
Abstract
Environments where lignocellulosic biomass is naturally decomposed are sources for discovery of new hydrolytic enzymes that can reduce the high cost of enzymatic cocktails for second-generation ethanol production. Metagenomic analysis was applied to discover genes coding carbohydrate-depleting enzymes from a microbial laboratory subculture using a mix of sugarcane bagasse and cow manure in the thermophilic composting phase. From a fosmid library, 182 clones had the ability to hydrolyse carbohydrate. Sequencing of 30 fosmids resulted in 12 contigs encoding 34 putative carbohydrate-active enzymes belonging to 17 glycosyl hydrolase (GH) families. One third of the putative proteins belong to the GH3 family, which includes β-glucosidase enzymes known to be important in the cellulose-deconstruction process but present with low activity in commercial enzyme preparations. Phylogenetic analysis of the amino acid sequences of seven selected proteins, including three β-glucosidases, showed low relatedness with protein sequences deposited in databases. These findings highlight microbial consortia obtained from a mixture of decomposing biomass residues, such as sugar cane bagasse and cow manure, as a rich resource of novel enzymes potentially useful in biotechnology for saccharification of lignocellulosic substrate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sugar cane bagasse is one of the most abundant agro-industry residues in Brazil and its high carbohydrate content could be used as a feedstock for second-generation ethanol production in biorefineries. It is estimated that, if nearly 90 % of the fermentable sugars in this feedstock could be recovered and fermented, then Brazilian ethanol production would increase by 50 % (Camargo 2005; Rabelo et al. 2011). However, the use of sugarcane biomass residue is still not economically viable for efficient ethanol fuel production, and the same applies to other sources of lignocellulosic biomass. Among the four stages of ethanol production from lignocellulosic waste—pretreatment, enzymatic hydrolysis, fermentation, and distillation (Balat and Balat 2009; Parawira and Tekere 2011)—the step involving enzymatic hydrolysis to release soluble sugars (Sun and Cheng 2002) remains cost prohibitive because of the large amount of enzymes necessary and the low activity of most of the enzymatic cocktails currently available (Del Pozo et al. 2012).
The identification of new hydrolytic enzymes or genetic improvement of existing ones could help lower the costs of enzyme production for biomass degradation, and there is considerable effort being directed toward improving cellulosic ethanol production by these and other means (Himmel et al. 2007; Duan et al. 2009; Li et al. 2009; Gnansounou and Dauriat 2010; Horn et al. 2012; Montella et al. 2015).
Metagenomics is a promising approach for functional analysis of the microbial genomes present in natural environments, especially the genomes of uncultured microbial species that represent an unexplored reservoir of new biomolecules (Uchiyama and Miyazaki 2009). Several studies have shown how metagenomic libraries provide a vast pool of new genes encoding biotechnology products of interest (Lee et al. 2004; Yun et al. 2004; Kim et al. 2006; Duan and Feng 2010; Hjort et al. 2010). Among the diverse array of non-cultivable microorganisms there likely exist novel cellulases and hemicellulases with better catalytic performance, exhibiting higher affinity and activity toward cellulosic substrates and having desirable traits for industrial operational conditions such as high activity at temperatures of 45–55 °C. The metagenomic strategy is important for mining new genes from untapped feedstock sources to increase the final hydrolysis yield from cellulose and hemicellulose.
The successful discovery of biomass-degrading genes from various environmental samples including cow rumen (Hess et al. 2011; Lopes et al. 2015), compost soils (Pang et al. 2009), composts (Matsuzawa et al. 2015), sugar cane bagasse (Kanokratana et al. 2015) and termite gut (Warnecke et al. 2007) encouraged us to investigate complex and unexploited microbial communities obtained by mixing different samples enriched for microorganisms capable of degrading cellulose. In addition, the screening of microbial genomes in their natural environments can accelerate the discovery of new genes. Thus, the possibility of isolating carbohydrate catabolic enzymes from a metagenomic library constructed from a thermophilic microbial consortium was evaluated. The consortium was derived from naturally degrading sugarcane bagasse associated with cow manure in a thermophilic composting phase. Functional screening was employed and led to the identification and phylogenetic analysis of seven novel putative protein sequences, including β-glucosidase and α-xylosidase that are essential to enzymatic conversion of lignocellulosic biomass to fermentable sugars for bioethanol production.
Materials and methods
Total DNA extraction and metagenomic fosmid library construction
The sample used in this study was obtained by Souza (2012) and consisted of a microbial community as source of metagenomic DNA. Briefly, residues of decomposing sugarcane bagasse and cow manure were inoculated in flasks containing cellulose-peptone solution and incubated at 55 °C under static aerobic conditions with a strip of Whatman filter paper as an indicator of cellulase activity (Souza 2012). When degradation of the filter paper strip was detected, 1.0 mL of suspension was subcultured into fresh medium with 1 g of sugarcane bagasse. This strategy favoured the presence of lignocellulosic activity.
Total DNA from the microbial consortium was extracted using a protocol described by Stevenson and Weimer (2007). Twenty-five milliliters of subcultured microbial community were centrifuged and cells were lysed using beads and SDS (20 %). DNA was isolated by purification using phenol/chloroform extraction followed by alcohol precipitation. The pellet of DNA was resuspended in TE (10 mM Tris/HCl, 1 mM EDTA, pH 8.0), treated with RNAse at 37 °C for 2 h and stored at −20 °C.
Cloning of metagenomic DNA into the vector Fosmid pCC2FOS (Epicentre®, Madison, WI, USA) was performed according to the manufacturer’s instructions. Briefly, the metagenomic DNA was size selected by preparative pulsed field (Pulsed-field CHEF DRIII System—Bio-Rad, Hercules, CA, USA) gel electrophoresis at an angle of 120°, 6 V cm−1, 0.5 s—0.5 s switch time, 5 h at 14 °C. DNA fragments of about 40 kb were excised from the gel and their ends repaired using the End-Repair Enzyme Mix before ligation into the vector. CopyControl fosmids containing the inserts were packaged with MaxPlax Lambda Packaging Extract and used to infect the Escherichia coli EPI300-T1® plating strain. Infected cells were spread on Luria–Bertani (LB) plates (10 g L−1 bactotryptone; 5 g L−1 yeast extract; 5 g L−1 NaCl) supplemented with 12.5 μg mL−1 chloramphenicol and incubated at 37 °C overnight to select for the CopyControl Fosmid clones. Clones were transferred to 96-well plates containing LB medium and stored after growth at −80 °C in the presence of glycerol (20 % v/v).
