
Abstract
The relationships among 214 wild-living sika deer from five locations in Germany and two in Lower Austria were examined using mitochondrial DNA (mtDNA) control region sequence. A total of 18 haplotypes are grouped consistently into two major divergent clades, A and B, which differ by a mean of 8.4% sequence divergence. Recently introduced sika deer showed a complex pattern of population structuring, which probably results from historical vicariance in at least two unknown source populations from southeastern Asia (as previously described by morphological and mtDNA findings), and subsequent population admixture as a result of human-mediated restocking. A strong genetic differentiation among populations was indicated by a global ΦST value of 0.78 reflecting mainly the differential distribution of clades A and B haplotypes. There was no association between related haplotypes and their distribution among local populations. These indicate that genealogy is a better predictor of the genetic affinity among most sika deer populations than their present-day locations. The abundant mitochondrial divergence we observed, may reflect a subspecies differentiation and could be associated with phenotypic differences among the introduced sika deer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The sika deer Cervus nippon Temminck, 1836 is a typical member of the old world deer subfamily Cervinae (Groves and Grubb 1987; Pitra et al. 2004). It is original native to East Asia, ranging from the southern Ussuri district of eastern Siberia to Korea, Manchuria and Eastern China, and parts of Vietnam. It is found also on Taiwan and Japan and some other smaller western Pacific islands. Up to 13 geographically separated subspecies are described (Whitehead 1993). The sika deer was raised in captivity for antlers for the Chinese medicine market for many years, but uncontrolled hunting and habitat loss either has, or is, driving them to extinction in the wild. Currently, China has some 600,000 in captivity, and less than 600 (and declining) in three isolated populations in the wild. They are extinct in the wild in Vietnam and Taiwan. Both countries have native sika deer in captivity, Vietnam about 3,000 in various locations, and Taiwan about 400 in two locations. They are also extinct in South Korea, and apparently no original stock is in captivity. Their status in North Korea is not known. Substantial wild populations still occur in Far East Russia (northern end of the distribution is in Sikhote-Alin Nature Reserve) and the Japanese archipelago. In modern times, wild-living populations of sikadeer are known to have been established in the British Isles, several countries of mainland Europe (Austria, Germany, Denmark, Poland, Hungary, France), several US states (Virgina, Maryland, Oklahoma, Texas), New Zealand and Australia. In many cases, they were originally introduced as ornamental animals in parkland, but establishing themselves in the wild.
The taxonomic status and interpretation of the history of wild-living sika deer in Germany and Austria is contentious. However, for convenience, we will describe the history of wild-living sikadeer in both countries primarily based on the traditional view of earlier zoologists (e.g. Koenig 1922; Uloth 1991; Eick 1998). Sika deer were first brought to Germany by the animal dealer Carl Hagenbeck and to Austria as a present of the Japanese emperor in 1893 and 1907, respectively (Eick 1990b, a; 1998). These introduced animals were subsequently bred and held in parks. Later, sikadeer were either deliberately released or escaped from deer parks and became part of the indigenous fauna. In Germany today, there are five geographically distinct populations that can be traced back to enclosures raised sika deer: Möhnesee (from 1893), Ostangeln (from 1928), Hüttener und Duvenstedter Berge (from 1964), Weserbergland (from 1933), and Hochrhein (from 1911). Several times thereafter, new individuals were introduced into some of these populations (Eick 1990a). In Austria, two populations of free ranging sika deer occur in Persenbeug–Ostrong (from 1907) and in the Tullner Donauauen (from 1920) located in the valley of the river Danube. Both populations can be traced back to animals from enclosures that were released to the wild during and after World War II. The founder animals were of Japanese origin and were imported from Podebrady in Bohemia (Eick 1990b). Spring animal number in both populations combined is estimated at 700–1,100 (Bauer 2001).
Population sizes of sika deer in Schleswig–Holstein range between 150 and 200 individuals in Ostangeln and were estimated at about 60 individuals in the Hüttner und Duvenstedter Berge. In North Rhine–Westphalia, it is attempted to stabilise the spring populations size in the Möhnesee area at 500 individuals and that in Weserbergland at 80 individuals. Population size in Baden–Württemberg was estimated at 400 animals (Ueckermann 1992). The goal to prevent population sizes from increasing required intermitted reduction culls in the Möhnesee area and Hochrhein where sika deer occur sympatric with red deer.
