235–240
Haversen L, Ohlsson BG, Hahn-Zoric M, Hanson LA, Mattsby-Baltzer I (2002) Lactoferrin down-regulates the LPS-induced cytokine production in monocytic cells via NF-kappa B. Cell Immunol 220:83–95
Togawa J, Nagase H, Tanaka K, Inamori M, Nakajima A, Ueno N, Saito T, Sekihara H (2002) Oral administration of lactoferrin reduces colitis in rats via modulation of the immune system and correction of cytokine imbalance. J Gastroenterol Hepatol 17:1291–1298
Zimecki M, Artym J, Chodaczek G, Kocieba M, Kruzel M (2005) Effects of lactoferrin on the immune response modified by the immobilization stress. Pharmacol Rep 57:811–817
Golstein P, Kroemer G (2007) Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 32:37–43
Festjens N, Vanden Berghe T, Vandenabeele P (2006) Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 1757:1371–1387
Lohmann C, Muschaweckh A, Kirschnek S, Jennen L, Wagner H, Hacker G (2009) Induction of tumor cell apoptosis or necrosis by conditional expression of cell death proteins: analysis of cell death pathways and in vitro immune stimulatory potential. J Immunol 182:4538–4546
Degterev A, Yuan J (2008) Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol 9:378–390
Li HN, Barlow PG, Bylund J, Mackellar A, Bjorstad A, Conlon J, Hiemstra PS, Haslett C, Gray M, Simpson AJ, Rossi AG, Davidson DJ (2009) Secondary necrosis of apoptotic neutrophils induced by the human cathelicidin LL-37 is not proinflammatory to phagocytosing macrophages. J Leukoc Biol 86:891–902
Patel VA, Longacre A, Hsiao K, Fan H, Meng F, Mitchell JE, Rauch J, Ucker DS, Levine JS (2006) Apoptotic cells, at all stages of the death process, trigger characteristic signaling events that are divergent from and dominant over those triggered by necrotic cells: Implications for the delayed clearance model of autoimmunity. J Biol Chem 281:4663–4670
Gallucci S, Matzinger P (2001) Danger signals: SOS to the immune system. Curr Opin Immunol 13:114–119
Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, Devera ME, Liang X, Tor M, Billiar T (2007) The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev 220:60–81
Javaherian K, Liu JF, Wang JC (1978) Nonhistone proteins HMG1 and HMG2 change the DNA helical structure. Science 199:1345–1346
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195
Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ (2001) HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med 164:1768–1773
Semino C, Angelini G, Poggi A, Rubartelli A (2005) NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood 106:609–616
Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA (2008) Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29:21–32
Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22:5551–5560
Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A (2002) The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 3:995–1001
Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T (2007) A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J 26:1129–1139
Treutiger CJ, Mullins GE, Johansson AS, Rouhiainen A, Rauvala HM, Erlandsson-Harris H, Andersson U, Yang H, Tracey KJ, Andersson J, Palmblad JE (2003) High mobility group 1 B-box mediates activation of human endothelium. J Intern Med 254:375–385
Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, Lepantalo M, Carpen O, Parkkinen J, Rauvala H (2004) Regulation of monocyte migration by amphoterin (HMGB1). Blood 104:1174–1182
Semino C, Ceccarelli J, Lotti LV, Torrisi MR, Angelini G, Rubartelli A (2007) The maturation potential of NK cell clones toward autologous dendritic cells correlates with HMGB1 secretion. J Leukoc Biol 81:92–99
Messmer D, Yang H, Telusma G, Knoll F, Li J, Messmer B, Tracey KJ, Chiorazzi N (2004) High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol 173:307–313
Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ (2000) High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 192:565–570