Estimation of the size of the metagenomic library was carried out with ten randomly selected clones. The fosmid DNA from each clone was extracted using a Wizard Plus DNA Purification kit (Promega, Madison, WI, USA), and digested using 10 U NotI restriction enzyme (Promega) at 37 °C overnight. The restriction fragments of the fosmid clones were separated by preparative pulsed field gel electrophoresis at an angle of 120°, 6 Vcm−1, 0.5 s—0.5 s switch time, 10.5 h at 14 °C.
Functional screening
Clones from the metagenomic library were pre-cultured in 96-wells microplates containing 150 µL of liquid LB medium supplemented with chloramphenicol (12.5 µg mL−1) and incubated at 37 °C on a rotary shaker (200 rpm) for 16 h. After growth, the clones were plated on different media for lignocellulose degradation activity screening using a 96 pin microplate replicator model 140500 (Boekel Scientific, Fearsteville, PA, USA). All functional screening in solid medium was carried out in triplicate.
Screening for cellulolytic activity was performed using the method described by Kasana et al. (2008) on agar CMC medium (0.2 % NaNO3, 0.1 % K2HPO4, 0.05 % MgSO4, 0.05 % KCl, 0.2 % carboxymethylcellulose sodium, 0.02 % peptone and 1.7 % agar). Three microliters of culture were transferred to agar CMC plates using a replicator. Following a 48-h incubation, Gram's iodine solution (6.6 g L−1 KCl and 3.33 g L−1 iodine) was spread on the plate for 3–5 min to visualize the results. Formation of a clear zone around the colony indicated the presence of cellulolytic enzymes.
Screening for xylanases was carried out according to the method described by Teather and Wood (1982), with modifications. Cells were inoculated on plates containing 1.5 % agar and 0.1 % xylan (w/v), followed by incubation at 37 °C for 48 h, after which the plates were stained with a solution of Congo Red (0.1 %) for 20 min and rinsed with 1 M NaCl for 20 min. Positive clones were identified by a clear zone around colonies.
Screening for β-glucosidases was carried out according to two methods. In the first, described by Eberhart et al. (1964), clones were cultured on LB agar plates containing 0.2 % esculin (w/v), 0.05 % ferric ammonium citrate (w/v), and 12.5 µg mL−1 chloramphenicol at 37 °C overnight. Those colonies forming clear black halos were selected as positive colonies. In the second method, described by Del Pozo et al. (2012), clones were cultured on LB agar plates containing 12.5 µg mL−1 chloramphenicol at 37 °C overnight, after which the plates were covered with an agar buffer substrate solution (40 mL of 50 mM C2H3NaO2, pH 5.6, 0.4 % agarose and 5 mg mL−1 of pNPβG as substrates). Positive clones were identified by formation of a yellow color.
Enzymatic activities from positive clones identified in the functional screening were confirmed by enzymatic assays for CMCase, β-glucosidase and xylanase. CMCase activity was analysed by the DNS (3,5-dinitrosalicylic acid) method that measures sugars released during enzymatic hydrolysis of cellulose, as described by Miller (1959), with the reaction mix adapted from Ghose (1987). Xylanase activity was spectrophotometrically measured by the DNS method using an adapted protocol from Bailey et al. (1992). In both cases, one unit of enzyme activity was defined as the amount of enzyme necessary to release one µmol of reduced sugar per gram of protein per minute under assay conditions.
β-glucosidase activity was determined according to Chen et al. (1994). One unit of β-glucosidase activity was defined as the amount of enzyme required to hydrolyse 1 µmol of substrate per minute, under assay conditions, per milligram of protein. Total protein concentration was determined according to Bradford (1976).
Thirty positive clones were selected for sequencing based on functional screening, and their fosmid DNA was digested with 10 units of BglII restriction enzyme (Promega) at 37 °C overnight to confirm that each fosmid was unique. Fosmid DNAs were pooled in equal amounts before sequencing using a HiSeq 2000 with the 100-bp paired-end protocol at Macrogen Inc. (http://dna.macrogen.com).
Sequence and phylogenetic analysis of fosmid DNA
A sequence quality check was performed using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Reads were filtered before assembly such that for a pair of Paired-Ended reads each read should have >90 % of bases with base quality ≥Q20. Reads matching the fosmid vector sequence were removed and the remaining reads were assembled into contigs using the SOAPdenovo2 method (http://soap.genomics.org.cn/soapdenovo.html).
Contig sequences were screened for Open Reading Frames (ORFs) in the PRODIGAL program (Prokaryotic Dynamic Programming Genefinding Algorithm) (http://prodigal.ornl.gov) (Meyer et al. 2008). The predicted genes were submitted to the MG-RAST metagenomic online server (Meyer et al. 2008) for both taxonomic and functional annotation. Taxonomic annotation was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa and Goto 2000). Functional annotation was performed using the subsystems technology, in which genes are classified in a hierarchical structure in which all genes required for a specific function are grouped into subsystems (Aziz et al. 2008). All annotation was done using standard MG-RAST (Meyer et al. 2008) cutoff values: E-value cutoff of 1E-5 and a minimum identity cutoff of 60 %.
To identify carbohydrate-active enzymes, the amino acid sequences were submitted to the CAZymes Analysis Toolkit (CAT) (http://mothra.ornl.gov/cgi-bin/cat/cat.cgi) (Cantarel et al. 2009) for annotation. Enzymes were searched using the sequence similarity-based annotation against the entire non-redundant sequences of the CAZy database, with the optional Pfam-based annotation that uses association rules inferred considering the CAZy database using the association rule-learning algorithm. Then, 14 pairs of primers were designed to amplify cellulase and hemicellulase genes using the ORF sequences identified by the PRODIGAL program. To amplify those glycoside hydrolase genes, pools of fosmid DNA were used as template, followed by individual PCRs to identify which clones were harboring which genes. In the negative PCR controls water replaced DNA sample in the reaction. PCR conditions were as follows: 95 °C for 120 s, followed by 30 cycles of 95 °C for 60 s, 61 °C for 45 s and 72 °C for 85 s, with a final annealing at 72 °C for 5 min. PCR was performed using GoTaq® DNA Polymerase (Promega). Pairs of primers and corresponding encoded enzyme for each gene are listed in Table 1.
In this dataset, two contigs (both >30 kb) containing carbohydrate-active enzymes were selected for further analysis. Translated ORF sequences were searched against the Non-redundant Protein Sequences (nr) and the Clusters of Orthologous Groups databases at NCBI to predict their functions.
To study the evolutionary relationships among the putative proteins identified in Contigs 31 and 61, the amino acid sequences of the identified ORFs encoding carbohydrate-active enzymes were compared to sequences from the Non-redundant Protein Sequences (nr) database at NCBI using the BLASTP algorithm (Altschul et al. 1990). Sequences with high identity, as identified by the search, were imported into MEGA 6 software (Tamura et al. 2013), aligned using Clustal W (Larkin et al. 2001), and calculation of the phylogenetic trees was based on these sequence alignments using the neighbor-joining method. To check the robustness of the resulting tree and the statistical significance levels of the interior nodes, bootstrap analysis with 1000 replicates was carried out and values above 70 % were reported.
