
Abstract
The Eriophyoidea, notable for specific morphological characters (four-legged mites) and gall-formation in host plants (gall mites), is one of the most species-rich superfamilies of Acari. Monophyly of the superfamily Eriophyoidea is accepted by all acarologists; however, monophyly of most genera has not been evaluated in a molecular phylogenetic network. Furthermore, most eriophyoid mites, especially species in the genus Diptilomiopus, are morphologically similar, challenging their identification. Here we test the phylogeny and cryptic diversity of Diptilomiopus species using fragments of two mitochondrial (COI and 12S) and two nuclear (18S and 28S) genes. Our results revealed the monophyly of Diptilomiopus. Sequence distance, barcode gap, and species delimitation analyses of the COI gene allowed us to resolve cryptic diversity of Diptilomiopus species. Additionally, we supposed that characteristics of genu fused with femur on both legs and seta ft′ absent on leg II evolved only once within Diptilomiopus, which are potential morphological synapomorphies. In contrast, characteristics of both setae ft′ and ft″ divided into a short branch and a long branch were supposed evolving multiple times independently. Our findings contribute to the understanding of phylogeny and morphological evolution of Diptilomiopus species and provide a DNA-based approach for species delimitation of Diptilomiopus mites.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Eriophyoidea is one of the most species-rich superfamilies of Acari, consisting of more than 4400 described species (Zhang 2011). Eriophyoid mites are notable for their specific and distinct morphological characters, e.g., two pairs of legs (four-legged mites), very small body size (200 µm long on average), fusiform or vermiform body shape, and ringed opisthosoma (Amrine et al. 2003). They are totally phytophagous, having high host-plant specialization and specificity (Skoracka et al. 2010). Some are of economic importance, e.g., Aceria tosichella Keifer (wheat curl mite), a major pest of the world's grain industry (Navia et al. 2013). Most eriophyoid mites are vagrant on their host plant, whereas some induce galls (gall mites), erinea, large buds, curved leaves, or dried branches (Petanovic and Kielkiewicz 2010).
The Eriophyoidea comprises three families: Phytoptidae, Eriophyidae and Diptilomiopidae. The genus Diptilomiopus, established by Nalepa (1916), belongs to the Diptilomiopidae (Amrine et al. 2003). Based on morphological characters, 116 Diptilomiopus species have been described worldwide (Table S1). The majority of these species have been recorded in the Oriental realm (Capinera 2008), whereas only a few have been reported from the remaining realms, i.e., three in the Palearctic realm, two in the Nearctic realm, three in the Afrotropical realm, and two in the Australasian realm (Fig. 1).

Global records/localities map of Diptilomiopus species. The red, blue, pink, black, and green dots represent the records/localities where Diptilomiopus species have been found in the Palearctic, Nearctic, Afrotropical, Oriental, and Australasian realms, respectively. (Color figure online)
Current eriophyoid mite taxonomy relies mostly on a few external morphological characters (Amrine et al. 2003), of which the pattern of prodorsal shield is widely used for species delimitation. The prodorsal shield of eriophyoid mites may have one median line, two admedian lines and several submedian lines (Fig. 2); however, Diptilomiopus species typically have a network-like prodorsal shield pattern (median, admedian and submedian lines are connected by short transverse lines; Fig. 2), which provides limited information to differentiate one species from another, challenging the species delimitation in Diptilomiopus.

Patterns of prodorsal shield in Diptilomiopus species. aD. milletus, bD. rotundus, cD. octandrus, dD. nobilus, eD. broussonetus, fD. fortunus,gD. sabahus, hD. callicarpus, iD. melastomae, jD. keningaus, kD. ligustri, lD. bischofiae; m, median line; am, admedian line; sm, submedian line
Previous molecular studies of eriophyoid mites were performed on the mitochondrial genomes (Xue et al. 2016), high-level (superfamily) phylogenetic positions of Eriophyoidea (Xue et al. 2017; Klimov et al. 2018), low-level (subfamily, tribe or genus) phylogeny of Eriophyoidea (Li et al. 2014a; Chetverikov et al. 2015), and genetic diversity of Ac. tosichella (Eriophyidae) (Skoracka et al. 2018) and Tetra pinnatifidae Xue et al. (Eriophyidae) (Li et al. 2014b). By using the mitochondrial cytochrome oxidase subunit I (COI) gene and nuclear D2 region of 28S (28S) rDNA, Lewandowski et al. (2014) revealed the genetic and morphological diversity of Trisetacus species (Phytoptidae), Cvrković et al. (2016) revealed the cryptic speciation of Phytoptus avellanae s.l. Nalepa (Phytoptidae), Duarte et al. (2019) revealed Abacarus species on sugarcane plants, Skoracka and Dabert (2010) revealed the Abacarus hystrix (Nalepa) complex (Eriophyidae), and Skoracka et al. (2013) revealed the Ac. tosichella complex (Eriophyidae). By using the COI, 18S and 28S genes, Guo et al. (2015) revealed the protogyne and deutogyne of Tegolophus celtis Guo et al. (Eriophyidae). These studies show that the COI gene can well resolve the identification and classification of species within Phytoptidae and Eriophyidae. However, no molecular studies have been performed on the genera in Diptilomiopidae, and the phylogeny and genetic diversity of Diptilomiopus species are largely unknown.
