
Abstract
The incidence of melanoma continues to dramatically increase in most Western countries with predominantly Caucasian populations. However, only limited therapies for the metastatic stage of the disease are currently available. The main purpose of this study is to determine approaches that can substantially increase radiosensitivity of melanoma cells. The PI3K-AKT, NF-κB and COX-2 pathways, which are involved in the radioprotective response, are highly active in melanoma cells. Pharmacological suppression of COX-2 and PI3K-AKT, or RNAi-mediated knockdown of COX-2, substantially increased levels of G2/M arrest of the cell cycle and decreased clonogenic survival of gamma-irradiated melanomas, predominantly via a necrotic mechanism. On the other hand, resveratrol, a polyphenolic phytoalexin, selectively targets numerous cell signaling pathways, decreasing clonogenic survival primarily via an apoptotic mechanism. In melanoma cells, resveratrol inhibits STAT3 and NF-κB-dependent transcription, culminating in suppression of cFLIP and Bcl-xL expression, while activating the MAPK- and the ATM-Chk2-p53 pathways. Resveratrol also upregulates TRAIL promoter activity and induces TRAIL surface expression in some melanoma cell lines, resulting in a rapid development of apoptosis. Sequential treatment of melanoma cells, first with γ-irradiation to upregulate TRAIL-R surface expression, and then with resveratrol to suppress antiapoptotic proteins cFLIP and Bcl-xL and induce TRAIL surface expression, had dramatic effects on upregulation of apoptosis in some melanoma lines, including SW1 and WM35. However, for melanoma lines exhibiting suppressed translocation of TRAIL to the cell surface, a necrotic mechanism of cell death was primarily involved in radiation response. Hence, surface expression of TRAIL induced by resveratrol appears to be a decisive event, one which determines an apoptotic versus a necrotic response of melanoma cells to sequential treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ionizing radiation and chemotherapy are two predominant therapeutic modalities used for cancer treatment. Cytotoxic effects of most conventional anti-cancer drugs (for example, cisplatin and etoposide) and ionizing radiation are mediated through pleotropic mechanisms, which include interaction and damage of genomic DNA followed by activation of DNA damage induced signaling pathways (both p53-dependent and p53-independent). These pathways culminate in cell cycle arrest and/or induction of cell death by apoptosis, necrosis, autophagy or mitotic catastrophe [1, 2]. In this scenario, the Bcl-2 family members with prosurviving functions, such as Bcl-xL, play a crucial role by neutralizing function of the proapoptotic BH3-only proteins, resulting in an inhibition of transmission of an apoptotic signal to the executor proteins BAX and BAK and suppression of the mitochondrial death pathway [3]. A role of transcription factor NF-κB, which controls expression of Bcl-xL and many other proteins with survival functions, is well established for positive regulation of radioprotection in normal and cancer cells [4]. On the other hand, alternative therapies, which have been suggested for induction of apoptosis in cancer cells, are often based on direct activation of extrinsic death signaling pathways. Examples of this strategy include the use of recombinant death ligands of the TNF superfamily, such as Fas Ligand and TRAIL, or the correspondent agonistic monoclonal antibodies to the death receptors, Fas, TRAIL-R1/DR4 or TRAIL-R2/DR5 [5, 6]. Targeting TRAIL-receptor mediated signaling pathways for induction of apoptosis is currently under evaluation in multiple clinical trials for several types of cancer [7, 8]. Finally, combined modality treatments, which include γ-irradiation and specific inhibitions of the cell survival pathways, appear to be promising approaches for the control and suppression of cancer development [9, 10].
Melanoma, the most aggressive form of skin cancer, is known to be relatively resistant to conventional radiotherapy and chemotherapy. In the USA, approximately 60,000 new cases were diagnosed and 8,100 deaths occurred in 2007 (ACS, 2007). However, only limited therapies for the metastatic stage of the disease are currently available. Various attempts have been made to achieve high levels of apoptosis in response to treatment for this type of cancer. The main purpose of this study is to ascertain approaches that can substantially increase radiosensitivity of melanoma cells by inhibiting activity/expression levels of the main regulators of the general cell survival, COX-2 [11], AKT [12] or by simultaneous expression of endogenous death ligand and death receptor, TRAIL and TRAIL-Receptor [5].
Most melanomas possess non-mutated wild-type p53, which functions as a transcription factor controlling TRAIL-R2/DR5, FAS, BAX, BID, PUMA, NOXA and p21-WAF gene expression and mediating the cell cycle arrest or/and apoptosis [13, 14]. However, p53 does not appear to play a direct role in executive regulation of apoptosis via p53-dependent BH3-only proteins (PUMA, NOXA) or p53-dependent BAX in many melanoma lines. This lack of proapoptotic p53 influence is due to downstream protective mechanisms, such as cancer-specific suppression of mitochondrial function and the mitochondrial death pathway [15, 16] that was often observed in advanced melanomas [17].
Materials and methods
Materials
Resveratrol (RV) was obtained from Sigma (St. Louis, MO, USA). Human soluble Killer-TRAIL (recombinant) was purchased from Alexis (San Diego, CA, USA). JNK inhibitor SP600125 and IKK-NF-κB inhibitor BAY 11-7082 were obtained from Biomol (Plymouth Meeting, PA, USA); PI3K inhibitor LY294002, MEK inhibitor U0126, MAPK p38 inhibitor SB203580 and caspase inhibitors zVAD-fmk were purchased from Calbiochem (La Jolla, CA, USA). COX-2 inhibitor NS398 was obtained from Cayman Chemical Company (Ann Arbor, Michigan, USA).