Nucleotide sequence accession numbers
Sequences were deposited in the Sequence Read Archive database under accession number SRR3310160 (http://trace.ncbi.nlm.nih.gov/Traces/sra/?run=SRR3310160).
Results
Metagenomic library screening for clones expressing lignocellulolytic activity and general characteristics
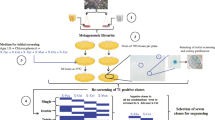
To identify novel genes coding for cellulolytic enzymes from new sources, a fosmid metagenomic library was constructed with total DNA isolated from a thermophilic microbial consortium originated from sugar cane bagasse and cow manure in a thermophilic composting phase (Souza 2012). A total of 135,000 clones was obtained from the metagenomic library. Ten randomly chosen fosmid clones were analysed by restriction enzyme digestion to determine the average size of the DNA inserts; this was calculated to be about 26 kb, with the full library harboring about 3.5 Gbp of metagenomic DNA.
About 5 % of the clones from the library were screened for lignocellulolytic activity on solid medium. Of the 6720 clones screened, 159 and 9 clones showed a clear zone surrounding the colonies to indicate CMCase and xylanase activity, respectively, and 14 clones showed a black zone around the colonies to indicate β-glucosidase activity. These results represent a hit rate of about 42, 746 and 480 tested clones for CMCase, xylanase and β-glucosidase activity, respectively. Positive clones for laccase activity were not found in this screening. In choosing fosmids for sequencing, the functional screening test on solid plates was repeated to confirm the positive clones, followed by enzymatic assay for CMCase, xylanase and β-glucosidase, with the highest clone reaching 204.57, 1689.45 and 1.34 U mg−1 specific activity, respectively (Fig. S1). The aim of this enzymatic assay was to rank the fosmids according to their activity and then reduce the number of fosmids for sequencing. Fosmid DNA from selected positive clones of 9 xylanases, 12 β-glucosidases (two of the original 14 with lowest activities—lower than 0.001 U mg−1—were discarded) and 9 cellulases (chosen for their stronger halo formation and CMCase activity) were extracted and digested with the same enzyme to confirm that all inserted DNA fragments were different (so as to avoid sequencing identical DNA fragments).
Fosmid sequence, phylogenetic and functional analyses
Thirty fosmids were selected for sequencing based on their positive results for CMCase, xylanase and β-glucosidase as indicated above. 9,256,230 high-quality reads were generated in this study, of which 2,996,474 matched fosmid vector sequence and were discarded from further analysis. The remaining 6,259,756 reads (average length of 101 bp) were assembled by using the SOAPdenovo2 method.
Sixty-seven contigs (average length of 16,743 bp) were generated after assembly, with the longest contig being 63,351 bp and the shortest 259 bp. The N50 (which means that half of all bases reside in contigs of this size or longer) was 33,374 bp. The average GC content of the reads was 59.33 %.
Sequences of the 67 contigs were screened for ORFs using the PRODIGAL program and annotated by the MG-RAST program. Based on the KEGG protein database used by MG-RAST, of the 1100 ORFs submitted to MG-RAST, 98.3 % of the annotated ORFs showed homology to proteins found in Bacteria, 0.39 % to Eukarya, 0.13 % to Archaea and 1.18 % was not assigned to any microbial group (Table S1). Within the Bacteria domain, the most abundant phylum was Firmicutes (55.2 %), with Clostridia being the most abundant class (90.8 %) and comprising mostly the order Clostridiales (93.88 %); Proteobacteria was the next most abundant phylum (39.47 %), with Gammaproteobacteria (68.92 %) being the most abundant class, comprising mostly the order Chromatiales (52.45 %) (Fig. 1).

ORF distribution according with their taxonomic classification using the KEGG database in the MG-RAST program (Meyer et al. 2008). The tree was constructed at the order level and it is colored at the phylum level. The stacked bars indicate the abundance of ORFs assigned to each order in the analysis considering all 67 contigs (red bars) and the 12 selected contigs (green bars). (Color figure online)
The amino acid sequences conceptually translated from the 67 contigs were used to predict a function based on homology by the MG-RAST platform using the SEED subsystem (Overbeek et al. 2005). Seven hundred and nineteen ORFs were assigned to 25 functional groups (subsystems). Clustering-based subsystems and carbohydrate metabolism had the largest quantity of annotated reads assigned, representing 15.43 and 12.38 % of the total of ORFs, respectively (Fig. 2; Table S2). Genes associated with miscellaneous (10.15 %), amino acid and derivatives (8.48 %), and cofactors, vitamins and prosthetic groups (7.78 %), and membrane transport (5.70 %) were the next most prevalent functional groups in the assigned annotated reads (Fig. 2; Table S2). Those groups accounted for more than 50 % of the hits. Based on the MG-RAST subsystem classification, most of the annotated ORFs (10.98 % of all ORFs) fell into the clustering-based subsystem, and were related to an uncharacterised second level which includes the category sugar utilization in thermotogales (3.47 % of all ORFs) (Tables S3, S4) containing such genes as β-glucosidase, α-galactosidase, endo-1,4-β-xylanase, xylose transporter and others. The carbohydrate subsystem, the second largest category, was also dominated by the uncharacterised category at the second level (containing 18.38 % of the ORFs) (Table S5), which includes the category sugar utilization in thermotogales metioned above. The next subcategories in the carbohydrate subsystem are enzymes involved in central carbohydrate metabolism (13.97 % of ORFs within the category), di- and oligosaccharides (13.24 %), CO2 fixation (12.50 %) and monosaccharides utilization (10.29 %) (Table S5). The subsystem amino acid and derivatives was the third most predominant, containing genes related to metabolism of lysine, threonine, methionine and cysteine (27.94 % of ORFs in this category), Branched-chain amino acids (20.59 %), alanine, serine and glycine (16.18 %), aromatic amino acids and derivatives (11.76 %) and other amino acids (23.52 %) (Table S6).