This has prompted government interest in active management of the Sika deer in relation to welfare, damage to forestry and agriculture, the genetic impact of introduced sika deer on the native red deer (Cervus elaphus) population and conservation reasons. However, little is known about the current population dynamics of sika deer and the genetic status of this population was uncertain at the outset of the study. Moreover, historical translocation programmes often failed to consider the inter-relatedness of genetic variability, stock source and number of founding individuals. Natural or human-assisted colonization of a new area (a founder event) with a small number of founders may result in a loss of genetic diversity, as well as rapid differentiation between source and founder populations, through random sampling error (a founder effect) (Nei et al. 1975). Even where relatively large founding populations are provided, survival of translocated individuals is often low (Nielsen and Brown 1988), reducing the effective population size. In addition, introduced or restocked populations that experienced severely reduced census size may show the effects of genetic bottlenecks. Finally, the use of geographically distant source populations or the restocking of disparate areas with common stocks can alter patterns of genetic distance between populations or, in extreme cases, be detrimental to population viability (Templeton 1986).
Using DNA sequences from the mitochondrial control region, the aim of the project was to determine levels of genetic differentiation within and among local populations and if genetically unique populations are present which might require special consideration. The control region is a hypervariable, non-coding segment of the mitochondrial genome that is often used in the studies of genetic population structure (Avise 1994). A further goal of the present study was to examine the effects of founder events on genetic variation in the wild-living sika deer. Results are discussed in the context of the current taxonomic controversy together with their implications for the continued protection and management of the sika deer populations in Germany and Austria.
Material and methods
Population samples
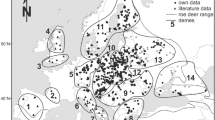
A total of 214 sika deer samples were collected from seven localities in Germany and Austria (Fig. 1 and Table 1). All samples were collected either from tissue biopsies of freshly dead animals (liver, spleen or pancreas) or from dried ear samples. In order to obtain intra-specific divergence estimates, we analysed specimens of the Vietnam sika deer Cervus nippon pseudaxis, an endangered subspecies, held as purebred population in the Tierpark Berlin. The specimens are currently held in the vertebrate collection in the Institute for Zoo and Wildlife Research, Berlin, Germany under the accession numbers SIKA1-215.

Sika deer sample locations in Germany and Lower Austria. Frequency distribution of the clade A and B haplotypes in each population is indicated in pie charts
DNA isolation and processing
Total genomic DNA was extracted by using standard commercial kits (genomic DNA Purification Kit, King Fisher/Thermolabsystems). A 601 bp fragment of the hypervariable part I of the mitochondrial control region (HV1) was amplified and sequenced using the primers SIKACTR1L (5′-GACGCTTATTAATATAGTTCC-3′) and SikaCTR4H (5′-AAAAAAGAACCAGATGTCTG-3′). PCR reaction mixtures contained 0.8 U AmpliTaq DNA Polymerase (Perkin–Elmer), 10 mM Tris–HCl (pH 8.3), 50 mM KCl, 2 mM MgCl2, 200 μM dNTPs, 10– 50 pmol of each primer and 100 ng of DNA in a final volume of 50 μL. Reaction mixtures were subjected to the following PCR cycling protocol: 1× (94°C: 3 min), 35× (94°C: 15 s; 50°C: 20 s; 72°C: 90 s), 1× (72°C: 7 min). All PCR products were purified (Quiaquik PCR Ourification Kit, Quiagen) and directly sequenced using the fluorescent Prism BigDye Terminator Cycle Sequencing kit (Applied Biosystems) according to the manufacturers instructions followed by product separation on the automated 3100 Genetic Analyzer (Applied Biosystems). Sequences were deposited in the GenBank database (accession numbers AY823717–AY823735). The alignments of the mtDNA haplotypes are shown in Table 1.
Data analyses
Mitochondrial DNA sequences were aligned using CLUSTALX (Thompson et al. 1997), visually checked and trimmed to the same length. Initial sequence comparisons and measures of variability were performed using MEGA 2.0 (Kumar et al. 1993). Several population genetic parameters such as nucleotide diversity and haplotype diversity were estimated from the mtDNA dataset using DNASP 3.0 (Rozas et al. 2003).