Cell lines
Human melanoma cell lines LU1205 (also known as 1205lu), WM9 and WM35 [18], as well as HHMSX, LOX, and mouse melanoma line SW1 were maintained in a DMEM medium supplemented with 10% fetal bovine serum (FBS), l-glutamine and antibiotics.
Irradiation procedures
To determine sensitivity to γ-rays, plated melanoma cells were exposed to radiation from a Gammacell 40 137Cs irradiator (dose rate, 0.82 Gy/min) at Columbia University. Six to 24 h after irradiation, cells were analyzed by flow cytometry or underwent additional treatment.
FACS analysis of TRAIL and TRAIL-R2/DR5 levels
Surface levels of TRAIL and TRAIL-R2/DR5 on human melanomas were determined by staining with a PE-conjugated anti-human-TRAIL or anti-human DR5 mAb (R&D System, Minneapolis, MN, USA and eBioscience, San Diego, CA, USA) and subsequent flow cytometry. PE-conjugated nonspecific mouse IgG1 was used as an immunoglobulin isotype control. A FACS Calibur flow cytometer (Becton Dickinson, Mountain View, CA, USA) and the CellQuest program were used to perform flow cytometric analysis. All experiments were independently repeated 3–5 times.
Transfection and luciferase assay
A NF-κB luciferase reporter containing two κB binding sites, a Jun2-Luc reporter and empty vector tk-Luc [19], and a STAT-Luc reporter containing three repeats of GAS sites from the Ly6A/E promoter, were used to determine NF-κB, AP-1 and STAT transactivation, respectively. Additional reporter constructs used included: 1.5 kb TRAIL-promoter-Luc [20], 1 kb cFLIP-promoter-Luc [21, 22] and p53RE-Luc [23]. Transient transfection of different reporter constructs (1 μg) together with pCMV-βgal (0.25 μg) into 5 × 105 melanoma cells was performed using Lipofectamine (Life Technologies-Invitrogen, Carlsbad, CA, USA). Proteins were prepared for β-Gal and luciferase analysis 16 h after transfection. Luciferase activity was determined using the Luciferase assay system (Promega, Madison, WI, USA) and was normalized based on β-galactosidase levels. Since resveratrol was known to partially inhibit enzymatic activity of firefly luciferase [24], a ratio of the specific luciferase reporter activity to luciferase activity driven by the empty tk-Luc vector in a mock control culture was determined for analysis.
COX-2 suppression by RNAi
The pSUPER retro RNA interference (RNAi) system (Oligoengine, Seattle, WA, USA), which has been utilized for the production of small RNAi transcripts, was used to suppress cFLIP expression. Three variants of RNAi, of 19 nucleotides each, were designed to target human COX-2 and expressed using vector pSUPER.retro.puro (pSR-puro). RNAi COX-2-379 (CCUUCUCUAACCUCUCCUA) was most efficient in corresponding mRNA targeting.
Apoptosis studies
Cells were exposed to soluble TRAIL (50 ng/ml) alone, resveratrol (RV) (25–100 μM) or TRAIL (50 ng/ml) in combination with cycloheximide (2 μg/ml). Apoptosis was assessed by quantifying the percentage of hypodiploid nuclei undergoing DNA fragmentation or by quantifying the percentage of Annexin-V-FITC-positive cells (BD Pharmingen, San Diego, CA, USA). Flow cytometric analysis was performed on a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA) using the CellQuest program.
Clonogenic survival assay
Cells (500/plate) were placed in triplicate on 10 cm- plates 16 h before treatment. For analysis of clonogenic survival of melanoma cells after treatment (24 h) with TRAIL, resveratrol (RV) or their combination, colonies were stained with crystal violet solution 12 days after treatment. The percentage of colony-forming efficiency (relative to values of untreated control cells) was calculated.
Western blot analysis
Total cell lysates (50 μg protein) were resolved on 10% SDS-PAGE, and processed according to standard protocols. The antibodies (Abs) used for Western blotting included: monoclonal anti-β-Actin (Sigma, St. Louis, MO, USA), monoclonal anti-FLIP (NF6) (Axxora, San Diego, CA, USA), monoclonal anti-XIAP (BD Biosciences, San Jose, CA, USA), monoclonal anti-p21-WAF1 (Cell Signaling Beverly, MA, USA); polyclonal Abs to TRAIL (human), TRAIL-R1/DR4 and TRAIL-R2/DR5 (Axxora, San Diego, CA, USA); polyclonal Abs against: phospho-AKT (Ser473) and AKT; phospho-STAT3 (Tyr705) and STAT3; phospho-c-Jun (Ser73) and c-Jun; phospho-SAPK/JNK (Thr183/Tyr185) and JNK; phospho-p44/p42 MAP kinase (Thr202/Tyr204) and p44/p42 MAP kinase; phospho-p38 MAP kinase (Thr180/Tyr182) and p38 MAP kinase; phospho-FOXO3A (Ser318/321) and FOXO3A; BAX, phospho-p53 (Ser20) and p53 (Cell Signaling, Beverly, MA, USA). Optimal dilutions of primary Abs were 1:1000 to 1:5000. The secondary Abs anti-mouse or anti-rabbit) were conjugated to horseradish peroxidase (dilution 1:5000 to 1:10000); signals were detected using the ECL system (Amersham, Piscataway, NJ, USA).