Rouhiainen A, Tumova S, Valmu L, Kalkkinen N, Rauvala H (2007) Pivotal advance: analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin). J Leukoc Biol 81:49–58
Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P (2005) Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur J Immunol 35:2184–2190
Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H (2006) HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26:174–179
Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E (2004) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 279:7370–7377
Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ (2004) Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA 101:296–301
Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, Inoue K, Yamada S, Ijiri K, Matsunaga S, Nakajima T, Komiya S, Maruyama I (2003) High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum 48:971–981
Kokkola R, Sundberg E, Ulfgren AK, Palmblad K, Li J, Wang H, Ulloa L, Yang H, Yan XJ, Furie R, Chiorazzi N, Tracey KJ, Andersson U, Harris HE (2002) High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum 46:2598–2603
Nakamura H, Izumoto Y, Kambe H, Kuroda T, Mori T, Kawamura K, Yamamoto H, Kishimoto T (1994) Molecular cloning of complementary DNA for a novel human hepatoma-derived growth factor. Its homology with high mobility group-1 protein. J Biol Chem 269:25143–25149
Zhou Z, Yamamoto Y, Sugai F, Yoshida K, Kishima Y, Sumi H, Nakamura H, Sakoda S (2004) Hepatoma-derived growth factor is a neurotrophic factor harbored in the nucleus. J Biol Chem 279:27320–27326
Donato R (2003) Intracellular and extracellular roles of S100 proteins. Microsc Res Tech 60:540–551
Odink K, Cerletti N, Bruggen J, Clerc RG, Tarcsay L, Zwadlo G, Gerhards G, Schlegel R, Sorg C (1987) Two calcium-binding proteins in infiltrate macrophages of rheumatoid arthritis. Nature 330:80–82
Yang Z, Tao T, Raftery MJ, Youssef P, Di Girolamo N, Geczy CL (2001) Proinflammatory properties of the human S100 protein S100A12. J Leukoc Biol 69:986–994
Rothermundt M, Peters M, Prehn JH, Arolt V (2003) S100B in brain damage and neurodegeneration. Microsc Res Tech 60:614–632
Lackmann M, Cornish CJ, Simpson RJ, Moritz RL, Geczy CL (1992) Purification and structural analysis of a murine chemotactic cytokine (CP-10) with sequence homology to S100 proteins. J Biol Chem 267:7499–7504
Devery JM, King NJ, Geczy CL (1994) Acute inflammatory activity of the S100 protein CP-10. Activation of neutrophils in vivo and in vitro. J Immunol 152:1888–1897
Eue I, Pietz B, Storck J, Klempt M, Sorg C (2000) Transendothelial migration of 27E10 + human monocytes. Int Immunol 12:1593–1604
Kerkhoff C, Klempt M, Kaever V, Sorg C (1999) The two calcium-binding proteins, S100A8 and S100A9, are involved in the metabolism of arachidonic acid in human neutrophils. J Biol Chem 274:32672–32679
Aguiar-Passeti T, Postol E, Sorg C, Mariano M (1997) Epithelioid cells from foreign-body granuloma selectively express the calcium-binding protein MRP-14, a novel down-regulatory molecule of macrophage activation. J Leukoc Biol 62:852–858
Brun JG, Ulvestad E, Fagerhol MK, Jonsson R (1994) Effects of human calprotectin (L1) on in vitro immunoglobulin synthesis. Scand J Immunol 40:675–680
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901
Li Y, Barger SW, Liu L, Mrak RE, Griffin WS (2000) S100beta induction of the proinflammatory cytokine interleukin-6 in neurons. J Neurochem 74:143–150
Hu J, Castets F, Guevara JL, Van Eldik LJ (1996) S100 beta stimulates inducible nitric oxide synthase activity and mRNA levels in rat cortical astrocytes. J Biol Chem 271:2543–2547
Bianchi R, Adami C, Giambanco I, Donato R (2007) S100B binding to RAGE in microglia stimulates COX-2 expression. J Leukoc Biol 81:108–118
Adami C, Sorci G, Blasi E, Agneletti AL, Bistoni F, Donato R (2001) S100B expression in and effects on microglia. Glia 33:131–142