To assess the extent of differentiation within and among populations, an analysis of molecular variance (AMOVA) (Excoffier et al. 1992) was used to estimate Φ-statistics and M (absolute number of migrants exchanged between populations) values, whose statistical significance was tested using 10,000 permutations as implemented in ARLEQUIN 2.0 (Schneider et al. 2000). AMOVA is a hierarchical analysis analogous to analysis of variance (ANOVA) in which the correlations among genotype distances at various hierarchical levels are used as F-statistic analogues, designated as Φ-statistics. ΦST is the correlation of random genotypes within a population relative to that from the whole species and is analogous to FST of Wright (1951), ΦCT is the correlation of random genotypes within a group of populations relative to that drawn from the entire species and measures the proportion of genetic variation among groupings of populations. Alternate hypotheses of population structure as suggested by tree visualizations also were tested.
To select the model of DNA substitution that best fitted the data, a hierarchical likelihood ratio test approach (Posada and Crandall 1998) implemented in the program MrModeltest 1.03 (Nylander 2002) was used. The model selected was the Hasegawa et al. (1985) model of substitution with rate heterogeneity (HKY + Γ). Using this model, the transition/transversion ratio (ti/tv) and gamma-shape parameter (α) were 14.9 and 0.215 for the mtDNA dataset. The phylogenetic relationships between haplotypes were then reconstructed using the neighbor-joining (NJ) method (Saitou and Nei 1987) under the HKY + Γ model of evolution with the parameter estimates given above. Confidence in estimated relationships was determined using the bootstrap approach (Felsenstein 1985). Bootstrap values were obtained through 1,000 replicates incorporating the same model as above. Bootstrap analysis and phylogeny reconstruction were performed using Paup* version 4.01b (Swofford 1998).
Results
Control region characteristics
The length of the amplified mtDNA fragments varied considerably because sika deer examined in this study possessed either four, seven or eight tandemly repeated 37–40 nucleotide blocks within the control region sequences, a previously described characteristics of native sika deer (Nagata et al. 1999; Cook et al. 1999) and other cervid taxa (Douzery and Randi 1997). The final alignment, including the outgroup and the firstly four tandem repeat but excluding further repetitive sequences units, consisted of 445 characters. Of these 445 positions, 73 were variable and 45 were parsimony informative. Among the 214 individuals analysed in this study, 18 unique haplotypes were identified, suggesting at least 18 maternal lineages in the extant population. Most of them were singletons (i.e. the 11 haplotypes that were sampled only once). The number of repeats and the alignments of the mtDNA haplotypes including the outgroup are shown in Table 1.
Phylogenetic relationships among the mtDNA haplotypes
Two control region sequence clades were clearly apparent in the NJ tree (Fig. 2). One clade contained the haplotypes A1, A3, A4, A7–11 and A17, and was supported, respectively, in 60 and 83% of NJ bootstrap replicate trees. The second clade included the haplotypes B2, B5, B6, B12–16 and B18, and was supported in 97% bootstrap replicate trees. A mean of 3.2 and 4.9 sequence differences existed between any nine haplotypes within each major grouping, but 37.5 sequence differences were found between the haplotypes from the two sequence clades. Gamma-corrected HKY distances were 0.7 and 1.1% for haplotypes within clade A and B, respectively and 8.41±1.49% between clades. Much less structure was apparent within both clades, with most groupings having little statistical support. An exception to this was the strong support obtained for a subclade grouping haplotypes B5 and B6 (100%) and for the basal positioning of haplotype A1 (96%) within clade A. The clear separation between the major clades was also confirmed by the numbers of tandemly repeated blocks. The clade A haplotypes consistently contained four repeat blocks, whereas seven to eight repeat blocks were observed within the control region of the clade B haplotypes (Table 1). Surprisingly, the Vietnam sika was phylogenetically more related with the clade A individuals (89.0±2.8%) than with individuals from clade B (77.5±6.0%).

a Consensus neighbor-joining tree of sika deer mtDNA haplotypes (numbered as in Table 1). The model of site substitution was the HKY85 model with gamma distribution of rate across sites. Bootstrap values (%) based on 1,000 replicates are given above branches
Population affinity
The frequency of the haplotypes in each geographical location is given in Table 1. Three populations, Hüttener and Duvenstedter Berge, Arnsberg and Persenbeug–Ostrong were fixed for clade A haplotypes, the other local populations harboured a variable mixture of both most distinct haplogroups (Fig. 1). Accordingly, haplotype diversity within populations widely ranged from 0% in Hüttener and Duvenstedter Berge to 58.7% in Hochrhein (Table 1). A strong genetic differentiation among populations was indicated by a global ΦST value of 0.78 and 0.75, for distance or frequency-based analysis, respectively, both being significantly different from zero (P<0.00001). Significant structuring of genetic diversity was found among most local populations, even for population pairs separated by only dozens of kilometres, indicating low levels of gene flow (Table 2). These results reflect mainly the differential distribution of clades A and B haplotypes (Fig.1). The groupings of populations into two clades suggested by phylogenetic analyses resulted in a very high Φ CT value (=0.71, P=0.03). In contrast, groupings of populations according to strict geographic criteria gave a considerable decrease in the amount of variation partitioned among groups. For example, in the pair wise comparisons of North versus Central German and South German versus Lower Austrian groups the Φ CT values decreased to zero. These results indicate that genealogy is a better predictor of the genetic affinity among most sika deer populations in our survey than their present-day locations.