EMSA
Electrophoretic mobility shift assay (EMSA) was performed for the detection of NF-κB DNA-binding activity as previously described, using the labeled double-strand oligonucleotide AGCTTGGGGACTTTCCAGCCG. (Binding site is underlined). Ubiquitous NF-Y DNA-binding activity was used as an internal control [25].
Statistical analysis
Data were calculated as means and standard deviations. Comparisons of surviving fractions between treated and control groups were made by the Students’ t-tests. A P-value of 0.05 or less between groups was considered significant.
Results
Effects of suppression of PI3K-AKT and COX-2 on radiosensitivity of melanoma cells
Direct γ-irradiation of most melanoma cell lines is followed by marked G2/M arrest of the cell cycle and low levels of apoptosis 48 h after treatment (Fig. 1a for WM35 melanoma cells). Results of clonogenic survival assays 12 days after γ-irradiation demonstrated, however, that melanoma lines exhibit differential radiation resistance, which was relatively high for SW1 murine and HHSMS human metastatic melanoma cells, average for LU1205 human metastatic and WM35 human radial growth phase melanoma cells, and low for WM9 human metastatic melanoma cells (Fig. 1b and c). Most melanoma lines used in this study possess relatively high basal levels of activated phospho-Akt (Ser473), which is involved in protection against cell death (Fig. 1d). The only exception was WM9 line that exhibits low to average levels of phospho-AKT. High AKT activity, found in WM35, LU1205 and SW1, but not in WM9 cells, was correlated with higher levels radioresistance of WM35, LU1205 and SW1 cells as determined by clonogenic survival assays (Fig. 1b, c).

Radiation resistance of melanoma cell lines. (a) Cell cycle-apoptosis analysis of melanoma lines WM35 48 h after γ-irradiation using PI staining DNA and flow cytometry. Gamma-irradiation induces G2/M arrest of the cell cycle, while levels of apoptosis (Apop) are relatively low. (b, c) Clonogenic survival assay of human WM35, WM9, LU1205, HHMSX and mouse SW1 melanoma lines 12 days after γ-irradiation of 1.25–5 Gy. Error bars represent mean ± SD from four independent experiments. Results of a typical experiment are shown in (b). (d) Western blot analysis of phospho-AKT (Ser473), total AKT and COX-2 levels in melanoma cell lines
Low apoptotic levels 48 h after irradiation, but pronounced decreases in clonogenic survival 12 days after treatment indicate that non-apoptotic mechanisms are preferentially involved in regulation of radiosensitivity of melanoma cells. The early response to γ-radiation, originating from activation of the DNA damage signaling pathways, includes p53-Ser20 phosphorylation (Fig. 2a) via ATM-Chk2 pathway and correspondent upregulation of p53 transcription factor activity [26]. In addition, upregulation of NF-κB activity, a common protective reaction of cells to different types of stress conditions (Fig. 2b), was observed 3–6 h after γ-irradiation of melanoma cells [10]. However, no notable activation of procaspase-3 was detected in these conditions, confirming the absence of induction of pronounced apoptotic signaling 6 h after γ-irradiation (Fig. 2a). Besides active phospho-AKT and NF-κB, two main protein regulators of general cell survival were detected in most melanoma lines at average to high levels: COX-2 and Bcl-xL (Figs. 1d and 2a).

Inhibition of COX-2 and PI3K-AKT activities further upregulates the G2/M arrest WM35 melanoma cells following γ-irradiation. (a) Western blot analysis of indicated proteins 6 h after irradiation. (b) NF-κB DNA-binding activity determined by EMSA 6 h after γ-irradiation of WM35 cells. (c) Effects of γ-radiation, COX-2 inhibitor NS398 (50 μM) and PI3K-AKT inhibitor LY294002 (50 μM) alone or in combination on the cell cycle of WM35 melanoma. Cells were stained with PI and analyzed by flow cytometry. Results of a typical experiment (one of three) are presented. (d) Effects of NS398 and LY294002 (50 μM) on γ-irradiation-induced death of WM35 cells. Apoptosis-necrosis analysis was performed by Annexin V-FITC + PI staining and the flow cytometry. Results of a typical experiment (one of four) are presented
To decrease radioresistance of WM35 and LU1205 cells, we used inhibition of enzymatic activity of PI3K-AKT or COX-2, which were constituently active in WM35 and LU1205 cells (Figs. 1d, 2a), in combination with γ-irradiation. An inhibitor of COX-2 (NS398, 50 μM) or PI3K-AKT (LY294002, 50 μM) was added to WM35 and LU1205 cell cultures immediately after γ-irradiation. Both inhibitors substantially increased levels of the G2/M arrest in irradiated WM35 (Fig. 2c) and LU1205 cells (data not shown). Furthermore, NS398 modestly enhanced upregulation of apoptosis in irradiated WM35 cells, while LY294002, which additionally to PI3K targets PI3K-related kinases, ATM, ATR and DNA-PK, was more efficient at increasing the percentage of cells in G2/M arrest (Fig. 2c). Staining WM35 melanoma cells with Annexin-V-FITC + PI and the subsequent FACS analysis determined preferentially necrotic response to γ-irradiation in the presence of COX-2 and PI3K-AKT inhibitors (Fig. 2d). Annexin-V-positive (apoptotic) cells were also observed at low levels 6 h after treatment WM9 and LU125 cells (data not shown). Furthermore, clonogenic survival was substantially decreased for irradiated melanoma cells in the presence of NS398 or LY294002 12 days after treatment, further suggesting the occurrence of a mixed type of slow cell death, principally by necrosis, but also involving apoptosis (Fig. 3a–c). Since WM9 cells were intrinsically more radiosensitive, both inhibitors notably accelerated cell death after irradiation, including after doses as low as 1.25 Gy (Fig. 3b).