In this study, we sequenced two mitochondrial (COI and 12S) and two nuclear (18S and 28S) gene fragments of representative species (25 terminals). By constructing phylogenetic trees and using integrative taxonomy approaches, we attempted to (1) test the monophyly of Diptilomiopus, (2) resolve the delimitation of Diptilomiopus species and cryptic diversity, and (3) reveal the morphological evolution of legs and demonstrate synapomorphies of Diptilomiopus.
Materials and methods
Specimen collection and morphological identification
Twenty-five Diptilomiopus samples were collected randomly on host plants with the help of a hand-lens (30 ×) in China and Malaysia in 2017 and 2018 (Table 1). Some mite specimens were used immediately for DNA extraction, whereas the remaining were preserved in 100% ethanol at − 20 °C prior to DNA extraction. Specimens of each species were also slide mounted as vouchers, using modified Berlese medium (Amrine and Manson 1996) for morphological checking with a Zeiss A2 microscope equipped with the AxioCam MRc camera. Microphotographs were taken with a Zeiss A2 research microscope with phase contrast or differential interference, using × 100 oil magnification; the microscope was connected to a computer using Axiovision image analysis software. The morphological terminology used herein follows that of Lindquist (1996); the generic classification is made according to Amrine et al. (2003). All of the specimens and vouchers were deposited in the Arthropod/Mite Collection of the Department of Entomology, Nanjing Agricultural University (NJAU), Jiangsu Province, China (Zhang 2018).
DNA extraction and PCR amplification
Genomic DNA was extracted from one specimen for each sample of eriophyoid mites, using a DNeasy Blood and Tissue Kit (Qiagen), following a previously reported modified protocol (Dabert et al. 2008). We amplified the fragments of two mitochondrial genes (COI and 12S) and two nuclear genes (18S and 28S) using published or modified PCR primer pairs for each fragment (Table 2). The PCR cycling conditions were as follows: 3 min of denaturation at 94 °C; 35 cycles of 30 s of denaturation at 94 °C, 30 s of annealing at 42–55 °C (depending on the primers) and 1 min of extension at 72 °C; 5 min final extension at 72 °C; holding at 4 °C. Each PCR contained 12.5 µl of PCR SuperMix (Transgen Biotech, Beijing, China), 2 µl of template DNA, and 0.4 µM of each primer, in a total volume of 25 µl. PCR products were visualized on a 1% agarose gel. Products were purified and sequenced in both directions at General Biosystems (Anhui, China) on an ABI 3730XL DNA Analyzer (Applied Biosystems).
Data matrices and sequence alignments
Sequences of four gene (COI, 12S, 18S and 28S) fragments of 25 Diptilomiopus samples representing 17 morphospecies were blasted in GenBank and checked for possible contaminants. All the sequences were deposited in GenBank under accession numbers: MK440001–MK440064, and MK516816–MK516842 (Table 1). The sequences of four outgroups (Nematalycidae), five phytoptid mites, four eriophyid mites and one diptilomiopid species were retrieved from GenBank (Table 1). Three rRNA genes were aligned individually using MAFFT v.7.423 web server (Katoh and Standley 2013) (http://mafft.cbrc.jp/alignment/server/) with G-INS-i strategy for global homology and manually inspected before concatenation. For COI, a preliminary alignment was generated using ClustalW in MEGA 6.0 (Tamura et al. 2013). Large gaps and ambiguous sites were deleted manually. Alignments of individual genes were concatenated in Geneious v.8.1.9 (Kearse et al. 2012). The final concatenated DNA dataset consisted of 4989 bp: 18S rRNA = 2297 bp, 28S rRNA D2–D5 = 916 bp, 28S rRNA D9–D10 = 752 bp, COI = 658 bp, and 12S rRNA = 366 bp. We analyzed the dataset as nucleotide sequences. Dataset partitioning was performed by PartitionFinder 2 (Lanfear et al. 2017), based on an initial total of five data blocks (18S, 28S D2–D5, 28S D9–D10, COI, and 12S). Models were predicted by PartitionFinder 2 using the Bayesian information criterion (BIC). PartitionFinder used unlinked branch lengths, the greedy search algorithm for nucleotide sequences and the MrBayes model. The GTR + G substitution model was chosen by PartitionFinder as the best for two partitions (18S + 28S D2–D5 + 28S D9–D10 and COI + 12S).
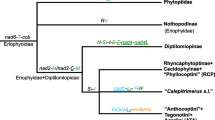
To test the monophyly and its phylogenetic position of Diptilomiopus within Eriophyoidea, we constructed an additional data matrix including four species of Nematalycidae, eight species of Phytoptidae, 21 species of Eriophyidae, and 30 species of Diptilomiopidae (Table S2). The nucleotide sequences of 18S rRNA of these species were retrieved from GenBank. Alignments were performed by MAFFT, and the substitution model (GTR + G) was predicted by PartitionFinder.
Phylogenetic analyses
Phylogenetic analyses were conducted using maximum likelihood (ML) and Bayesian inference (BI) methods. ML analyses were performed using the GTRGAMMAI model in RAxML-HPC-PTHREADS (Stamatakis 2006) implemented in raxmlGUI 1.3 (Silvestro and Michalak 2011). Clade support was assessed using a nonparametric bootstrapping with 1000 replicates. Nodes supported by bootstrap values (BSP) ≥ 70% were considered strongly supported (Hillis and Bull 1993). BI analyses were performed with MrBayes v.3.2.2 (Ronquist et al. 2012). For MrBayes v.3.2.2, we used separate data partitions plus mixed models and conducted two independent runs each with four Markov Chains Monte Carlo (one cold chain and three heated chains). The combined dataset was run for 20 million generations, with trees sampled every 1000 generations. The convergence of the parameter estimates was performed with Tracer v.1.6. A conservative burn-in of 25% was then applied. The consensus tree was edited with FigTree1.4.0. Nodes supported by posterior probabilities (BPP) ≥ 95% were considered strongly supported (Alfaro 2003).