Adami C, Bianchi R, Pula G, Donato R (2004) S100B-stimulated NO production by BV-2 microglia is independent of RAGE transducing activity but dependent on RAGE extracellular domain. Biochim Biophys Acta 1742:169–177
Willoughby KA, Kleindienst A, Muller C, Chen T, Muir JK, Ellis EF (2004) S100B protein is released by in vitro trauma and reduces delayed neuronal injury. J Neurochem 91:1284–1291
Ellis EF, Willoughby KA, Sparks SA, Chen T (2007) S100B protein is released from rat neonatal neurons, astrocytes, and microglia by in vitro trauma and anti-S100 increases trauma-induced delayed neuronal injury and negates the protective effect of exogenous S100B on neurons. J Neurochem 101:1463–1470
Frosch M, Strey A, Vogl T, Wulffraat NM, Kuis W, Sunderkotter C, Harms E, Sorg C, Roth J (2000) Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum 43:628–637
Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C (1997) Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem 272:9496–9502
Whitaker-Azmitia PM, Murphy R, Azmitia EC (1990) Stimulation of astroglial 5-HT1A receptors releases the serotonergic growth factor, protein S-100, and alters astroglial morphology. Brain Res 528:155–158
Ciccarelli R, Di Iorio P, Bruno V, Battaglia G, D’Alimonte I, D’Onofrio M, Nicoletti F, Caciagli F (1999) Activation of A(1) adenosine or mGlu3 metabotropic glutamate receptors enhances the release of nerve growth factor and S-100beta protein from cultured astrocytes. Glia 27:275–281
Pinto SS, Gottfried C, Mendez A, Goncalves D, Karl J, Goncalves CA, Wofchuk S, Rodnight R (2000) Immunocontent and secretion of S100B in astrocyte cultures from different brain regions in relation to morphology. FEBS Lett 486:203–207
Foell D, Frosch M, Sorg C, Roth J (2004) Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin Chim Acta 344:37–51
Bukau B, Weissman J, Horwich A (2006) Molecular chaperones and protein quality control. Cell 125:443–451
Xiao Q, Mandal K, Schett G, Mayr M, Wick G, Oberhollenzer F, Willeit J, Kiechl S, Xu Q (2005) Association of serum-soluble heat shock protein 60 with carotid atherosclerosis: clinical significance determined in a follow-up study. Stroke 36:2571–2576
Njemini R, Lambert M, Demanet C, Mets T (2003) Elevated serum heat-shock protein 70 levels in patients with acute infection: use of an optimized enzyme-linked immunosorbent assay. Scand J Immunol 58:664–669
Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK (2000) Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol 12:1539–1546
Mambula SS, Calderwood SK (2006) Heat induced release of Hsp70 from prostate carcinoma cells involves both active secretion and passive release from necrotic cells. Int J Hyperthermia 22:575–585
Barreto A, Gonzalez JM, Kabingu E, Asea A, Fiorentino S (2003) Stress-induced release of HSC70 from human tumors. Cell Immunol 222:97–104
Lancaster GI, Febbraio MA (2005) Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem 280:23349–23355
Clayton A, Turkes A, Navabi H, Mason MD, Tabi Z (2005) Induction of heat shock proteins in B-cell exosomes. J Cell Sci 118:3631–3638
Mambula SS, Calderwood SK (2006) Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol 177:7849–7857
Altmeyer A, Maki RG, Feldweg AM, Heike M, Protopopov VP, Masur SK, Srivastava PK (1996) Tumor-specific cell surface expression of the-KDEL containing, endoplasmic reticular heat shock protein gp96. Int J Cancer 69:340–349
Srivastava PK (2000) Heat shock protein-based novel immunotherapies. Drug News Perspect 13:517–522
Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK (2000) HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med 6:435–442
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK (2002) Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 277:15028–15034
Singh-Jasuja H, Scherer HU, Hilf N, Arnold-Schild D, Rammensee HG, Toes RE, Schild H (2000) The heat shock protein gp96 induces maturation of dendritic cells and down-regulation of its receptor. Eur J Immunol 30:2211–2215