Inhibition of COX-2 and PI3K-AKT decreased melanoma cell survival following γ-irradiation. (a–c) Clonogenic survival assay of WM35, WM9 and LU1205 cells 12 days after γ-irradiation of 1.25–5 Gy alone or in combination with either NS398 (50 μM) or LY294002 (50 μM). Error bars represent mean ± SD from three independent experiments
To obtain additional data supporting our hypothesis that COX-2 suppression upregulates radiation-induced level of the G2/M arrest of the cell cycle, we established LU1205, WM9 and WM35 mass cultures with partial suppression of COX-2 expression levels by specific RNAi (Fig. 4a and data not shown). Basal and radiation-induced (2.5–5 Gy) levels of total p53 and phospho-p53 (Ser20) were substantially increased after COX-2 knockdown (Fig. 4a) followed by a corresponding increase in p53-dependent transcription as observed by luciferase reporter analysis (data not shown). Radiation-induced levels of G2/M arrest also were correspondently higher in cells with COX-2 knockdown (Fig. 4b). No additional effects were detected in upregulation of TRAIL-R2/DR5 surface expression after irradiation of COX-2 knockdown cells (Fig. 4c). On the other hand, clonogenic survival analysis demonstrated a pronounced decrease in survival of melanoma cells with suppressed COX-2 after γ-irradiation (Fig. 4d). Similar to results of pharmacological inhibition of COX-2 activity, COX-2 knockdown increased levels of mixed, mainly non-apoptotic, death of LU1205, WM9 and WM35 melanoma cells (Fig. 4d and data not shown). Hence, these data demonstrated substantial effects resulting from suppression of both COX-2 activity and expression levels and culminating in decreased clonogenic survival of irradiated melanoma cells.

RNAi-mediated knockdown of COX-2 expression substantially increased levels of the G2/M arrested metastatic melanoma cells and decreased cancer cell survival. (a) Suppression COX-2 expression levels by specific RNAi affect p53 (P-Ser20) basal levels in LU1205 melanoma cells. (b) Effects of COX-2 knockdown on the cell cycle in LU1205 and WM9 melanoma cells. PI staining and FACS analysis were used. (c) Effects of γ-irradiation on surface TRAIL-R2/DR5 levels in the control and COX-2 knockdown LU1205 cells. Immunostaining with anti-DR5-PE and FACS analysis were used. (d) Clonogenic survival assay of the control and COX-2 knockdown LU1205 and WM9 cells after γ-irradiation. Error bars represent mean ± SD from three independent experiments
TRAIL-induced apoptosis in γ-irradiated melanoma cells
By default, necrosis is accompanied acutely by severe inflammation, and often chronically by fibrosis or even organ damage, while apoptosis represents a more organized and physiologically rapid form of cell death [27]. Could it be possible to control the mechanism of death after γ-irradiation of melanoma cells? As a rule, the extrinsic death pathway can be activated in melanoma cells after appropriate treatment. Based on our previous results [10], we induced TRAIL-mediated apoptosis 16 h after γ-irradiation melanoma cells. This approach was very effective for treatment of WM35 and LU1205 cells by exogenous recombinant TRAIL, due to an additional upregulation of TRAIL-R2/DR5 surface levels by γ-irradiation (Fig. 5a). As expected, the presence of cycloheximide (CHX) substantially increased TRAIL-mediated apoptosis (Fig. 5b and data not shown). Clonogenic survival assay further confirmed high effectiveness of TRAIL treatment for downregulation of survival of irradiated WM35 and LU1205 melanoma cells (Fig. 5c). For intrinsically TRAIL-sensitive WM9 cells, we observed only a small additional benefit for upregulation of TRAIL-mediated apoptosis and downregulation of cell survival after irradiation (Fig. 5c).

Combined treatment of melanoma lines with γ-irradiation and TRAIL. (a) Effect of γ-irradiation on TRAIL-R2/DR5 surface expression. Immunostaining with anti-DR5-PE and FACS analysis were used. (b). Upregulation of TRAIL-induced apoptosis in WM35 melanoma cells after sequential treatment with γ-irradiation (5 Gy) and 16 h after irradiation with recombinant TRAIL (50 ng/ml) for an additional 24 h. Cell cycle-apoptosis analysis was performed using PI staining DNA and flow cytometry. (c) Clonogenic survival assay of WM35, WM9 and LU1205 cells 12 days after treatment with recombinant TRAIL (50 ng/ml) alone or in combination with γ-irradiation of 1.25–5 Gy. Error bars represent mean ± SD from three independent experiments
Taken together, results obtained with metastatic LU1205 cells were relatively similar to those obtained with radial growth phase WM35 cells (Fig. 5). Both cell lines display average levels of radiosensitivity that could be enhanced by combined treatment with γ-irradiation and COX-2 or PI3K-AKT inhibitors via necrotic pathways. However, γ-irradiation substantially decreased clonogenic survival of LU1205 and WM35 after the subsequent treatment with recombinant TRAIL via apoptotic pathway. Although inhibitors of general cell survival and recombinant TRAIL decrease clonogenic survival of irradiated melanoma cells at potentially similar levels, there could be a clinically relevant difference between necrotic cancer cell death after γ-irradiation in the presence of NS398 or LY294002 and TRAIL-mediated apoptosis of pre-irradiated melanoma cells. Could endogenous TRAIL be induced on the surface of TRAIL-Receptor positive melanoma cells after proper treatment for the subsequent initiation TRAIL-mediated apoptosis?