Relative distribution (in percentage of annotated ORFs) of the metabolic subsystems (using SEDD subsystems in the MG-RAST program) detected in the fosmid library. In blue, ORFs detected in all 67 contigs. In red, ORFs detected in the 12 selected contigs containing carbohydrate-degrading enzymes. (Color figure online)
Contig analysis of clones associated with carbohydrate hydrolysis
To search for genes related to hydrolysis of carbohydrate, analysis of the amino acid sequences of ORFs from all 67 contigs was performed using the Carbohydrate-Active enZYme Database (CAZy). This analysis identified 188 gene modules across 45 families of glycosyl hydrolases, glycosyltransferases and carbohydrate esterases, with 74 genes in 26 families of glycosyl hydrolases. Twelve contigs containing 34 genes coding for carbohydrate-degrading enzymes, which are interesting in terms of the saccharification step of cellulose- and hemicellulose-deconstruction for bioethanol production, were detected and manually annotated by BLASTp. Table 2 summarizes the information about the glycoside hydrolase families detected. The most similar protein and the microbial class for each ORF in those 12 contigs were assigned.
The ORFs detected in those 12 contigs were predominantly affiliated to organisms from the Firmicutes (76.38 % of ORFs in the 12 contigs) and Proteobacteria (12.56 %) phyla. At the hierarchical class level, Clostridia (87.5 %) and Gamma-proteobacteria (44 %) were the most frequent classes found within the Firmicutes and Proteobacteria phyla, respectively. ORFs in the 12 selected contigs represent 36.7 % of all ORFs identified in Firmicutes considering the 67 contigs, demonstrating an enrichment of genes affiliated to this phylum in these contigs (Table S1).
A closer look at the annotation of the ORFs from the 12-selected contigs by using MG-RAST (Fig. 2) showed that their subsystem abundances were the same in those 67 contigs, but most ORFs (24.22 % of all ORFs in the 12 contigs) were assigned within the carbohydrate subsystem (Fig. 2). Those ORFs represent 43.8 % of all ORFs in the 67 contigs identified within this subsystem (Table S2), in addition to representing 94.4 and 78.57 % of the ORFs assigned in the categories di- and oligosaccharides and monosaccharides, respectively (Table S5). Furthermore, the 12 selected contigs harbor 72 % of all ORFs assigned to the category sugar utilization in Thermotogales.
To confirm the correct assembly of the data 14 pairs of primers (Table 1) targeting the 34 genes encoding carbohydrate-degrading enzymes were designed. Only two pairs of primers (P4 and P6) failed to amplify the corresponding putative α-N-arabinofuranosidase and β-xylosidase genes, respectively. Because PCR amplification using the other 12 pairs of primers was successful, it was possible to detect which fosmids contained each gene (Fig. S2) (because the 30 fosmid DNAs had been pooled before sequencing). Although the sequenced fosmids were selected by their positive result for CMCase (9 fosmids), xylanase (9 fosmids), and β-glucosidase (12 fosmids) activity, PCR amplification was detected only in clones selected by their β-glucosidase activity. Besides these genes, one α-l-fucosidase, one glycosyl hydrolase from the GH130 family, and one α-xylosidase gene were amplified.
Two contigs (Contig 31 and 61) were selected for further analysis based on their length and the presence of genes coding for putative glycoside hydrolases but with low identity to similar proteins in databases. The assembled sequences of each one were 42,007 bp (Contig 31) and 30,498 bp (Contig 61) (Fig. 3). Of the 31 ORFs in Contig 31, 10 showed ≤60 % identity with any known gene, whereas 7 showed ≥80 % identity. For the 33 ORFs in Contig 61, 17 showed ≤60 % identity with any known gene and only 5 showed ≥80 % identity with proteins in the database. All ORFs of Contig 31 and Contig 61 were assigned to the Firmicutes phylum. From the 14 pairs of primers used to confirm the assembly data, three pairs were designed for ORFs in Contig 31 (ORF 31_21; 31_24; 31_25) and two pairs for those in Contig 61 (ORF 61_21; 61_23) (Tables 1, 2). All five of these predicted proteins show low identity to proteins in the database. For example, the protein assigned to ORF 31_25 shows 53 % identity with a β-glucosidase of Roseburia intestinalis, and the protein assigned to ORF61_21 shows 39 % identity with a glucan 1,3-β-glucosidase of Azoarcus sp. (Table 2). Contig 31 contains 31 ORFs, 4 positive- and 27 negative-stranded, whereas Contig 61 contains 33 ORFs, 5 positive- and 28 negative-stranded. All ORFs from these two contigs were also searched against the Non-redundant protein Sequences (nr) and the COG databases to predict functional categories (Fig. 3). The ORFs were classified into functional categories, with 9 (29 %) and 4 (12 %) classified into the G category (carbohydrate transport and metabolism), and 12 (38.7 %) and 10 (30 %) classified into the S category (function unknown) for Contig 31 and Contig 61, respectively (Fig. 3). In Contig 61, seven (21 %) ORFs were classified in the L category (replication, recombination and repair). The rest of the categories were represented by ≤3 ORFs each in the two contigs.

Schematic map of ORFs identified in Contig 31 (a) and Contig 61 (b). The ORFs are colored and labeled according to the COG functional categories as E (amino acid transport and metabolism), G (carbohydrate transport and metabolism), I (lipid metabolism), K (transcription), L (replication, recombination and repair), M (cell wall/membrane/envelope biogenesis), O (post-translational modification, protein turnover, chaperone functions), P (inorganic ion transport and metabolism), R (general function prediction only), S (function unknown), and T (signal transduction mechanisms). ORFs labeled with an X had no match in the protein or nucleotide databases. Each ORF was assigned a number shown below each map and the asterisks indicate those ORFs coding carbohydrate-degrading enzymes identified by the CAZy toolkit (Meyer et al. 2008)
Phylogenetic analyses were done for those enzymes in Contigs 31 and 61 identified as carbohydrate-active enzymes by the CAZy tool kit (Table 2). A comparison of the β-glucosidases of these sequences with those previously deposited in the GenBank database revealed the three enzymes to be distributed in different groups supported by high bootstrap values, and two of those enzymes (Contig 31_25 and Contig 61_33) form a separate branch from other sequences from bacteria belonging to the Firmicutes phylum (Fig. 4a), whereas the putative β-glucosidase encoded by ORF 61_21 clusters with β-glucosidase sequences derived from bacteria of the genera Azoarcus (Betaproteobacteria class), Haloplasma (unclassified Bacteria) and Carnobacterium (Bacilli class) (Fig. 4a).

Neighbor-joining phylogenetic trees based on deduced amino acid sequences of seven carbohydrate-active enzymes identified by the CAZy toolkit in Contigs 31 and 61. a β-glucosidase; b α-l-fucosidase; c α-xylosidase; d α-galactosidase. Bootstrap values are shown for nodes with over 70 % support. The sequences found in this study are in bold script. The accession numbers for each sequence extracted from the GenBank database are after each sequence name
In the phylogenetic analysis using amino acid sequences from the putative α-l-fucosidases identified in Contig 31, the two sequences clustered with sequences of α-l-fucosidases derived from bacteria of the Clostridia, Bacilli, and Alpha- and Beta-proteobacteria classes (Fig. 4b), which belong to the Firmicutes and Proteobacteria phyla. Although they are phylogenetically related, their low identity with known proteins in databases suggests they are novel proteins. Phylogenetic reconstruction using the amino acid sequences of the ORFs encoding the putative α-xylosidase and α-galactosidase demonstrated that they are closely related to proteins derived from bacteria of the Clostridium genus, but form distinct branches (Fig. 4c, d).