Singh-Jasuja H, Toes RE, Spee P, Munz C, Hilf N, Schoenberger SP, Ricciardi-Castagnoli P, Neefjes J, Rammensee HG, Arnold-Schild D, Schild H (2000) Cross-presentation of glycoprotein 96-associated antigens on major histocompatibility complex class I molecules requires receptor-mediated endocytosis. J Exp Med 191:1965–1974
Arnold-Schild D, Hanau D, Spehner D, Schmid C, Rammensee HG, de la Salle H, Schild H (1999) Cutting edge: receptor-mediated endocytosis of heat shock proteins by professional antigen-presenting cells. J Immunol 162:3757–3760
Doody AD, Kovalchin JT, Mihalyo MA, Hagymasi AT, Drake CG, Adler AJ (2004) Glycoprotein 96 can chaperone both MHC class I- and class II-restricted epitopes for in vivo presentation, but selectively primes CD8 + T cell effector function. J Immunol 172:6087–6092
van Eden W, van der Zee R, Prakken B (2005) Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol 5:318–330
Kingston AE, Hicks CA, Colston MJ, Billingham ME (1996) A 71-kD heat shock protein (hsp) from Mycobacterium tuberculosis has modulatory effects on experimental rat arthritis. Clin Exp Immunol 103:77–82
Tang D, Kang R, Xiao W, Wang H, Calderwood SK, Xiao X (2007) The anti-inflammatory effects of heat shock protein 72 involve inhibition of high-mobility-group box 1 release and proinflammatory function in macrophages. J Immunol 179:1236–1244
Basu S, Binder RJ, Ramalingam T, Srivastava PK (2001) CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14:303–313
Binder RJ, Han DK, Srivastava PK (2000) CD91: a receptor for heat shock protein gp96. Nat Immunol 1:151–155
Becker T, Hartl FU, Wieland F (2002) CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes. J Cell Biol 158:1277–1285
Berwin B, Hart JP, Rice S, Gass C, Pizzo SV, Post SR, Nicchitta CV (2003) Scavenger receptor-A mediates gp96/GRP94 and calreticulin internalization by antigen-presenting cells. EMBO J 22:6127–6136
Delneste Y, Magistrelli G, Gauchat J, Haeuw J, Aubry J, Nakamura K, Kawakami-Honda N, Goetsch L, Sawamura T, Bonnefoy J, Jeannin P (2002) Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity 17:353–362
Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S (2005) Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis 182:1–15
Theriault JR, Mambula SS, Sawamura T, Stevenson MA, Calderwood SK (2005) Extracellular HSP70 binding to surface receptors present on antigen presenting cells and endothelial/epithelial cells. FEBS Lett 579:1951–1960
Theriault JR, Adachi H, Calderwood SK (2006) Role of scavenger receptors in the binding and internalization of heat shock protein 70. J Immunol 177:8604–8611
Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H (2001) Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem 276:31332–31339
Shi Y, Evans JE, Rock KL (2003) Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425:516–521
Shi Y, Zheng W, Rock KL (2000) Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc Natl Acad Sci USA 97:14590–14595
Shi Y, Rock KL (2002) Cell death releases endogenous adjuvants that selectively enhance immune surveillance of particulate antigens. Eur J Immunol 32:155–162
Behrens MD, Wagner WM, Krco CJ, Erskine CL, Kalli KR, Krempski J, Gad EA, Disis ML, Knutson KL (2008) The endogenous danger signal, crystalline uric acid, signals for enhanced antibody immunity. Blood 111:1472–1479
Shi Y, Galusha SA, Rock KL (2006) Cutting edge: elimination of an endogenous adjuvant reduces the activation of CD8 T lymphocytes to transplanted cells and in an autoimmune diabetes model. J Immunol 176:3905–3908
Dalbeth N, Haskard DO (2005) Mechanisms of inflammation in gout. Rheumatology (Oxford) 44:1090–1096
Abramson S, Hoffstein ST, Weissmann G (1982) Superoxide anion generation by human neutrophils exposed to monosodium urate. Arthritis Rheum 25:174–180
Chen L, Hsieh MS, Ho HC, Liu YH, Chou DT, Tsai SH (2004) Stimulation of inducible nitric oxide synthase by monosodium urate crystals in macrophages and expression of iNOS in gouty arthritis. Nitric Oxide 11:228–236