Effects of resveratrol (RV) on TRAIL surface expression and the main signaling pathways in melanoma cells
Our recent finding demonstrates that resveratrol (RV), a polyphenolic phytoalexin, may accelerate exogenous TRAIL induced death in many melanoma cell lines [17]. Furthermore, RV by itself was an efficient inducer of apoptotic death in some cancer cell lines [28], including melanoma lines [17]. We observed that RV upregulated TRAIL gene promoter activity and induced TRAIL surface expression that was quite diverse in different melanoma lines (Fig. 6a and b). We chose the SW1 melanoma cells, due to its intrinsically high level of radioresistance (Fig. 1b), but high sensitivity to RV for further investigations of the interaction of RV and TRAIL mediated signaling.

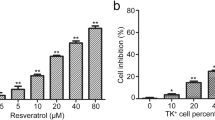
Effects of resveratrol (RV) on cellular proteins controlling cell survival and apoptosis in SW1 melanoma cells. (a) Effects of RV on NF-κB-, AP-1, STAT- and p53-dependent luciferase reporter activities, TRAIL and FLIP promoter activities. (b) Effects of RV on TRAIL surface expression in SW1 melanoma cells. Immunostaining with anti-TRAIL-PE mAb and FACS analysis were used. (c) Effects of RV (50 μM) on basal nuclear NF-κB and NF-Y activities in SW1 cells determined by EMSA 6 h after treatment. Positions of DNA-binding complexes are indicated. Free labeled probes are not shown. Western blot analysis was performed for detection of total and phospho-protein levels of STAT3, FOXO-3A, ERK1/2, JNK1/2, cJun, p38MAPK, ATF2, TRAIL, TRAIL-R, cFLIP, phospho-p53 (Ser20), total p53 and p21-WAF 6 h after treatment with RV. Western blot analysis of Bcl-xL, XIAP and β-actin levels 16 h after treatment with RV. (d) Levels of apoptosis induced by RV (50 μM) in melanoma lines 16 h after treatment. (e) Effects of anti-TRAIL (5 μg/ml) and anti-TNF (5 μg/ml) inhibitory mAbs on RV-induced apoptosis in SW1 cells. PI staining DNA and flow cytometry analysis were used for cell cycle-apoptosis analysis. (f) Clonogenic survival assay for combined treatment of SW1 cells by RV (0–100 μM) and Bay 11-7082 (5 μM). Error bars represent mean ± SD from three independent experiments
There is a profound qualitative similarity in action of RV on signaling pathways in human LU1205 [29] and murine SW1 melanoma cells (Fig. 6c). Similarly to human LU1205 cells, RV treatment downregulated NF-κB activity, phospho-Tyr-STAT3 and phospho-Ser-FOXO3A levels, while activated MAPK’s (ERK, p38 and JNK) and their targets cJun and ATF2 in murine SW1 cells. There was a notable increase in AP-1/Jun2-dependent and p53-dependent transcription and simultaneous decrease in STAT3- and NF-κB-mediated transcription in SW1 cells following RSV treatment (Fig. 6a). RV caused opposite effects on promoter activity of TRAIL (upregulation) and cFLIP (downregulation) (Fig. 6a) that was accompanied by downregulation of cFLIP-L protein levels (Fig. 6c). Furthermore, Bcl-xL substantially decreased, while phospho-p53 (Ser20) levels and levels of its target p21-WAF further increased in SW1 cells after RSV treatment (Fig. 6a and c).
In order to determine the role of RV in elevated activation of the MAPK pathways (MEK-ERK, MKK6-p38-ATF2, MKK4/7-JNK-cJun) in both LU1205 and SW1 melanoma cell lines, we used specific pharmacological inhibitors: U0126 (10 μM) for MEK-ERK, SB203580 (10 μM) for p38 and SP600125 (20 μM) for JNK. Inhibition of RV-induced MEK-ERK or p38 MAPK activation substantially accelerated apoptotic response of SW1 cells to RV (25–50 μM), while inhibition of JNK activity did not cause pronounced changes in apoptotic levels induced by RV (data not shown). These data clearly demonstrated a prosurvival function of ERK1/2, and especially MAPK p38 activation, following RV treatment of LU1205 and SW1 cells. JNK activation by RV in melanoma cells appears to play dual proapoptotic and prosurvival roles and suppression of JNK does not notably change the life-death balance in RV-treated melanoma cells. Hence, RV via its effects on the main signaling pathways in melanoma cells resulting in strong downregulation of levels of antiapoptotic proteins, such as cFLIP-L (caspase-8 inhibitor) and Bcl-xL (Fig. 6c), can sensitize these cells to TRAIL-mediated apoptosis [17]. Knockdown of cFLIP levels by the specific RNAi also sensitizes SW1 cells to TRAIL-mediated apoptosis (data not shown).