Discussion
The use of function-based metagenomic approaches to search for novel lignocellulosic enzymes have led to the discovery of novel β-galactosidases/α-arabinopyranosidases (Beloqui et al. 2010), cellulases (Duan et al. 2009; Kim et al. 2008; Lopes et al. 2015; Pandey et al. 2016), xylanases (Hu et al. 2008; Kanokratana et al. 2015), xylose isomerases (Parachin and Gorwa-Grauslund 2011), β-xylosidase/α–arabinofuranosidase (Matsuzawa et al. 2015) and β-glucosidases (Del Pozo et al. 2012; Biver et al. 2014), supporting this strategy as a powerful activity-based screening tool to identify entirely new classes of gene sequences encoding new or known functions (Handelsman 2004; Parachin and Gorwa-Grauslund 2011). Several sources of potential cellullose- and hemicellulose-depleting microbial communities are being used for metagenomic analysis. A microbial consortium was reproduced by mixing two natural environmental sources: sugarcane bagasse and cow manure (Souza 2012). This study focused on sequencing and the identification of new enzymes using bioinformatics, but enzymatic assays (Fig. S1) were performed using cell-extracts from fosmid clones with the purpose to select clones for sequencing. Considering that each fosmid contained a different number of genes, and the cellulolytic genes were not isolated we cannot compare our results with others studies that cloned individual genes in appropriated expression vectors.
Consistent with the origin of the consortium (rich in lignocellulosic materials) and the conditions of its reproduction in the laboratory, a high hit rate in the screening was possible by favouring the presence of lignocellulosic activity. It is a common practice for enhancing the desired functions in a microbial community to induce the growth of specific microorganisms by applying pre-enrichment methods to the sample to produce an increased screening hit rate (Cowan et al. 2005). For example, Grant et al. (2004) used metagenomic DNA of cultures grown in medium containing carboxymethylcellulose as the only carbon source, and they observed that the number of glycosyl hydrolases detected was about four times higher than the number identified in metagenomic libraries constructed with DNA taken directly from environmental samples. Usually, the rates of positive hits using functional metagenomic approaches in search of novel cellulases and hemicellulases are low. From a fosmid metagenomic library constructed with DNA samples isolated from soils from a wetland, the number of positive hits for β-glucosidase activity was just 5 from the 14,000 clones screened (Kim et al. 2007). Feng et al. (2009) found 11 positive clones for cellulase activity of 32,500 clones screened from a cosmid metagenomic library originated from rabbit cecum contents. In a metagenomics for genes in the sheep microbiome the average of positive hits per sample were 69, 42 and 13 lignocellulases, amylases and other carbohydrate active enzymes, respectively, and the screening of 100,000 clones from a metagenomic library derived from sugarcane bagasse generated only 5 positive hits. Hit rates for the metagenomic library constructed in this study were 42 (159 positives in 6720 clones), 746 (9 in 6720) and 480 (14 in 6720) tested clones for CMCase, xylanase, and β-glucosidase activity, respectively, demonstrating that the approach was successful and could improve the recovery of genes from metagenomics studies.
Analysis of 1100 ORFs detected after fosmid sequencing and sequence annotation showed dominance of bacterial phyla Firmicutes (55.2 %) and Proteobacteria (39.47 %), representing 98.3 % of the annotated ORFs (Fig. 1). These results are consistent with literature data, in that Firmicutes and Bacteroidetes are reported to be abundant in environment involved with cellulose degradation such as: the thermophilic composting phase (Li et al. 2014), gut of termites (Makonde et al. 2013), cattle feces (de Oliveira et al. 2013), in the bovine rumen (Jami and Mizrahi 2012) and in the sheep microbiome (Lopes et al. 2015). In sugarcane bagasse Proteobacteria were found to be especially abundant, followed by Firmicutes (Rattanachomsri et al. 2011).
Concerning their functional roles, the majority of ORFs were classified in clustering-based subsystems and carbohydrate metabolism (Fig. 2), both including the category sugar utilization in Thermotogales, which comprises genes encoding putative β-glucosidase, α-galactosidase, and endo-1,4-β-xylanase enzymes. Genes characterised in the clustering-based subsystems category usually cluster together in genomic regions and they are functionally coupled (Lu et al. 2012). In addition, the order Thermotogales is represented by anaerobic, thermophilic and hyperthermophilic microorganisms (Huber and Stetter 1992). It is known that in anaerobic microorganisms there is an entity called the cellulosome which is an extracellular enzyme complex consisting of a scaffold and enzymes capable of degrading plant cell walls, whereas in aerobic bacteria several individual cellulases are secreted and act synergistically to hydrolyse plant cell walls (Doi and Kosugi 2004). Some cellulases, hemicellulases and other carbohydrate-active enzymes work in concert to facilitate the degradation of carbohydrates. The predominance of ORFs assigned to anaerobic microorganisms in this study might result from the presence of carbohydrate-depleting enzyme genes colocalized in genomic regions that may codify for cellulosomal enzymes. For example, in Contig 31 (Fig. 3a) there are nine linked genes related to carbohydrate transport and metabolism, six of which belong to glycoside hydrolase families (Table 2), and additional 12 genes of unknown function.
The same profile of predominant phyla and functional groups was observed in the 12 contigs selected for the presence of carbohydrate-active enzymes. The main difference concerned the carbohydrate functional group, which was more prevalent than the clustered-based subsystems (Fig. 2). Thirty-four ORFs (present in the 12 contigs) encode putative proteins related to glycosyl hydrolases distributed in 17 GH families, especially the GH3 (35 %) and GH130 (9 %) families. Sixteen protein sequences shared ≤60 % identity with the most similar protein sequence deposited in databases, with six of them (ORFs: 31_15; 31_21; 31_24; 31_25; and ORFs: 61_21; 61_33) (Table 1) likely derived from representatives of the Firmicutes. Among detected enzymes there are five β-glucosidases from GH3 family, an important enzyme class needed to hydrolyse cellulose but is usually present with low activity in commercial cocktails of cellulases (Del Pozo et al. 2012). One putative β-glucosidases found in this study shared only 43 % identity (at protein level) with a similar enzyme of Carnobacterium maltaromaticum, a bacterium of Firmicutes phylum and Bacilli class, and another β-glucosidase shared only 41 % identity with a similar enzyme of Clostridium saccharoperbutylacetonicum, from class Clostridia within Firmicutes (Table 2). Studies of diverse novel cellulases have shown that these enzymes have evolved independently and are unrelated in sequence and structure (Sukharnikov et al. 2011), characterising them as a large class of nonhomologous isofunctional enzymes (Omelchenko et al. 2010). Diversity of domain architectures of cellulases, even within the same protein family, as well as differences of sequence and structure can complicate the identification of novel enzymes (Sukharnikov et al. 2011), but supports the use of functional metagenomic approaches for discovering improved (for biotechnological purposes) and new hydrolase enzymes. Ferrer et al. (2012) isolated and characterised a novel multifunctional enzyme from the GH43 family and suggested that diversity of polymeric substrates imposed on a complex microbial community may drive the evolution of this enzyme category.