Discussion
Because mtDNA is maternally inherited, the number and frequency distribution of mtDNA haplotypes revealed here defines the basic structure of the female component of the sika deer populations and provides testimony to population history and founder events. Assuming a generation time of 4 years for the female sika (based on Forestry Commission records; G. Swanson and D. Hendry, unpublished data), we suggested that there were about 28 generations since the population was founded. Given the very recent introduction, it is unlikely that the observed haplotypes reflect new mutations, and thus the current size of 18 haplotypes estimates the minimum number of female individuals that founded the wild-living sika deer populations in Germany and Austria. However, our analysis showed that the present-day population contains highly divergent sequences, defining at least two distinct haplotype clades (Fig. 2). Additionally, genetic differentiation occurred among closely spaced populations and a hierarchical geographical structure of local populations was not evident. We interpret this pattern as due to a strong degree of genetic divergence between at least two source populations that were sampled for translocation. Assuming a substitution rate of 1.11–1.13% per million years for the highly variable control region of deer (Randi et al. 2001), the origin of the net divergence between the major haplotype clades A and B (8.41±1.49%) traced back to 3.8–3.7 million years ago. Although the number of introduced individuals was relatively low, the intra-clade divergences of 0.7 and 1.1% in the haplogroups A and B, respectively, were in the range of 0.2–5% (average divergence of 2.4%) that was observed in the native sika population on Hokkaido Island of Japan (Nagata et al. 1998). This suggests that the underlying structure of the sika population in Germany and Austria is derived from ancient diversification processes rather than from drift and bottlenecks alone in the contemporary population. An other example that low numbers of founding individuals are not ultimately associated with a significant reduction in genetic diversity (founder effect) is the restoration of white-tailed deer in Mississippi (DeYoung et al. 2003). The history and genetic consequences of the introduction of sika deer to the UK where only one haplotype clade based on control region sequences was observed (Goodman et al. 2001) seems to be quite different from the situation in Germany and Austria.
The existence of two genetically distinct lineages in Germany and Austria may reflect a subspecies differentiation and could be associated with phenotypic differences among the introduced sika deer that were often reported by earlier taxonomists (Eick 1990b). Currently, however, there are no data to specify genetic contributions to morphological differences or to subspecies differentiation. Comparative molecular and morphological analyses on individuals representing divergent phenotypes and on congeneric species should be undertaken to further investigate the phylogeography and systematics of the introduced sika deer.
References
Avise JC (1994) Molecular markers, natural history and evolution. Chapman& Hall, New York
Bauer K (2001) Sikahirsch. In: Spitzenberger F (eds) Die Säugetierfauna Österreichs. Bundesministerium für Land- und Forstwirtschaft, Umwelt und Wasserwirtschaft. Grüne Reihe, 13:698–700
Cook CE, Wang Y, Sensabaugh G (1999) A mitochondrial control region and cytochrome b phylogeny of sika deer (Cervus nippon) and report of tandem repeats in the control region. Mol Phylogenet Evol 12:47–56
DeYoung RW, Demarais S, Honeycutt RL, Rooney AP, Gonzales RA, Gee KL (2003) Genetic consequences of white-tailed deer (Odocoileus virginianus) restoration in Mississippi. Mol Ecol 12:3237–3252
Douzery E, Randi E (1997) The mitochondrial control region of Cervidae: evolutionary patterns and phylogenetic content. Mol Biol Evol 14:1154–1166
Eick E (1990a) Anthropogen begründete Bestände. Österreich Kap. 2.2 A/K In: Eick E, König R, Schwyn G, Baumgardt A (eds) Sika –it Cervus nippon Temminck, 1838, Vol. 1, 2. Aufl, Internationale Gesellschaft Sikawild, Möhnesee-Körbecke, 1993, p 3