Genetic distance, barcode gap discovery, and species delimitation
Sequence genetic distances were calculated for COI, 18S and 28S (Table 1) using MEGA 6.0 (Tamura et al. 2013) under the Kimura two-parameter (K2P) model (Kimura 1980). The substitution model was chosen by d: transitions + transversions. Heatmaps were drawn by R v.3.5.2 (R Core Team 2018). Pairwise distances of 12S gene were not measured because only a few Diptilomiopus species were successfully sequenced.
Barcode gap was analyzed by Automatic Barcode Gap Discovery (ABGD) (Puillandre et al. 2012) web server https://bioinfo.mnhn.fr/abi/public/abgd/ using X value 0.9 and K2P distance. We applied ABGD to each of the three genes (COI, 18S, 28S) with the following Pmax settings: 0.002–0.130 in COI, 0.001–0.033 in 18S, 0.001–0.023 in 28S, which were consistent with the range of intraspecific distances of each gene dataset (Table S3).
All phylogenetic trees constructed from the concatenated dataset showed that the Diptilomiopus was a monophyletic clade. We therefore used Diptilomiopus species as a reduced dataset (excluding outgroups and other eriophyoid mite species) for species delimitation analysis. The General Mixed Yule Coalescent (GMYC) model identifies the transition points between inter-and intraspecific processes on an ultrametric tree (Pons et al. 2006). Ultrametric trees were constructed in BEAST v.1.8.0 (Drummond and Rambaut 2007) through the CIPRES Science Gateway (Miller et al. 2010), using GTR + G model, a Yule speciation prior and a lognormal uncorrelated relaxed clock. As COI is one of protein coding genes in the mitochondrial genome, the 1st, 2nd and 3rd codon positions have different evolutionary rates. The COI data were either not partitioned or partitioned into 1st, 2nd and 3rd codon positions. Two independent runs of 100 million generations were executed with sampling every 1000 generations. Post burn-in trees were merged and re-sampled at a lower frequency (every 10,000 generations) using the LogCombiner of BEAST. The final ultrametric trees were entered into R (R Core Team 2018) package splits v.1.0–19 (Ezard et al. 2014) with the single threshold option as recommended by Fujisawa and Barraclough (2013). bPTP analyses were performed in the bPTP server (http://species.h-its.org/) (Zhang et al. 2013) with default values. We used MrBayes v.3.2.2 to reconstruct input trees of COI, 18S and 28S to bPTP. The reduced datasets were run for 5 million generations, with trees sampled every 5000 generations.
Results
Molecular phylogeny of Diptilomiopus species
The ML and BI analyses showed very similar topologies (Figs. 3, 4, S1–S4). Our results demonstrate that the Diptilomiopus is monophyletic with strong support (BSP > 95, BPP = 1) based on the dataset of nucleotide sequences of a single gene (18S) or multiple genes (18S, 28S, COI, and 12S) (Figs. 3, 4, S1–S4). Monophyly of the Diptilomiopus ligustri Wang et al. group was recovered with strong support (BSP > 87, BPP = 1) based on the dataset of a single gene (18S) (Figs. 3, S2) and multiple genes (Figs. 4, S4). The Diptilomiopus bischofiae Li et al. group is monophyletic with strong support (BSP = 100, BPP = 1) based on the dataset of multiple genes (Figs. 4, S4) or a single gene (Figs. 3, S2). Two representatives of Diptilomiopus fortunus Liu et al. were grouped with strong support (BSP = 100, BPP = 1) (Figs. S1–S4). Except for these species groups, some clades within Diptilomiopus was observed with low or very low support; species from the same regions (China or Malaysia) or having characteristics of divided setae ft′ and ft″ were not grouped (Figs. 3, 4).

Phylogenetic trees inferred from nucleotide sequences of 18S gene using maximum likelihood method. Branch lengths presented here follow the maximum likelihood analysis using the best partition found by PartitionFinder. Nodes, marked with a blue dot, indicate bootstrap values (BSP) ≥ 70%. Red stars indicate the species having divided seta ft′ and ft″. (Color figure online)

Phylogenetic trees inferred from nucleotide sequences of two mitochondrial (COI and 12S) and two nuclear (18S and 28S) gene fragments using maximum likelihood method. Nodes, marked with a blue disc, indicate bootstrap values (BSP) ≥ 70%. Characters of potential synapomorphies were traced: genu on both legs, scapular tubercles, scapular setae (sc), network prodorsal shield design, shape of the empodium (em), tarsal setae ft′ and ft″, setae 1b and setae c2. (Color figure online)
Genetic distance and molecular delimitation of Diptilomiopus species
The pairwise K2P interspecies distances of COI gene fragment ranged from 25.3 to 41.3%, and the intraspecies variation ranged from 0.2 to 13% (Fig. 5a, Table S3). The greatest genetic intraspecies distances occurred in D. fortunus, possibly indicating cryptic species. For 18S, the interspecies distances ranged from 1.6 to 14%, and the intraspecies variation ranged from 0.0 to 3.3% (Fig. 5b, Table S3). The greatest genetic intraspecies and interspecies distances occurred in D. ligustri (YN253). For 28S, the interspecies distances ranged from 2.4 to 30.4%, and the intraspecies variation ranged from 0.0 to 2.3% (Fig. 5c, Table S3). The greatest genetic intraspecies variation occurred in D. ligustri (YN253).