Most ORFs in Contig 31 and Contig 61 were encoded on the same strand (Fig. 3), indicating the existence of possible clusters of genes coding for enzymes involved in polysaccharide degradation. Clostridium cellulovorans, C. acetobutylicum and C. cellulolyticum contain unlinked genes that encode cellulosomal enzymes, but contain large gene clusters with related organization (Tamaru et al. 2000; Nolling et al. 2001; Belaich et al. 2002). More than 60 % of ORFs in Contig 31 and almost 50 % in Contig 61 were related to carbohydrate transport and metabolism, and ‘unknown function’ categories. All ORFs in those two contigs that could not be assigned to any function showed homology with hypothetical proteins of different microorganisms (Tables S7, S8). Three genes of Contig 31 (ORFs 31_21; 31_24; 31_25) were found in the fosmid clone FG8 (Table 2; Fig. S2), and two genes of Contig 61 (ORF 61_21 and ORF 61_33) were found in the fosmid clone FG6. These results might indicate that Contigs 31 and 61 represent a genomic region from single microorganisms from the microbial consortium. Considering the presence of genes coding for putative transposases in these contigs (three genes in each contig) and large sizes of the two contigs (42 Mbp for Contig 31 and 30.5 Mbp for Contig 61) it is reasonable to hypothesize that these contigs contain sequences that could have been horizontally transferred among bacteria. Accordingly, C. cellulovorans contains a transposase gene at the 3′ end of a cluster of genes involved in cellulose degradation, indicating that lateral gene transfer might have occurred (Tamaru et al. 2000).
Comparison of all amino acid sequences from proteins of Contigs 31 and 61 with those previously deposited in the GenBank database confirmed the existence of multiple differences among the newly discovered proteins and known sequences. Moreover, enzymes from the same contig clustered separately and with proteins from different bacteria. Although some enzymes (such as the putative α-galactosidase encoded by ORF 31_28) showed higher identity with enzymes from Clostridium the comparison of the entire sequence of Contig 31 against the Complete Genome NCBI database revealed that only 3 % of sequences from Contig 31 matched sequences in the genome of Clostridium thermocellum and 10 % the genome of Clostridium stercorarium. Considering Contig 61, 5 % of sequences had 80 % identity with the genome of C. stercorarium. Taken together, the phylogenetic analyses and the low identity values (Tables S7, S8) indicate that enzymes identified in this work are distantly related to known proteins and could represent new enzymes for biotechnological purposes such as cellulose degradation.
In conclusion, screening of a metagenomic library based on function revealed 182 positive clones with gene products able to hydrolyse polysaccharide. Sequencing results of 30 positive fosmids proved the feasibility of finding new genes for this purpose by using functional metagenomics applied to a complex microbial community obtained from decomposing sugarcane bagasse and cow manure. This result was confirmed by a more detailed analysis of 12 selected contigs containing seven carbohydrate-active enzymes sharing low identity with protein sequences in databases. Thus, such reproducible mixed cultures could serve as reservoirs of enzymes for future applications in biomass-degradation for biofuels production, including cloning genes into expression systems to obtain hydrolytic enzymes secreted on an industrial scale with no cellular lysis, or for use in yeast transformants themselves for simultaneous saccharification and fermentation.
References
Altschul S, Gish W, Miller W, Myers E, Lipman D (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O (2008) The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi:10.1186/1471-2164-9-75
Bailey MJ, Biely P, Poutanen K (1992) Interlaboratory testing of methods for assay of xylanase activity. J Biotechnol 23:257–270. doi:10.1016/0168-1656(92)90074-J
Balat M, Balat H (2009) Recent trends in global production and utilization of bioethanol fuel. Appl Energy 86:2273–2282. doi:10.1016/j.apenergy.2009.03.015
Belaich A, Parsiegla G, Gal L, Villard C, Haser R, Belaich JP (2002) Cel9 M, a new family 9 cellulase of the Clostridium cellulolyticum cellulosome. J Bacteriol 184:1378–1384. doi:10.1128/JB.184.5.1378-1384.2002
Beloqui A, Nechitaylo TY, López-Cortés N, Ghazi A, Guazzaroni ME, Polaina J, Strittmatter AW, Reva O, Waliczek A, Yakimov MM, Golyshina OV, Ferrer M, Golyshin PN (2010) Diversity of glycosyl hydrolases from cellulose depleting communities enriched from casts of two earthworm species. Appl Environ Microbiol 76:5934–5946. doi:10.1128/AEM.00902-10
Biver S, Stroobants A, Portetelle D, Vandenbol M (2014) Two promising alkaline β-glucosidases isolated by functional metagenomics from agricultural soil, including one showing high tolerance towards harsh detergents, oxidants and glucose. J. Ind Microbiol Biotechnol 41:479–488. doi:10.1007/s10295-014-1400-0
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254. doi:10.1016/0003-2697(76)90527-3
Camargo PD (2005) Força verde: um novo campo para a indústria química. Revista Brasileira de Engenharia Química: 18–21
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D233–D238. doi:10.1093/nar/gkn663
Chen H, Li X, Ljundahl LG (1994) Isolation and properties of an extracellular beta-glucosidase from the polycentric rumen Fungus Orpinomyces sp. strain PC-2. Appl Environ Microbiol 60:64–70
Cowan D, Meyer Q, Stafford W, Muyanga S, Cameron R, Wittwer P (2005) Metagenomic gene discovery: past, present and future. Trends Biotech 23:321–329. doi:10.1016/j.tibtech.2005.04.001
de Oliveira MNV, Jewell KA, Freitas FS, Benjamin LA, Tótola MR, Borges AC, Moraes CA, Suen G (2013) Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Vet Microbiol 164:307–314. doi:10.1016/j.vetmic.2013.02.013