Resveratrol (RV)-induced TRAIL-mediated cell suicide in some melanoma lines
Human melanocytes and most metastatic human melanomas are relatively resistant to RV-induced apoptotic stimuli (at RV doses of 25–100 μM). In contrast, SW1 mouse melanoma cells were sensitive to RV-induced death signaling, and quickly developed apoptosis at average to high levels 16 h after treatment (Fig. 6d). In addition, levels of apoptosis further increased 40 h after treatment. A clonogenic survival assay confirmed 75–90% death of SW1 melanoma cells 12 days after treatment with 50–100 μM RV (data not shown). There are at least two possible mechanisms that could account for the hypersensitivity of SW1 cells to RV: (i) dose–dependent down-regulation STAT3- and NF-κB-dependent transcription (Fig. 6a and c), which control expression genes with survival functions including cFLIP-L and Bcl-xL [30, 31]; (ii) upregulation of TRAIL promoter activity (via the JNK-cJun pathway) and increased surface expression of endogenous TRAIL induced by RV in a dose–dependent manner (Fig. 6b). In conditions of suppression of STAT3-dependent gene expression and a partial suppression of NF-κB-dependent gene expression, endogenously produced TRAIL, via autocrine/paracrine action, induces a receptor-mediated death signaling cascade in TRAIL-R-positive SW1 cells, ultimately resulting in suicide of these cells. Levels of RV-induced death are correlated with surface expression of endogenous TRAIL following RV treatment of melanoma cell lines (Fig. 6b and d).
RV-induced death of SW1 cells could be blocked by an inhibitory antibody to TRAIL (5 μg/ml) introduced into the culture medium, but not by anti-TNF mAb, which actually promoted RV-induced apoptosis (Fig. 6e). These data indicate that endogenous TRAIL is the critical factor of RV-induced apoptosis in SW1 cells. On the other hand, endogenous TNFα appears to protect against TRAIL-mediated apoptosis via NF-κB activation. We used a pharmacological inhibitor of IKK-NF-κB activation, Bay 11-7082 (5 μM) fur further suppression of basal NF-κB activity in RV-treated melanoma cells. Bay 11-7082, after addition to the cell culture, notably increased RV-induced apoptotic levels, highlighting a protective role of NF-κB against RV-induced apoptosis (Fig. 6f). Taken together, results obtained indicate that proapoptotic effects of RV correlates with negative regulation of STAT3-, NF-κB-mediated transcription, the correspondent suppression of expression of antiapoptotic proteins and, finally, with induction of surface expression of TRAIL. This expression was substantially higher in the sensitive melanoma line SW1, compared to WM35 or LU1205 (Fig. 6b). We have not found a perfect human analog of SW1 melanoma, although LOX human melanoma cells exhibit average levels of endogenous TRAIL expression and TRAIL-mediated apoptosis following RV treatment [17]. RV by itself does not upregulate surface expression of death receptors and requires an additional pretreatment of cancer cells with γ-irradiation for establishing the most optimal conditions for induction of apoptosis.
Synergistic effects of sequential treatment of SW1 melanoma cells by γ-radiation and RV
SW1 cells are relatively radioresistant, while WM35 and LU1205 cells are notably less resistant to γ-radiation (Fig. 1c). In contrast, RV induced higher levels of apoptosis 16 after treatment in SW1 than in WM35 and LU1205 cells (Fig. 7a and data not shown). As expected, γ-irradiation of SW1 cells increased TRAIL-R, but did not affect TRAIL surface expression (Fig. 7b). Finally, sequential treatment of SW1 cells first by γ-radiation to increase surface expression of TRAIL-R, and then 16 h after irradiation with RV for an additional 24 h to induce endogenous TRAIL translocation to the cell surface and to suppress anti-apoptotic cFLIP and Bcl-xL, results in a strong upregulation of RV-induced apoptosis (Fig. 7a) and dramatic downregulation of SW1 clonogenic survival (Fig. 7c). Furthermore, clonogenic survival assays demonstrated substantially decreased survival for WM35 cells after combined treatment by radiation and RV (Fig. 7d). LU1205 cells were more resistant to RV treatment, which did not notably change levels of radiation-induced cell death (Fig. 7e). Optimized strategy requires treatment of LU1205 cells with a combination of TRAIL+RV to achieve effective killing [14].

Synergistic effects of RV and γ-irradiation on regulation of cell death in SW1 mouse and WM35 human melanomas. (a) Effects of γ-irradiation (5 Gy), RV (50 μM) and combined treatment on the cell cycle and apoptosis in SW1 cells. Cells were stained by PI 40 h after treatment. RSV was added 16 h after irradiation for an additional 24 h. Apoptosis (Apop) levels were determined as the percentage of cells with hypodiploid content of DNA in the pre-G0/G1 region using flow cytometry; % cells at the distinct phases of the cell cycle is indicated. Results of typical experiments (one of three) are presented. (b) Effects of γ-irradiation on TRAIL and TRAIL-R surface expression in SW1 cells. (c) Clonogenic survival assay of SW1 cells 12 days after indicated treatments: RV (25–100 μM) and γ-irradiation (2.5 Gy and 5 Gy) were used. Error bars represent mean ± SD from three independent experiments. (d, e) Clonogenic survival assay of WM35 and LU1205 cells 12 days after indicated treatments. Error bars represent mean ± SD from three independent experiments
Critical questions remain regarding: (i) how RV induces endogenous TRAIL protein transport to the cell surface; and (ii) why this process is so different in distinct melanoma lines. These are under current investigation in our laboratory.