Heatmap of pairwise genetic distances, DNA barcode and ABDG delimitation of Diptilomiopus species. a COI gene, b 18S gene, c 28S gene
ABGD of COI delimited 12 initial partitions with prior intraspecific divergences (P) varying from 0.2 to 13% (Fig. 5a). Barcode gaps were observed at K2P distances of 4–11% and 13–26% (Fig. 5a). Initial partitions were identical at 12 molecular operational taxonomic units (MOTUs), corresponding to our morphologically identified 12 species. For 18S, ABGD delimited initial partitions of 19 MOTUs with P varying from 0.1 to 1.03% (Fig. 5b), which was consistent with our 18 a prior morphospecies, except populations of D. ligustri were split into two groups, ML + YN, GX + S6 + YN253. No barcode gap was observed. For 28S, ABGD delimited 17 MOTUs with P varying from 0.1 to 1.15% (Fig. 5c), corresponding to our morphologically identified 17 species. No barcode gap was observed.
The single-threshold GMYC analyses of COI gene identified 13 entities with significant support (not partitioned, p = 0.02; partitioned by codon, p = 0.004), which were consistent with our morphologically identified 12 species, except D. fortunus KK19 and D. fortunus KK21a which were inferred as distinct species (Figs. S5A, S5B). For 18S, the single-threshold GMYC analyses gave the same results as the ABGD analysis (see above) (Fig. S5C). For 28S, the single-threshold GMYC analyses identified 19 entities with significant support (p = 0.002), which were inconsistent with our morphologically identified 17 species, as populations of D. ligustri were split into two entities (ML + YN, GX + S6 + YN253) and also populations of D. bischofiae were split into two entities (S11, BY + WM) (Fig. S5D). bPTP analyses of COI, 18S and 28S genes resulted in similar species delimitation as the GMYC analyses (Fig. S5).
Morphological evolution of legs in Diptilomiopus species
We mapped some morphological characters (at the generic or species level) on the phylogenetic tree (Fig. 4), i.e., genu on both legs, scapular tubercles, scapular setae (sc), network prodorsal shield design, shape of the empodium (em), tarsal setae ft′ and ft″, setae 1b and setae c2, and found that Diptilomiopus species were united by some genetic morphological characters (genu fused with femur on both legs, scapular tubercles and setae absent, empodium divided, setae 1b and setae c2 absent). Intriguingly, the species sharing divided tarsal setae ft′ and ft″ (D. octandrus, D. milletus, D. fortunus, D. callicarpus, D. keningaus, D. retusus, and D. sabahus) were not grouped (Fig. 4), indicating these characteristics may have evolved multiple times independently. However, characteristics of genu fused with femur on both legs and tarsal setae ft″ absent from leg II were suggested evolving only once, indicating they are morphological synapomorphies of Diptilomiopus (Fig. 4).
Discussion
The Eriophyoidea comprises more than 357 genera (Zhang 2011); however, the monophyly of these genera has seldom been tested by morphological characters or molecular approaches (Lewandowski et al. 2014). Herein, we inferred the phylogeny of Diptilomiopus by nucleotide sequences of multiple genes for the first time. Our phylogenetic results demonstrated the monophyly of Diptilomiopus with strong support (BSP > 95, BPP = 1) (Figs. 3, 4, S2, S4). All Diptilomiopus species in our molecular analyses were collected from the Oriental realm. Therefore, more Diptilomiopus species, especially from the remaining realms, should be explored and included in future analyses.
In addition to molecular evidence, the monophyly of Diptilomiopus might be supported by some morphological synapomorphies: genu fused with femur on leg I and leg II, seta ft′ absent on leg II, scapular setae sc absent, empodium divided, coxal setae 1b absent, and setae c2 absent from the opisthosoma (Fig. 4). The combination of character states – genu fused with femur on both legs and seta ft′ absent on legs II – are specific to Diptilomiopus species and were not found in other extant species of Eriophyoidea (Amrine et al. 2003) or fossil species of Triasacaroidea (Sidorchuk et al. 2015). We propose that these characters have evolved and occurred only once within Diptilomiopus, indicating they are synapomorphies (Fig. 4). However, new genera of eriophyoid mites were consistently erected in recent years, and more new genera, having those characters, cannot be ruled out in future studies. The remaining potential synapomorphies were found in the species of more than one genus of Eriophyoidea or Triasacaroidea. For instance, scapular setae sc absent is characteristic for Cecidophyini tribe (Eriophyidae), which includes 11 genera (Amrine et al. 2003), and for Calacarini tribe (Eriophyidae), which includes 15 genera (Amrine et al. 2003; Huang and Wang 2004; Xue et al. 2007; Chandrapatya et al. 2016); and it is characteristic for four genera (Pseudocalepitrimerus, Knorella, Schizacea, and Namengia) of Acaricalini (Eriophyidae) (Amrine et al. 2003; Zhao et al. 2018), 14 genera of Diptilomiopinae (Diptilomiopidae) (Amrine et al. 2003), and two genera (Asetacus and Sakthirhynchus) of Rhyncaphytoptinae (Diptilomiopidae) (Amrine et al. 2003). A deeply divided empodium is the main character of the subfamily Diptilomiopinae that differentiates it from the other subfamily Rhyncaphytoptinae (Amrine et al. 2003). However, this is also characteristic of Acaricalini tribe (Eriophyidae), which comprises 23 extant genera (Amrine et al. 2003; Flechtmann 2004; Chandrapatya et al. 2016; Chetverikov et al. 2018; Zhao et al. 2018), and of two fossil genera (Triasacarus and Minyacarus) (Sidorchuk et al. 2015). The character of coxal setae 1b absent or setae c2 absent was consistently found in many genera of Eriophyidae and Diptilomiopidae.