di Giovine FS, Malawista SE, Thornton E, Duff GW (1991) Urate crystals stimulate production of tumor necrosis factor alpha from human blood monocytes and synovial cells. Cytokine mRNA and protein kinetics, and cellular distribution. J Clin Invest 87:1375–1381
Di Giovine FS, Malawista SE, Nuki G, Duff GW (1987) Interleukin 1 (IL 1) as a mediator of crystal arthritis. Stimulation of T cell and synovial fibroblast mitogenesis by urate crystal-induced IL 1. J Immunol 138:3213–3218
Guerne PA, Terkeltaub R, Zuraw B, Lotz M (1989) Inflammatory microcrystals stimulate interleukin-6 production and secretion by human monocytes and synoviocytes. Arthritis Rheum 32:1443–1452
Terkeltaub R, Zachariae C, Santoro D, Martin J, Peveri P, Matsushima K (1991) Monocyte-derived neutrophil chemotactic factor/interleukin-8 is a potential mediator of crystal-induced inflammation. Arthritis Rheum 34:894–903
Murakami Y, Akahoshi T, Hayashi I, Endo H, Hashimoto A, Kono S, Kondo H, Kawai S, Inoue M, Kitasato H (2003) Inhibition of monosodium urate monohydrate crystal-induced acute inflammation by retrovirally transfected prostaglandin D synthase. Arthritis Rheum 48:2931–2941
Murakami Y, Akahoshi T, Kawai S, Inoue M, Kitasato H (2002) Antiinflammatory effect of retrovirally transfected interleukin-10 on monosodium urate monohydrate crystal-induced acute inflammation in murine air pouches. Arthritis Rheum 46:2504–2513
Terkeltaub R, Baird S, Sears P, Santiago R, Boisvert W (1998) The murine homolog of the interleukin-8 receptor CXCR-2 is essential for the occurrence of neutrophilic inflammation in the air pouch model of acute urate crystal-induced gouty synovitis. Arthritis Rheum 41:900–909
Ryckman C, Gilbert C, de Medicis R, Lussier A, Vandal K, Tessier PA (2004) Monosodium urate monohydrate crystals induce the release of the proinflammatory protein S100A8/A9 from neutrophils. J Leukoc Biol 76:433–440
Rouleau P, Vandal K, Ryckman C, Poubelle PE, Boivin A, Talbot M, Tessier PA (2003) The calcium-binding protein S100A12 induces neutrophil adhesion, migration, and release from bone marrow in mouse at concentrations similar to those found in human inflammatory arthritis. Clin Immunol 107:46–54
Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL (2006) MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 116:2262–2271
Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R (2005) Innate immunity conferred by toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum 52:2936–2946
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440:237–241
Di Virgilio F (2000) Dr. Jekyll/Mr. Hyde: the dual role of extracellular ATP. J Auton Nerv Syst 81:59–63
Grierson JP, Meldolesi J (1995) Shear stress-induced [Ca2+]i transients and oscillations in mouse fibroblasts are mediated by endogenously released ATP. J Biol Chem 270:4451–4456
Burnstock G (2006) Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev 58:58–86
Ferrari D, Chiozzi P, Falzoni S, Hanau S, Di Virgilio F (1997) Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J Exp Med 185:579–582
MacDonald PE, Braun M, Galvanovskis J, Rorsman P (2006) Release of small transmitters through kiss-and-run fusion pores in rat pancreatic beta cells. Cell Metab 4:283–290
Eltzschig HK, Eckle T, Mager A, Kuper N, Karcher C, Weissmuller T, Boengler K, Schulz R, Robson SC, Colgan SP (2006) ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res 99:1100–1108
Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S (2001) Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci 21:1975–1982
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758
Wu LJ, Vadakkan KI, Zhuo M (2007) ATP-induced chemotaxis of microglial processes requires P2Y receptor-activated initiation of outward potassium currents. Glia 55:810–821