Discussion
Scientific observations indicate that the incidence of melanoma has significantly increased over the last 40 years in the USA and worldwide. The probability of an American developing melanoma jumped from 1 in 1,500 in 1960, to 1 in 68 in 2000, and is projected to increase to 1 in 50 by the year 2010 [32, 33]. There is a lack of effective treatments for individuals with advanced disease, and the key to improve survival remains early diagnosis and treatment [34, 35]. Ionizing radiation is not widely used for treatment of metastatic melanomas due to relative radioresistance and prevalent genomic instability of this type of cancer. The complementary approach, based on the combined treatment of γ-irradiation and inhibitors of cell survival pathways, was quite successfully used for treatment of many types of cancer [36], and, as demonstrated by the present study, has potential for use in the treatment of metastatic melanoma. One serious problem for the clinical application of this approach is the intransigence of melanoma to p53-dependent apoptotic mechanisms. Since p53 mutations are relatively rare in melanomas [37], wild-type p53 operates as an important transcription factor, controlling transcription of genes whose proteins are directly involved in cell cycle regulation and apoptosis. However, its downstream proapoptotic function could be systematically suppressed due to aberrancies in melanoma cellular physiology. For example, a blockage of mitochondrial function [15] and the mitochondrial death pathway in metastatic tumors, including advanced melanomas, can silence the proapoptotic function of p53. Thus, melanoma cells, injured by γ-radiation and arrested at the G2/M phase of the cell cycle, are destined to die slowly by a necrotic mechanism. As we observed in the present study, inhibitors of COX-2 and PI3K-AKT may facilitate this process, still continuing to use a necrotic mechanism.
Furthermore, one of the critical factors that determine the efficacy of a given anticancer therapy is its ability to maximize cancer cell death, while minimizing effects on normal cells, thereby enhancing the therapeutic ratio. However, cytotoxic chemotherapy and ionizing radiation not only affect tumors, but also normal cells, especially dividing lymphocytes that are required to develop an antitumor immune response. In contrast to these conventional therapies, TRAIL, via interaction with the correspondent TRAIL-R1 and TRAIL-R2 on surface of cancer cells, may induce a fatal signaling cascade in cancer cells and have only minimal cytotoxic effects in normal cells [7, 8]. Unfortunately, most human melanomas are resistant to TRAIL [38]. During the last several years we and others used different approaches to enhance sensitivity of melanoma cells to TRAIL. Recently, we revealed that resveratrol (RV) in combination with TRAIL may efficiently increase levels of TRAIL-mediated apoptosis in melanoma cells [29], as it was previously observed to do for gliomas and prostate cancer [39, 40]. The striking feature of RV treatment for some melanoma lines, which was elucidated in the present study, was induction of apoptotic death by itself, or in combination with γ-irradiation. These effects of RV appear to be based on several critical targets of its action: (1) downregulation of NF-κB and STAT3-dependent gene expression controlling expression of numerous genes involved in cell survival [31, 40]; (2) the subsequent negative regulation of cFLIP and Bcl-xL gene expression and protein stability in melanoma cells; (3) the positive regulation of TRAIL gene expression and TRAIL protein transport to the cell surface. The melanoma lines resistant to radiation-induced cell death, such as SW1 and HHMSX, have substantial deficiency in mitochondrial function and the mitochondrial death pathway. Despite inherent mitochondrial deficiencies, we have observed, however, that the extrinsic DR5-mediated death pathway could still be upregulated in these cells, using pretreatment with γ-irradiation that was followed by RV treatment, which induced endogenous TRAIL translocation and efficient apoptosis. This finding provides a potentially important rational approach to more efficacious melanoma treatment. We suggest that elucidation of mechanisms of RV-induced TRAIL expression and translocation will yield new insight for the treatment of resistant metastatic melanomas. Taken together, results of our study demonstrated effectiveness of combined treatment of melanoma cells with: (i) γ-irradiation combined with chemical suppression of COX-2 or PI3K-AKT; (ii) γ-irradiation combined with TRAIL; (iii) γ-irradiation with the addition of RV for radiosensitization of radioresistant melanoma lines. By the use of TRAIL and RV as an accelerating agent, the preference for an apoptotic mechanism of cell death is largely achieved.