Most eriophyoid mites have normal tarsal seta ft′ and seta ft″ on legs I and II (Fig. 6). Intriguingly, seta ft′ and seta ft″, divided into a short branch and a long branch, respectively (Fig. 6), has been found in a few Diptilomiopus species (Craemer et al. 2017). These divided setae were not observed in four fossil species (Schmidt et al. 2012; Sidorchuk et al. 2015). Moreover, the Diptilomiopus species, sharing these characters, were not grouped in our phylogenetic topologies (Figs. 3, 4), indicating that divided seta ft′ and seta ft″ were evolved at the species level.

Hypothesized schematic evolutionary route of legs. a Legs of Epitrimerus gaotainensus,b legs of D. fortunus, c legs of D. rotundus; L1, leg I; L2, leg II; fm, femur; ge, genu; tb, tibia; t, tarsus; bv, femural seta; l″, genual seta; l', tibial seta; ft′, tarsal seta ft′; ft″, tarsal seta ft″; u', seta u'; em, empodium; ω, solenidion.
The genus Diptilomiopus comprises 116 currently described species (Table S1). Morphological similarity challenges the delimitation of Diptilomiopus species. Our ABGD, GMYC and bPTP results showed that most of our tested morphospecies of Diptilomiopus, except three (D. fortunus, D. ligustri and D. bischofiae), were resolved by fragments of COI, 18S or 28S genes. However, those resolved morphospecies were based on single sequences from one population. More Diptilomiopus species, especially species from different populations, are needed in future collections. Inferred by different methods (ABGD, GMYC or bPTP) or genes (COI, 18S or 28S), three species (D. fortunus, D. ligustri and D. bischofiae) showed inconsistency between morphospecies and MOTUs, indicating the presence of cryptic species. However, monophyly of each of these species was always recovered in our ML and BI trees. Further, most eriophyoid mites were reported having high host-plant specificity (Skoracka et al. 2010). The various population of each of our tested species of D. fortunus, D. ligustri or D. bischofiae were all collected from the same corresponding host plant (i.e., one host plant species per mite species; Table 1). We therefore suggest that the high sequence genetic diversity (distance) within populations of those three mite species may be an effect of host-constrained isolation, which leads to incomplete lineage sorting (Toews and Brelsford 2012). It is widely accepted that simply relying on one approach to delimitate species, especially when they are highly morphologically similar or have two forms (protogyne an deutogyne in eriophyoid mites), is problematic (Cvrković et al. 2016; Guo et al. 2015; Skoracka et al. 2013). Herein, we underline the integrative taxonomy approach, combining morphological characters and molecular approaches, in resolving species delimitation of eriophyoid mites.
Eriophyoid mites are distributed worldwide; however, most genera are distributed regionally (Amrine and Stasny 1994) due to low dispersal ability (Michalska et al. 2010), high host-plant specificity (Skoracka et al. 2010), or possibly uneven regional field surveys. Similarly, most Diptilomiopus species have been reported from the Oriental realm, only a few have been recorded in the remaining realms (Fig. 1). Consistent with a previous hypothesis of Craemer et al. (2017), the most parsimonious explanation for this uneven distribution could be that Diptilomiopus species originated in the Oriental realm, and some dispersed to the remaining realms. Eriophyoid mites have a low positive dispersal ability (Sabelis and Bruin 1996). Long-distance dispersal is achieved by aerial dispersal (Zhao and Amrine 1997), phoresy on host-specific insects (Sabelis and Bruin 1996; Liu et al. 2016), or probable transportation of host plants (Craemer et al. 2017). If the Oriental origin of Diptilomiopus is true, then Diptilomiopus species should occasionally occur in the Palearctic realm, Nearctic realm, Ethiopian realm, and Australasian realm due to such dispersal modes. Additional field surveys of Diptilomiopus spp. worldwide are obviously necessary to decipher the biogeographical distribution and dispersal routes of Diptilomiopus species.
Conclusions
In this study, we demonstrated that the genus Diptilomiopus is monophyletic by multiple lines of evidence from molecular approaches and morphological synapomorphies. Most Diptilomiopus species are highly similar in morphology, what may hinder their correct indentification and induce species complexes. We provide an integrative taxonomic approach to the resolution of cryptic Diptilomiopus species or other eriophyoid mite complexes. These findings highlight the cryptic species diversity within Diptilomiopus; more descriptions of new Diptilomiopus species and new findings related to their biogeographical distributions are expected.