Del Pozo MV, Fernandéz-Arrojo L, Gil-Martínez J, Montesinos A, Chernikova TN, Nechitaylo TY, Waliszek A, Tortajada M, Rojas A, Huws SA, Golyshina OV, Newbold CJ, Polaina J, Ferre M, Golyshin PN (2012) Microbial β-glucosidases from cow rumen metagenome enhance the saccharification of lignocellulose in combination with commercial cellulase cocktail. Biotechnol Biofuels 5:1–13. doi:10.1186/1754-6834-5-73
Doi RH, Kosugi A (2004) Cellulosomes: plant-cell-wall-degrading enzyme complexes. Nat Rev Microbiol 2:541–551. doi:10.1038/nrmicro925
Duan CJ, Feng JX (2010) Mining metagenomes for novel cellulase genes. Biotechnol Lett 32:1765–1775. doi:10.1007/s10529-010-0356-z
Duan CJ, Xian L, Zhao GC, Feng Y, Pang H, Bai XL, Tang JL, Ma QS, Feng JX (2009) Isolation and partial characterization of novel genes encoding acidic cellulases from metagenomes of buffalo rumens. J Appl Microbiol 107:245–256. doi:10.1111/j.1365-2672.2009.04202.x
Eberhart B, Cross DF, Chase LR (1964) Beta-glucosidase system of Neurospora crass. I. beta-glucosidase and cellulose activities of mutant and wild-type strains. J Bacteriol 87:761–770
Feng Y, Duan CJ, Pang H, Mo XC, Wu CF, Yt Y, Hu YL, Wi J, Tang JL, Feng JX (2009) Cloning and identification of novel cellulase genes from uncultured microorganisms in rabbit cecum and characterization of the expressed cellulases. Appl Microbiol Biotechnol 75:319–328. doi:10.1007/s00253-006-0820-9
Ferrer M, Ghazi A, Beloqui A, Vieites JM, López-Cortéz N, Marín-Navarro J, Necgutaylo TY, Guazzaroni ME, Polaina J, Waliczek A, Chernikova TN, Reva ON, Golyshina OV, Golyshin PN (2012) Functional metagenomics unveils a multifunctional glycosyl hydrolase from the family 43 catalysing the breakdown of plant polymers in the calf rumen. PLoS One 7:e38134. doi:10.1371/journal.pone.0038134
Ghose TK (1987) Measurement of cellulase activity. Pure Appl Chem 59:257–268
Gnansounou E, Dauriat A (2010) Techno-economic analysis of lignocellulosic ethanol: a review. Bioresour Technol 101:4980–4991. doi:10.1016/j.biortech.2010.02.009
Grant S, Sorokin DY, Grant WD, Jones BE, Heaphy S (2004) A phylogenetic analysis of Wadi el Natrun soda lake cellulase enrichment cultures and identification of cellulase genes from these cultures. Extremophiles 8:421–429. doi:10.1007/s00792-004-0402-7
Handelsman J (2004) Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68:669–685. doi:10.1128/MMBR.68.4.669-685.2004
Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G, Luo S, Clark DS, Chen F, Zhang T, Mackie RI, Pennacchio LA, Tringle SG, Visel A, Woyke T, Wang Z, Rubin EM (2011) Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331:463–467. doi:10.1126/science.1200387
Himmel ME, Ding SY, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315:804–807. doi:10.1126/science.1137016
Hjort K, Bergstrom M, Adesina MF, Jansson JK, Smalla K, Sjöling S (2010) Chitinase genes revealed and compared in bacterial isolates, DNA extracts and a metagenomic library from a phytopathogen-suppressive soil. FEMS Microbiol Ecol 71:197–207. doi:10.1111/j.1574-6941.2009.00801.x
Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VGH (2012) Novel enzymes for the degradation of cellulose. Biotechnol Biofuels 5:45. doi:10.1186/1754-6834-5-45
Hu Y, Guimin Z, Aiying L, Jing C, Lixin M (2008) Cloning and enzymatic characterization of a xylanase gene from a soil-derived metagenomic library with an efficient approach. Appl Microbiol Biotechnol 80:823–830. doi:10.1007/s00253-008-1636-6
Huber R, Stetter KO (1992) The order Thermotogales. In: Rosenberg E (ed) The Prokaryotes. Springer, Berlin, pp 3809–3815
Jami E, Mizrahi I (2012) Composition and similarity of bovine rumen microbiota across individual animals. PLoS One 7:e33306. doi:10.1371/journal.pone.0033306
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Kanokratana P, Eurwilaichitr L, Pootanakit K, Champreda V (2015) Identification of glycosyl hydrolases from a metagenomic library of microflora in sugarcane bagasse collection site and their cooperative action on cellulose degradation. J Biosci Bioeng 119:384–391. doi:10.1016/j.jbiosc.2014.09.010
Kasana RC, Salwan R, Dhar H, Dutt S, Gulati A (2008) A rapid and easy method for the detection of microbial cellulases on agar plates using Gram’s iodine. Curr Microbiol 57:503–507. doi:10.1007/s00284-008-9276-8
Kim YJ, Choi GS, Kim SB, Yoon GS, Kim YS, Ryu YW (2006) Screening and characterization of a novel esterase from a metagenomic library. Protein Expr Purif 45:315–323. doi:10.1016/j.pep.2005.06.008
Kim SJ, Lee CM, Kim MY, Yeo YS, Yoon SH, Kang HC, Koo BS (2007) Screening and characterization of an enzyme with beta-glucosidase activity from environmental DNA. J Microbiol Biotechnol 17:905–912
Kim SJ, Chang-Muk L, Bo-Ram H, Min-Yong K, Yun-Soo Y, Sang-Hong Y, Bon-Sung K, Hong-Ki J (2008) Characterization of a gene encoding cellulase from uncultured soil bacteria. FEMS Microbiol Lett 282:44–51. doi:10.1111/j.1574-6968.2008.01097.x
Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWillian H, Valentin F, Wallace I, Wilm A, Lopez R, Thompson J, Gibson T, Higgins D (2001) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi:10.1093/bioinformatics/btm404
Lee SW, Won K, Lim HK, Kim JC, Choi GJ, Cho KY (2004) Screening for novel lipolytic enzymes from uncultured soil microorganisms. Appl Microbiol Biotechnol 65:720–726. doi:10.1007/s00253-004-1722-3
Li LL, McrCorkle SR, Monchy S, Taghavi S, van der Lelie D (2009) Bioprospecting metagenomes: glycosyl hydrolases for converting biomass. Biotechnol Biofuels 2:10. doi:10.1186/1754-6834-2-1
Li R, Li L, Huang R, Sun Y, Mei X, Shen B, Shen Q (2014) Variations of culturable thermophilic microbe numbers and bacterial communities during the thermophilic phase of composting. World J Microbiol Biotechnol 30:1737–1746. doi:10.1007/s11274-013-1593-9