Idzko M, Dichmann S, Ferrari D, Di Virgilio F, la Sala A, Girolomoni G, Panther E, Norgauer J (2002) Nucleotides induce chemotaxis and actin polymerization in immature but not mature human dendritic cells via activation of pertussis toxin-sensitive P2y receptors. Blood 100:925–932
Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232
Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Melchiorri L, Baricordi OR, Di Virgilio F (1997) Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J Immunol 159:1451–1458
la Sala A, Sebastiani S, Ferrari D, Di Virgilio F, Idzko M, Norgauer J, Girolomoni G (2002) Dendritic cells exposed to extracellular adenosine triphosphate acquire the migratory properties of mature cells and show a reduced capacity to attract type 1 T lymphocytes. Blood 99:1715–1722
la Sala A, Ferrari D, Corinti S, Cavani A, Di Virgilio F, Girolomoni G (2001) Extracellular ATP induces a distorted maturation of dendritic cells and inhibits their capacity to initiate Th1 responses. J Immunol 166:1611–1617
Hasko G, Kuhel DG, Salzman AL, Szabo C (2000) ATP suppression of interleukin-12 and tumour necrosis factor-alpha release from macrophages. Br J Pharmacol 129:909–914
Bulanova E, Budagian V, Orinska Z, Hein M, Petersen F, Thon L, Adam D, Bulfone-Paus S (2005) Extracellular ATP induces cytokine expression and apoptosis through P2X7 receptor in murine mast cells. J Immunol 174:3880–3890
Ferrari D, Wesselborg S, Bauer MK, Schulze-Osthoff K (1997) Extracellular ATP activates transcription factor NF-kappaB through the P2Z purinoreceptor by selectively targeting NF-kappaB p65. J Cell Biol 139:1635–1643
Ferrari D, Stroh C, Schulze-Osthoff K (1999) P2X7/P2Z purinoreceptor-mediated activation of transcription factor NFAT in microglial cells. J Biol Chem 274:13205–13210
Robson SC, Wu Y, Sun X, Knosalla C, Dwyer K, Enjyoji K (2005) Ectonucleotidases of CD39 family modulate vascular inflammation and thrombosis in transplantation. Semin Thromb Hemost 31:217–233
Picher M, Burch LH, Hirsh AJ, Spychala J, Boucher RC (2003) Ecto 5′-nucleotidase and nonspecific alkaline phosphatase. Two AMP-hydrolyzing ectoenzymes with distinct roles in human airways. J Biol Chem 278:13468–13479
Kaisho T, Akira S (2006) Toll-like receptor function and signaling. J Allergy Clin Immunol 117:979–987 (quiz 988)
Ni H, Capodici J, Cannon G, Communi D, Boeynaems JM, Kariko K, Weissman D (2002) Extracellular mRNA induces dendritic cell activation by stimulating tumor necrosis factor-alpha secretion and signaling through a nucleotide receptor. J Biol Chem 277:12689–12696
Kariko K, Ni H, Capodici J, Lamphier M, Weissman D (2004) mRNA is an endogenous ligand for toll-like receptor 3. J Biol Chem 279:12542–12550
Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, Akira S, Kelly KM, Reeves WH, Bauer S, Krieg AM (2005) Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves toll-like receptors 7 and 8. J Exp Med 202:1575–1585
Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T (2008) Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol 9:1179–1188
Hassfeld W, Steiner G, Studnicka-Benke A, Skriner K, Graninger W, Fischer I, Smolen JS (1995) Autoimmune response to the spliceosome. An immunologic link between rheumatoid arthritis, mixed connective tissue disease, and systemic lupus erythematosus. Arthritis Rheum 38:777–785
Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, Kohn LD, Klinman DM (2001) Genomic DNA released by dying cells induces the maturation of APCs. J Immunol 167:2602–2607
Yasuda K, Ogawa Y, Yamane I, Nishikawa M, Takakura Y (2005) Macrophage activation by a DNA/cationic liposome complex requires endosomal acidification and TLR9-dependent and -independent pathways. J Leukoc Biol 77:71–79
Yasuda K, Rutz M, Schlatter B, Metzger J, Luppa PB, Schmitz F, Haas T, Heit A, Bauer S, Wagner H (2006) CpG motif-independent activation of TLR9 upon endosomal translocation of “natural” phosphodiester DNA. Eur J Immunol 36:431–436
Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, Bauer S, Hochrein H, Wagner H (2005) Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J Immunol 174:6129–6136