References
Alfaro ME (2003) Bayes or bootstrap? A simulation study comparing the performance of Bayesian Markov Chain Monte Carlo sampling and bootstrapping in assessing phylogenetic confidence. Mol Biol Evol 20:255–266. https://doi.org/10.1093/molbev/msg028
Amrine JW Jr, Manson DCM (1996) Preparation, mounting and descriptive study of eriophyoid mites. In: Lindquist EE, Sabelis MW, Bruin J (eds) Eriophyoid mites: their biology, natural enemies and control. World Crop Pests, vol 6. Elsevier, Amsterdam, pp 383–396
Amrine JW Jr, Stasny TA, Flechtmann CHW (2003) Revised keys to world genera of Eriophyoidea (Acari: Prostigmata). Indira Publishing House, Michigan
Amrine JW Jr, Stasny TA (1994) Catalog of the Eriophyoidea (Acarina: Prostigmata) of the world. Indira Publishing House, Michigan
Campbell CL, Tanaka N, White KH, Thorsness PE (1994) Mitochondrial morphological and functional defects in yeast caused by yme1 are suppressed by mutation of a 26S protease subunit homologue. Mol Biol Cell 5:899–905. https://doi.org/10.1091/mbc.5.8.899
Capinera JL (2008) Encyclopedia of entomology. Springer, Dordrecht
Chandrapatya A, Konvipasruang P, Amrine JW Jr (2016) New eriophyoid mites from Thailand. Syst Appl Acarol 21:55–78. https://doi.org/10.11158/saa.21.1.5
Chetverikov PE, Craemer C, Neser S (2018) New pseudotagmic genus of acaricaline mites (Eriophyidae, Acaricalini) from a South African palm Hyphaene coriacea and remarks on lateral opisthosomal spines and morphology of deutogynes in Eriophyoidea. Syst Appl Acarol 23:1073–1101. https://doi.org/10.11158/saa.23.6.6
Chetverikov PE, Cvrkovic T, Makunin A, Sukhareva S, Vidovic B, Petanovic R (2015) Basal divergence of Eriophyoidea (Acariformes, Eupodina) inferred from combined partial COI and 28S gene sequences and CLSM genital anatomy. Exp Appl Acarol 67:219–245. https://doi.org/10.1007/s10493-015-9945-9
Craemer C, Amrine JW Jr, Childers CC, Rogers ME, Achor DS (2017) A new eriophyoid mite species, Diptilomiopus floridanus (Acari: Eriophyoidea: Diptilomiopidae), from citrus in Florida, USA. Syst Appl Acarol 22:386–402. https://doi.org/10.11158/saa.22.3.5
Cvrković T, Chetverikov P, Vidović B, Petanović R (2016) Cryptic speciation within Phytoptus avellanae s.l. (Eriophyoidea: Phytoptidae) revealed by molecular data and observations on molting Tegonotus-like nymphs. Exp Appl Acarol 68:83–96. https://doi.org/10.1007/s10493-015-9981-5
Dabert J, Ehrnsberger R, Dabert M (2008) Glaucalges tytonis sp. n. (Analgoidea, Xolalgidae) from the barn owl Tyto alba (Strigiformes, Tytonidae): compiling morphology with DNA barcode data for taxon descriptions in mites (Acari). Zootaxa 1719:41–52
Dabert M, Witalinski W, Kazmierski A, Olszanowski Z, Dabert J (2010) Molecular phylogeny of acariform mites (Acari, Arachnida): strong conflict between phylogenetic signal and long-branch attraction artifacts. Mol Phylogenet Evol 56:222–241. https://doi.org/10.1016/j.ympev.2009.12.020
Duarte ME, de Mendonca RS, Skoracka A, Silva ES, Navia D (2019) Integrative taxonomy of Abacarus mites (Eriophyidae) associated with hybrid sugarcane plants, including description of a new species. Exp Appl Acarol 78: 373–401. https://doi.org/10.1007/s10493-019-00388-y
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. https://doi.org/10.1186/1471-2148-7-214
Ezard THG, Fujisawa TE, Barraclough TG (2014) Splits: species' limits by threshold statistics. R package version 1.0–19/r51. http://RForge.Rproject.org/projects/splits/
Flechtmann CHW (2004) Eriophyid mites (AcariL Eriophyidae) from brazilian sedges (Cyperaceae). Int J Acarol 30:157–164. https://doi.org/10.1080/01647950408684385
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Fujisawa T, Barraclough TG (2013) Delimiting species using single-locus data and the generalized mixed Yule coalescent approach: a revised method and evaluation on simulated data sets. Syst Biol 62:707–724. https://doi.org/10.1093/sysbio/syt033
Guo J-F, Li H-S, Wang B, Xue X-F, Hong X-Y (2015) DNA barcoding reveals the protogyne and deutogyne of Tegolophus celtis sp. nov. (Acari: Eriophyidae). Exp Appl Acarol 67:393–410. https://doi.org/10.1007/s10493-015-9953-9
Hillis DM, Dixon MT (1991) Ribosomal DNA: molecular evolution and phylogenetic inference. Q Rev Biol 66:411–453. https://doi.org/10.1086/417338
Hillis DM, Bull JJ (1993) An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol 42:182–192. https://doi.org/10.1093/sysbio/42.2.182
Huang K-W, Wang C-F (2004) Eriophyoid mites of Taiwan: descriptin of three species of Calacarini from Hueysuen (Acari: Eriophyoidea: Phyllocoptinae). Zootaxa 527:1–8
Kambhampati S, Smith PT (1995) PCR primers for the amplification of four insect mitochondrial gene fragments. Insect Mol Biol 4:233–236. https://doi.org/10.1111/j.1365-2583.1995.tb00028.x