Lopes LD, Lima AOS, Taketani RG, Darias P, Silva LRF, Romagnoli EM, Louvandini H, Abdalla AL, Mendes R (2015) Exploring the sheep rumen microbiome for carbohydrate active enzymes. A Van Leeuwenhoek 118:15–30. doi:10.1007/s10482-015-0459-6
Lu HP, Wang YB, Huang SW, Lin CY, Wu M, Hsieh CH, Yu HT (2012) Metagenomic analysis reveals a functional signature for biomass degradation by cecal microbiota in the leaf-eating flying squirrel (Petaurista alborufus lena). BMC Genomics 13:466. doi:10.1186/1471-2164-13-466
Makonde HM, Boga HI, Osiemo Z, Mwirichia R, Mackenzie LC, Goker M, Klenk HS (2013) 16S-rRNA-based analysis of bacterial diversity in the gut of fungus-cultivating termites (Microtermes and Odontotermes species). A Van Leeuw 104:869–883. doi:10.1007/s10482-013-0001-7
Matsuzawa T, Kaneko S, Yaoi K (2015) Screening, identification, and characterization of a GH43 family β-xylosidase/α-arabinofuranosidase from a compost microbial metagenome. Appl Microbiol Biotechnol 99:8943–8954. doi:10.1007/s00253-015-6647-5
Meyer F, Paarmann D, Souza MD, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA (2008) The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform 9:386. doi:10.1186/1471-2105-9-386
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anl Chem 31:426–428. doi:10.1021/ac60147a030
Montella S, Amore A, Faraco V (2015) Metagenomics for the development of new biocatalysts to advance lignocellulose saccharification for bioeconomic development. Crit Rev Biotechnol. doi:10.3109/07388551.2015.1083939
Nolling J, Breton G, Omelchenko MV, Makarova KS, Zeng Q, Gibson R, Lee HM, Dubois J, Qiu D, Hitti J, Wolf YI, Tatusov RL, Sabathe F, Doucette-Stamm L, Soucaille P, Daly MJ, Bennett GN, Koonin EV, Smith DR (2001) Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol 183:4823–4838. doi:10.1128/JB.183.16.4823-4838.2001
Omelchenko MV, Galperin MY, Wolf YI, Koonin EV (2010) Non-homologous isofunctional enzymes: a systematic analysis of alternative solutions in enzyme evolution. Biol Direct 5:31. doi:10.1186/1745-6150-5-31
Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, Crecy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Ruckert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V (2005) The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33:5691–5702. doi:10.1093/nar/gki866
Pandey S, Gulat S, Goyal E, Sing S, Kumar K, Nain L, Saxena AK (2016) Construction and screening of metagenomic library derived from soil for β-1,4-endoglucanase gene. Biocatal Agric Biotechnol 5:186–192. doi:10.1016/j.bcab.2016.01.008
Pang H, Zhang P, Duan CJ, Mo VX, Tang JL, Feng JX (2009) Identification of cellulase genes from the metagenomes of compost soils and functional characterization of one novel endoglucanase. Curr Microbiol 58:404–408. doi:10.1007/s00284-008-9346-y
Parachin NS, Gorwa-Grauslund MF (2011) Isolation of xylose isomerases by sequence- and function-based screening from a soil metagenomic library. Biotechnol Biofuels 4:1–10. doi:10.1186/1754-6834-4-9
Parawira W, Tekere M (2011) Biotechnological strategies to overcome inhibitors in lignocellulose hydrolysates for ethanol production: review. Crit Rev Biotechnol 31:20–31. doi:10.3109/07388551003757816
Rabelo SC, Fonseca NA, Andrade RR, Maciel Filho RR, Costa AC (2011) Ethanol production from enzymati/c hydrolysis of sugarcane bagasse pretreated with lime and alkaline hydrogen peroxide. Biomass Bioenergy 35:2600–2607. doi:10.1016/j.biombioe.2011.02.042
Rattanachomsri U, Kanokratana P, Eurwilaichitr L, Igarashi Y, Champreda V (2011) Culture-independent phylogenetic analysis of the microbial community in industrial sugarcane bagasse feedstock piles. Biosci Biotechnol Biochem 75:232–239. doi:10.1271/bbb.100429
Souza RA (2012) Obtenção de Inoculante e de Coquetel Enzimático Lignocelulolítico a partir de Comunidades Microbianas Termofílicas. 2012. 56f. Dissertação (mestrado). Universidade Federal de Viçosa, Viçosa
Stevenson DM, Weimer PJ (2007) Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl Microbiol Biotechnol 75:165–174. doi:10.1007/s00253-009-2033-5
Sukharnikov LO, Cantwell BJ, Podar M, Zhulin IB (2011) Cellulases ambiguous nonhomologous enzymes in a genomic perspective. Trends Biotechnol 29:473–479. doi:10.1016/j.tibtech.2011.04.008
Sun Y, Cheng J (2002) Hydrolysis of lignocellulosic materials for ethanol production: a review. Bioresour Technol 83:1–11. doi:10.1016/S0960-8524(01)00212-7
Tamaru Y, Karita S, Ibrahim A, Chan H, Doi RH (2000) A large gene cluster for the Clostridium cellulovorans cellulosome. J Bacteriol 182:5906–5910. doi:10.1128/JB.182.20.5906-5910.200
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Teather RM, Wood PJ (1982) Use of Congo red polysaccharide interactions complex formation between Congo red and polysaccharide in detection and assay of polysaccharide hydrolases. Methods Enzymol 160:59–74
Uchiyama T, Miyazaki K (2009) Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr Opin Biotechnol 20:616–622. doi:10.1016/j.copbio.2009.09.010
Warnecke F, Luginbuhl P, Ivanova N, Ghassemian M, Richardson TH et al (2007) Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450:560–565. doi:10.1038/nature06269
Yun J, Kang S, Park S, Yoon H, Kim MJ, Heu S, Ryu S (2004) Characterization of a novel amylolytic enzyme encoded by a gene from a soil-derived metagenomic library. Appl Environ Microbiol 70:7229–7235. doi:10.1128/AEM.70.12.7229-7235.2004
Acknowledgments
This work was supported by CAPES, CNPq and FAPEMIG Brazilian funding agencies.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Colombo, L.T., de Oliveira, M.N.V., Carneiro, D.G. et al. Applying functional metagenomics to search for novel lignocellulosic enzymes in a microbial consortium derived from a thermophilic composting phase of sugarcane bagasse and cow manure. Antonie van Leeuwenhoek 109, 1217–1233 (2016). https://doi.org/10.1007/s10482-016-0723-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-016-0723-4