Eick E (1990b) Anthropogen begründete Bestände. Deutschland Kap. 2.2 D/K In: Eick E, König R, Schwyn G, Baumgardt A (eds) Sika—Cervus nippon Temminck, 1838, Vol. 1, 2. Aufl, Internationale Gesellschaft Sikawild, Möhnesee-Körbecke, 1993, p 13
Eick E (1998) Das Sikawild. Deutscher Jagdschutz-Verband. Dieter Hoffmann, Mainz, p 19
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Goodman SJ, Tamate HB, Wison R, Nagata J, Tatsuzawa S, Swanson GN, Pemberton JM, McCullough DR (2001) Bottlenecks, drift and differentiation: the population struicture and demographic history of sika deer (Cervus nippon) in the Japanese archipelago. Mol Ecol 10:1357–1370
Groves CP, Grubb P (1987) Relationships of living deer. In: Wemmer CM (eds) Biology and management of the Cervidae. Smithsonian Institute Press, Washington
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Koenig A (1922) Einiges über Dybowskihirsche. Wild u. Hund 28:35
Kumar S, Tamura K, Nei M (1993) MEGA: molecular evolutionary genetics analysis, version 1.02. Pennsylvania State University, University Park
Nagata J, Masuda R, Kaji K, Kaneko M, Yoshida MC (1998) Genetic variation and population structure of the Japanese sika deer (Cervus nippon) in Hokkaido Island, based on mitochondrial D-loop sequences. Mol Ecol 7:871–877
Nagata J, Masuda R, Tamate HB et al (1999) Two genetically distinct lineages of the sika deer, Cervus nippon, in Japanese islands: Comparison of mitochondrial d-loop region sequences. Mol Phylogenet Evol 13:511–519
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Nielsen L, Brown RD (1988) Translocation of wild animals. Wisconsin Humane Society/Caesar Kleberg Wildlife Research Institute, Milwaukee/Kingsville
Nylander JAA (2002) MrModeltest v1.0b. Program distributed by the author. Department of Systematic Zoology, Uppsala University
Pitra C, Fickel J, Meijaard E, Groves CP (2004) Evolution and phylogeny of old world deer. Mol Phylogenet Evol 33:880–895
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Randi E, Mucci N, Claro-Hergueta F, Bonnet A, Douzery EJP (2001) A mitochondrial DNA control region phylogeny of the Cervinae: speciation in Cervus and implications for conservation. Anim Conserv 4:1–11
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schneider S, Kueffer JM, Roessli D, Excoffier L (2000) ARLEQUIN, version 2.01: A software for population genetic data analysis. University of Geneva, Geneva
Swofford DL (1998) PAUP* (Phylogenetic Analyses Using Parsimony), version 4.0d64 (PPC, test). Smithsonian Institution, Washington
Templeton AR (1986) Coadaptation and outbreeding depression. In: Soule ME (ed) Conservation biology: the science of scarcity and diversity. Sinauer Associates, Sunderland, pp 105–116
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 15:4876–4882
Ueckermann E (1992): Das Sikawild. Vorkommen, Naturgeschichte und Bejagung. 2. neubearb. u. erw. Aufl., P Parey, Hamburg und Berlin
Uloth W (1991): Zur Geschichte der Einbürgerung des Sikawildes—Versuch einer Klassifizierung und vorläufigen Wertung. In: Sika Cervus nippon Temminck, 1838; Kapitel 2.2-SU; Internat. Gesellschaft Sikawild; Möhnesee 1
Whitehead GK (1993) The Encyclopaedia of deer. Swan Hill Press, Shrewsbury
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Acknowledgments
This study would not have been possible without the help of many people to whom we express our sincere gratitude. Special thanks go to the International Sika Deer Society, whose chairmen and members supported the study in many ways. In particular, we would like to acknowledge the help by W.-H. von Rumohr, H.-P. Kuhr, H. Brühne, P. Wederhake, M. Strauß, S. Bertagnoli, Dipl. Ing. W. Reiter and Ing. I. Zinn. Dr. St. Rehbein also kindly participated in sample collection. The authors thank Miss A. Schmidt for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s10344-005-0116-x
Rights and permissions
About this article
Cite this article
Pitra, C., Lutz, W. Population genetic structure and the effect of founder events on the genetic variability of introduced sika deer, Cervus nippon, in Germany and Austria. Eur J Wildl Res 51, 95–100 (2005). https://doi.org/10.1007/s10344-005-0087-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10344-005-0087-y