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. https://doi.org/10.1007/BF01731581
Klimov PB, OConnor BM, Chetverikov PE, Bolton SJ, Pepato AR, Mortazavi AL, Tolstikov AV, Bauchan GR, Ochoa R (2018) Comprehensive phylogeny of acariform mites (Acariformes) provides insights on the origin of the four-legged mites (Eriophyoidea), a long branch. Mol Phylogenet Evol 119:105–117. https://doi.org/10.1016/j.ympev.2017.10.017
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34:772–773. https://doi.org/10.1093/molbev/msw260
Lewandowski M, Skoracka A, Szydlo W, Kozak M, Druciarek T, Griffiths DA (2014) Genetic and morphological diversity of Trisetacus species (Eriophyoidea: Phytoptidae) associated with coniferous trees in Poland: phylogeny, barcoding, host and habitat specialization. Exp Appl Acarol 63:497–520. https://doi.org/10.1007/s10493-014-9805-z
Li H-S, Xue X-F, Hong X-Y (2014a) Homoplastic evolution and host association of Eriophyoidea (Acari, Prostigmata) conflict with the morphological-based taxonomic system. Mol Phylogenet Evol 78:185–198. https://doi.org/10.1016/j.ympev.2014.05.014
Li H-S, Xue X-F, Hong X-Y (2014b) Cryptic diversity in host-associated populations of Tetra pinnatifidae (Acari: Eriophyoidea): What do morphometric, mitochondrial and nuclear data reveal and conceal? Bull Entomol Res 104:221–232. https://doi.org/10.1017/S0007485313000746
Lindquist EE (1996) External anatomy and notation of structures. In: Lindquist EE, Sabelis MW, Bruin J (eds) Eriophyoid mites: their biology, natural enemies and control. World Crop Pests, vol 6. Elsevier, Amsterdam, pp 3–31
Liu S, Li J, Guo K, Qiao H, Xu R, Chen J, Xu C, Chen J (2016) Seasonal phoresy as an overwintering strategy of a phytophagous mite. Sci Rep 6:25483. https://doi.org/10.1038/srep25483
Michalska K, Skoracka A, Navia D, Amrine JW Jr (2010) Behavioural studies on eriophyoid mites: an overview. Exp Appl Acarol 51:31–59. https://doi.org/10.1007/s10493-009-9319-2
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), 14 Nov 2010, New Orleans, pp 1–8
Nalepa A (1916) Neue Gallmilben (32. Fortsetzung). Anzeiger der kaiserlichen Akademie Wissenschaften. Mathematische-naturwissenschaftliche Klasse Wien 53:283–284
Navia D, de Mendonca RS, Skoracka A, Szydlo W, Knihinicki D, Hein GL, da Silva Pereira PR, Truol G, Lau D (2013) Wheat curl mite, Aceria tosichella, and transmitted viruses: an expanding pest complex affecting cereal crops. Exp Appl Acarol 59:95–143. https://doi.org/10.1007/s10493-012-9633-y
Petanovic R, Kielkiewicz M (2010) Plant-eriophyoid mite interactions: specific and unspecific morphological alterations. Part II. Exp Appl Acarol 51:81–91. https://doi.org/10.1007/s10493-009-9328-1
Pons J, Barraclough T, Gomez-Zurita J, Cardoso A, Duran D, Hazell S, Kamoun S, Sumlin W, Vogler A (2006) Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol 55:595–609. https://doi.org/10.1080/10635150600852011
Puillandre N, Lambert A, Brouillet S, Achaz G (2012) ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol 21:1864–1877. https://doi.org/10.1111/j.1365-294X.2011.05239.x
R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Sabelis MW, Bruin J (1996) Evolutionary ecology: Life history patterns, food plant choice and dispersal. In: Lindquist EE, Sabelis MW, Bruin J (eds) Eriophyoid mites: their biology, natural enemies and control. World Crop Pests, vol 6. Elsevier, Amsterdam, pp 329–366
Schmidt AR, Jancke S, Lindquist EE, Ragazzi E, Roghi G, Nascimbene PC, Schmidt K, Wappler T, Grimaldi DA (2012) Arthropods in amber from the Triassic Period. Proc Natl Acad Sci USA 109:14796–14801. https://doi.org/10.1073/pnas.1208464109
Sidorchuk EA, Schmidt AR, Ragazzi E, Roghi G, Lindquist EE (2015) Plant-feeding mite diversity in Triassic amber (Acari: Tetrapodili). J Syst Palaeontol 13:129–151. https://doi.org/10.1080/14772019.2013.867373
Silvestro D, Michalak I (2011) raxmlGUI: a graphical front-end for RAxML. Org Divers Evol 12:335–337. https://doi.org/10.1007/s13127-011-0056-0
Skoracka A, Dabert M (2010) The cereal rust mite Abacarus hystrix (Acari: Eriophyoidea) is a complex of species: evidence from mitochondrial and nuclear DNA sequences. Bull Entomol Res 100:263–272. https://doi.org/10.1017/S0007485309990216
Skoracka A, Kuczyński L, Szydło W, Rector B (2013) The wheat curl mite Aceria tosichella (Acari: Eriophyoidea) is a complex of cryptic lineages with divergent host ranges: evidence from molecular and plant bioassay data. Biol J Linnean Soc 109:165–180. https://doi.org/10.1111/bij.12024
Skoracka A, Lopes LF, Alves MJ, Miller A, Lewandowski M, Szydlo W, Majer A, Rozanska E, Kuczynski L (2018) Genetics of lineage diversification and the evolution of host usage in the economically important wheat curl mite, Aceria tosichella Keifer, 1969. BMC Evol Biol 18:122. https://doi.org/10.1186/s12862-018-1234-x