Decker P, Singh-Jasuja H, Haager S, Kotter I, Rammensee HG (2005) Nucleosome, the main autoantigen in systemic lupus erythematosus, induces direct dendritic cell activation via a MyD88-independent pathway: consequences on inflammation. J Immunol 174:3326–3334
Vanden Berghe T, Kalai M, Denecker G, Meeus A, Saelens X, Vandenabeele P (2006) Necrosis is associated with IL-6 production but apoptosis is not. Cell Signal 18:328–335
Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, Cidlowski JA, Ciechanover A, Cohen GM, De Laurenzi V, De Maria R, Deshmukh M, Dynlacht BD, El-Deiry WS, Flavell RA, Fulda S, Garrido C, Golstein P, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Jaattela M, Kepp O, Kimchi A, Klionsky DJ, Knight RA, Kornbluth S, Kumar S, Levine B, Lipton SA, Lugli E, Madeo F, Malomi W, Marine JC, Martin SJ, Medema JP, Mehlen P, Melino G, Moll UM, Morselli E, Nagata S, Nicholson DW, Nicotera P, Nunez G, Oren M, Penninger J, Pervaiz S, Peter ME, Piacentini M, Prehn JH, Puthalakath H, Rabinovich GA, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Scorrano L, Simon HU, Steller H, Tschopp J, Tsujimoto Y, Vandenabeele P, Vitale I, Vousden KH, Youle RJ, Yuan J, Zhivotovsky B, Kroemer G (2009) Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 16:1093–1107
Bell DA, Morrison B (1991) The spontaneous apoptotic cell death of normal human lymphocytes in vitro: the release of, and immunoproliferative response to, nucleosomes in vitro. Clin Immunol Immunopathol 60:13–26
Bell CW, Jiang W, Reich CF 3rd, Pisetsky DS (2006) The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 291:C1318–C1325
Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, Kalden JR, Schett G, Rovere-Querini P, Herrmann M, Voll RE (2008) Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 205:3007–3018
Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR (1998) Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum 41:1241–1250
Amoura Z, Piette JC, Chabre H, Cacoub P, Papo T, Wechsler B, Bach JF, Koutouzov S (1997) Circulating plasma levels of nucleosomes in patients with systemic lupus erythematosus: correlation with serum antinucleosome antibody titers and absence of clear association with disease activity. Arthritis Rheum 40:2217–2225
Rosen A, Casciola-Rosen L (1999) Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ 6:6–12
Casciola-Rosen LA, Anhalt G, Rosen A (1994) Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 179:1317–1330
Wu X, Molinaro C, Johnson N, Casiano CA (2001) Secondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens: implications for systemic autoimmunity. Arthritis Rheum 44:2642–2652
Casciola-Rosen L, Andrade F, Ulanet D, Wong WB, Rosen A (1999) Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J Exp Med 190:815–826
Berg CP, Stein GM, Keppeler H, Gregor M, Wesselborg S, Lauber K (2008) Apoptosis-associated antigens recognized by autoantibodies in patients with the autoimmune liver disease primary biliary cirrhosis. Apoptosis 13:63–75
Prasad S, Soldatenkov VA, Srinivasarao G, Dritschilo A (1998) Identification of keratins 18, 19 and heat-shock protein 90 beta as candidate substrates of proteolysis during ionizing radiation-induced apoptosis of estrogen-receptor negative breast tumor cells. Int J Oncol 13:757–764
Acknowledgments
We thank the members of the Wesselborg and the Herrmann lab for stimulating discussions and apologize to all colleagues in the field of engulfment and inflammation, whose work could not be cited here owing to space constraints. This work was supported by the DFG We 1801/2-4 and SFB 685.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peter, C., Wesselborg, S., Herrmann, M. et al. Dangerous attraction: phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis 15, 1007–1028 (2010). https://doi.org/10.1007/s10495-010-0472-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-010-0472-1