References
Debatin KM, Krammer PH (2004) Death receptors in chemotherapy and cancer. Oncogene 23:2950–2966
Okada H, Mak TW (2004) Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 4:592–603
Cory S, Adams JM (2005) Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell 8:5–6
Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441:431–436
Krammer PH, Kaminski M, Kiessling M, Gulow K (2007) No life without death. Adv Cancer Res 97C:111–138
Adams C, Totpal K, Lawrence D, Marsters S, Pitti R, Yee S, Ross S, Deforge L, Koeppen H, Sagolla M, Compaan D, Lowman H, Hymowitz S, Ashkenazi A (2008) Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell Death Differ 15:751–761
Ashkenazi A (2002) Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer 2:420–430
Schaefer U, Voloshanenko O, Willen D, Walczak H (2007) TRAIL: a multifunctional cytokine. Front Biosci 12:3813–3824
Chinnaiyan AM, Prasad U, Shankar S, Hamstra DA, Shanaiah M, Chenevert TL, Ross BD, Rehemtulla A (2000) Combined effect of tumor necrosis factor-related apoptosis-inducing ligand and ionizing radiation in breast cancer therapy. Proc Natl Acad Sci USA 97:1754–1759
Ivanov VN, Zhou H, Hei TK (2007) Sequential treatment by ionizing radiation and sodium arsenite dramatically accelerates TRAIL-mediated apoptosis of human melanoma cells. Cancer Res 67:5397–5407
Subbaramaiah K, Dannenberg AJ (2003) Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol Sci 24:96–102
Luo J, Manning BD, Cantley LC (2003) Targeting the PI3K-AKT pathway in human cancer: rationale and promise. Cancer Cell 4:257–262
Satyamoorthy K, Chehab NH, Waterman MJ, Lien MC, El-Deiry WS, Herlyn M, Halazonetis TD (2000) Aberrant regulation and function of wild-type p53 in radioresistant melanoma cells. Cell Growth Differ 11:467–474
Vousden KH, Lane DP (2007) p53 in health and disease. Nat Rev Mol Cell Biol 8:275–283
Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, Hu Y, Ramdas L, Hu L, Keating MJ, Zhang W, Plunkett W, Huang P (2006) Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol 175:913–923
Gogvadze V, Orrenius S, Zhivotovsky B (2008) Mitochondria in cancer cells: what is so special about them? Trends Cell Biol 18:165–173
Ivanov VN, Partridge MA, Johnson GE, Huang SX, Zhou H, Hei TK (2008) Resveratrol sensitizes melanomas to TRAIL through modulation of antiapoptotic gene expression. Exp Cell Res 314:1163–1176
Satyamoorthy K, DeJesus E, Linnenbach AJ, Kraj B, Kornreich DL, Rendle S, Elder DE, Herlyn M (1997) Melanoma cell lines from different stages of progression and their biological and molecular analyses. Melanoma Res 7(Suppl 2):S35–S42
van Dam H, Huguier S, Kooistra K, Baguet J, Vial E, van der Eb AJ, Herrlich P, Angel P, Castellazzi M (1998) Autocrine growth and anchorage independence: two complementing Jun-controlled genetic programs of cellular transformation. Genes Dev 12:1227–1239
Baetu TM, Kwon H, Sharma S, Grandvaux N, Hiscott J (2001) Disruption of NF-kappaB signaling reveals a novel role for NF-kappaB in the regulation of TNF-related apoptosis-inducing ligand expression. J Immunol 167:3164–3173
Bartke T, Siegmund D, Peters N, Reichwein M, Henkler F, Scheurich P, Wajant H (2001) p53 upregulates cFLIP, inhibits transcription of NF-kappaB-regulated genes and induces caspase-8-independent cell death in DLD-1 cells. Oncogene 20:571–580
Ricci MS, Jin Z, Dews M, Yu D, Thomas-Tikhonenko A, Dicker DT, El-Deiry WS (2004) Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol Cell Biol 24:8541–8555
Resnick-Silverman L, St Clair S, Maurer M, Zhao K, Manfredi JJ (1998) Identification of a novel class of genomic DNA-binding sites suggests a mechanism for selectivity in target gene activation by the tumor suppressor protein p53. Genes Dev 12:2102–2107
Bakhtiarova A, Taslimi P, Elliman SJ, Kosinski PA, Hubbard B, Kavana M, Kemp DM (2006) Resveratrol inhibits firefly luciferase. Biochem Biophys Res Commun 351:481–484
Ivanov VN, Hei TK (2004) Arsenite sensitizes human melanomas to apoptosis via tumor necrosis factor alpha-mediated pathway. J Biol Chem 279:22747–22758
Shiloh Y (2006) The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci 31:402–410
Nagata S (1999) Fas ligand-induced apoptosis. Annu Rev Genet 33:29–55
Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM (1997) Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275:218–220
Ivanov VN, Partridge MA, Johnson GE, Huang SXL, Zhou H, Hei TK (2008) Resveratrol sensitizes melanomas to TRAIL through modulation of antiapoptotic gene expression. Exp Cell Res 314:1163–1176
Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y (2004) Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res 24:2783–2840
Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, Carter B, Turkson J, Jove R (2006) Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther 5:621–629
Rigel DS, Friedman RJ, Kopf AW (1996) The incidence of malignant melanoma in the United States: issues as we approach the 21st century. J Am Acad Derm 34:839–847
Rigel DS (2002) The effect of sunscreen on melanoma risk. Derm Clin 20:601–606
Perlis C, Herlyn M (2004) Recent advances in melanoma biology. Oncologist 9:182–187
Atkins MB (2006) Cytokine-based therapy and biochemotherapy for advanced melanoma. Clin Cancer Res 12:2353s–2358s
Oehler C, Dickinson DJ, Broggini-Tenzer A, Hofstetter B, Hollenstein A, Riesterer O, Vuong V, Pruschy M (2007) Current concepts for the combined treatment modality of ionizing radiation with anticancer agents. Curr Pharm Des 13:519–535
Gwosdz C, Scheckenbach K, Lieven O, Reifenberger J, Knopf A, Bier H, Balz V (2006) Comprehensive analysis of the p53 status in mucosal and cutaneous melanomas. Int J Cancer 118:577–582
Hersey P, Zhang XD (2001) How melanoma cells evade trail-induced apoptosis. Nat Rev Cancer 1:142–150
Fulda S, Debatin KM (2004) Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res 64:337–346
Shankar S, Singh G, Srivastava RK (2007) Chemoprevention by resveratrol: molecular mechanisms and therapeutic potential. Front Biosci 12:4839–4854
Acknowledgments
We would like to thanks Drs. Z. Ronai, M. Herlyn, S. Y. Fuchs, H. B. Liberman and Y. Yin for discussion of this manuscript. This research was supported by funding from the National Institutes of Health Grants CA 49062 and ES 12888.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Johnson, G.E., Ivanov, V.N. & Hei, T.K. Radiosensitization of melanoma cells through combined inhibition of protein regulators of cell survival. Apoptosis 13, 790–802 (2008). https://doi.org/10.1007/s10495-008-0212-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0212-y