Skoracka A, Smith L, Oldfield G, Cristofaro M, Amrine JW Jr (2010) Host-plant specificity and specialization in eriophyoid mites and their importance for the use of eriophyoid mites as biocontrol agents of weeds. Exp Appl Acarol 51:93–113. https://doi.org/10.1007/s10493-009-9323-6
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. https://doi.org/10.1093/bioinformatics/btl446
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. https://doi.org/10.1093/molbev/mst197
Toews DP, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21:3907–3930. https://doi.org/10.1111/j.1365-294X.2012.05664.x
Whiting MF, Carpenter JC, Wheeler QD, Wheeler WC (1997) The Strepsiptera problem: phylogeny of the holometabolous insect orders inferred from 18S and 28S ribosomal DNA sequences and morphology. Syst Biol 46:1–68. https://doi.org/10.1093/sysbio/46.1.1
Xue X-F, Song Z-W, Amrine JW Jr, Hong X-Y (2007) Eriophyoid mites on coniferous plants in China with descriptions of a new genus and five new species (Acari: Eriophyoidea). Int J Acarol 33:333–345. https://doi.org/10.1080/01647950708683695
Xue X-F, Dong Y, Deng W, Hong X-Y, Shao R (2017) The phylogenetic position of eriophyoid mites (superfamily Eriophyoidea) in Acariformes inferred from the sequences of mitochondrial genomes and nuclear small subunit (18S) rRNA gene. Mol Phylogenet Evol 109:271–282. https://doi.org/10.1016/j.ympev.2017.01.009
Xue X-F, Guo J-F, Dong Y, Hong X-Y, Shao R (2016) Mitochondrial genome evolution and tRNA truncation in Acariformes mites: new evidence from eriophyoid mites. Sci Rep 6:18920. https://doi.org/10.1038/srep18920
Zhang J, Kapli P, Pavlidis P, Stamatakis A (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29:2869–2876. https://doi.org/10.1093/bioinformatics/btt499
Zhang Z-Q (2011) Animal biodiversity: an outline of higher-level classification and survey of taxonomic richness. Magnolia Press, Auckland
Zhang Z-Q (2018) Repositories for mite and tick specimens: acronyms and their nomenclature. Syst Appl Acarol 23:2432–2466. https://doi.org/10.11158/saa.23.12.12
Zhao S, Amrine JW Jr (1997) Investigation of snowborne mites (Acari) and relevancy to dispersal. Int J Acarol 23:209–213. https://doi.org/10.1080/01647959708683565
Zhao Y, Li W, Wang G-Q (2018) New asian phyllocoptines (Eriophyidae, Phyllocoptinae): descriptions of Namengia latifloris gen. nov. & sp. nov. (Acaricalini) and Petanovicia cathartica sp. nov. (Phyllocoptini) from northwest Laos. Syst Appl Acarol 23:2022–2032. https://doi.org/10.11158/saa.23.10.11
Acknowledgements
We would like to thank Mr. Jimmy Chew (Borneo Jungle Girl Camp, Keningau, Sabah, Malaysia) for kindly collecting mite specimens in Sabah, Malaysia. This research was funded by the National Natural Science Foundation of China (31970437).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material
10493_2019_443_MOESM1_ESM.pdf
Supplementary material 1 Phylogenetic trees inferred from nucleotide sequences of 18S gene using maximum likelihood method. Node numbers indicate maximum likelihood bootstrap proportion (BSP) (PDF 166 kb)
10493_2019_443_MOESM2_ESM.pdf
Supplementary material 2 Phylogenetic trees inferred from nucleotide sequences of 18S gene using Bayesian method. Node numbers indicate Bayesian posterior probabilities (BPP) (PDF 159 kb)
10493_2019_443_MOESM3_ESM.pdf
Supplementary material 3 Phylogenetic trees inferred from nucleotide sequences of two mitochondrial (COI and 12S) and two nuclear (18S and 28S) gene fragments using maximum likelihood method. Node numbers indicate maximum likelihood bootstrap proportion (BSP) (135 kb)
10493_2019_443_MOESM4_ESM.pdf
Supplementary material 4 Phylogenetic trees inferred from nucleotide sequences of two mitochondrial (COI and 12S) and two nuclear (18S and 28S) gene fragments using Bayesian method. Node numbers indicate Bayesian posterior probabilities (BPP) (134 kb)
10493_2019_443_MOESM5_ESM.pdf
Supplementary material 5 Phylogenetic trees of Diptilomiopus species inferred from nucleotide sequences of (A) COI (no partitioned), (B) COI (partitioned by three codons), (C) 18S and (D) 28S using Bayesian method. Node numbers (black) indicate Bayesian posterior probabilities (BPP). Branch numbers (red) indicate support values inferred by bPTP. Blue bars indicate delimitated species by analyses of GYMC and bPTP (440 kb)
Rights and permissions
About this article

Cite this article
Liu, Q., Yuan, YM., Lai, Y. et al. Unravelling the phylogeny, cryptic diversity and morphological evolution of Diptilomiopus mites (Acari: Eriophyoidea). Exp Appl Acarol 79, 323–344 (2019). https://doi.org/10.1007/s10493-019-00443-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-019-00443-8