
Abstract
The European roe deer (Capreolus capreolus) is one of the most numerous and widespread ungulate species in Europe, which has complicated the assessment of its genetic diversity on a range-wide scale. In this study, we present the mitochondrial DNA control region (mtDNA CR) genetic diversity and population structure of roe deer in Europe based on the analyses of 3010 samples, which were described as European roe deer individuals. Our analyses revealed two main diversity hotspots, namely Eastern and Central Europe. We proposed that these hotspots result from the Siberian roe deer (C. pygargus) mtDNA introgression and the secondary contact of mtDNA clades, respectively. Significantly lower values of genetic diversity (nucleotide and haplotype diversity) were recorded in the peripheral areas of the species’ range, including the southernmost parts of the Last Glacial Maximum (LGM) refugial areas. Roe deer population in Europe consists of 2–3 genetic groups according to SAMOVA, and 15–16 clusters identified by GENELAND. The main driver of roe deer population structure in the eastern parts of the continent has been introgression of mtDNA of C. pygargus. Spatial genetic analyses revealed a complex structure of roe deer on a pan-European scale, which presumably results from post-glacial recolonization of the continent from various parts of a large LGM refugial area by different roe deer mtDNA clades and haplogroups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The European roe deer (Capreolus capreolus) is a numerous and widely distributed ungulate species in Europe (Andersen et al. 1998; Lovari et al. 2016), occurring throughout the Western Palaearctic region, from the Iberian Peninsula eastwards to the Volga river and from Fennoscandia to southern Greece. It is also recorded in Turkey, Syria, Iran, and Iraq (Lovari et al. 2016). In Eastern Europe (the Volga–Don rivers region in Russia) the range of European roe deer overlaps with that of its sister species—the Siberian roe deer (C. pygargus) (Danilkin 1996, 2014).
Roe deer have been present in Europe for at least 600,000 years (Sommer et al. 2009) and throughout that period its population size has changed in time and space. During the Last Glacial Maximum (LGM, 26–19 Ka BP; Clark et al. 2009), roe deer survived in a large refugial area stretching from the Iberian Peninsula and southern France, to the Apennine Peninsula and the northern parts of Italy, the Balkan and the Carpathian regions, the northern shores of the Black Sea, as far as the Caucasus Mts. (Barros et al. 2020; Lorenzini et al. 2014; Plis et al. 2022; Randi et al. 2004; Sommer et al. 2009). After the LGM roe deer spread from refugia to most European countries and even to the northern parts of the continent, including the then existing Doggerland—a land bridge between the British Isles, Scandinavia, and mainland Europe. In recent centuries roe deer have been affected by hunting, which probably caused extinctions of local populations (e.g., in southern parts of the British Isles in the sixteenth century: Baker and Hoelzel 2013; in Scandinavia in the nineteenth century: Randi et al. 2004). The local extinctions of Finnish roe deer during the Little Ice Ages of the seventeenth and eighteenth centuries suggest that climate fluctuations continued to impact demography of the species throughout the Holocene (Pulliainen 1980). Population restoration occurred in many places due to natural processes (the Scandinavian Peninsula: Thulin 2006) as well as reintroductions (the British Isles: Baker and Hoelzel 2013; Finland: Pulliainen 1980, Helle 1996).
Demographic processes are important in population genetics and can influence various parameters such as genetic drift (Caballero 1994) or effective population size (Kimura 1955; Wright 1931). This can directly affect genetic diversity, which is one of the basic parameters describing the evolutionary status and condition of a population (Clark 2001). Species and populations that hold higher genetic variation can be better adapted to different environments and have a greater potential to recover after population declines (Woodruff 2001). High population genetic diversity is more likely in common species due to a combination of factors, such as larger local population sizes and higher levels of gene flow (Hague and Routman 2016).
Although roe deer is widely distributed species in Europe, not many studies have evaluated its genetic diversity on a range-wide scale. Previous analyses of incomplete mitochondrial DNA (D-loop or cytochrome b) revealed three clades among the European roe deer (Lorenzini et al. 2014; Plis et al. 2022; Randi et al. 2004) with a further subdivision into haplogroups, often geographically separated (Baker and Hoelzel 2014; Barros et al. 2020; Gentile et al. 2009; Mucci et al. 2012; Plis et al. 2022; Tsaparis et al. 2019). Studies which included the eastern part of the continent, revealed a wide area, where hybridization between Siberian and European roe deer led to introgression of the Siberian mtDNA into European roe deer populations (Lorenzini et al. 2014; Markov et al. 2016; Matosiuk et al. 2014; Olano-Marin et al. 2014; Zvychaynaya et al. 2013). The causes of this introgression are still under debate, but most probably both natural processes (long-term overlapping of the ranges of these two species, Matosiuk et al. 2014) and translocations of Siberian roe deer to Eastern Europe (Kashinina et al. 2018; Olano-Marin et al. 2014) resulted in the observed phylogenetic pattern. Matosiuk et al. (2014) stated that introgression of mtDNA did not play any adaptive role in roe deer populations, but it affected the genetic diversity of roe deer in Eastern Europe.
Most of the genetic studies concerning roe deer were performed at regional scales e.g. in Great Britain (Baker and Hoelzel 2014), Slovenia (Buzan et al. 2020, 2022), Poland (Matosiuk et al. 2014; Olano-Marin et al. 2014), Italy (Gentile et al. 2009; Mucci et al. 2012), Germany (Steinbach et al. 2018), Hungary and Bulgaria (Markov et al. 2016), Spain and Portugal (Barros et al. 2020; Royo et al. 2007). Although all these studies quantified genetic diversity of roe deer as high in local populations, an integrative assessment of genetic diversity pattern across the whole species range is still missing. According to our previous study, (Plis et al. 2022) there are contact zones of European and Siberian mtDNA lineages in the Central and Eastern part of the continent, as well as three clades and several haplogroups of C. capreolus. Based on those results, we hypothesised that the highest mtDNA genetic diversity of roe deer would occur in Central and Eastern Europe. To test this hypothesis, we evaluated the genetic diversity across the whole range of the European roe deer.
The objectives of the study were to: (1) describe the mtDNA diversity and its spatial pattern in roe deer throughout the species’ range in Europe; and (2) determine the population genetic structure of roe deer and define the factors affecting it.
Materials and methods
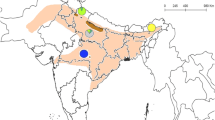
We analysed a fragment of the mtDNA control region (610 bp) of 3010 roe deer individuals. All 1469 sequences were obtained from our previous study (Plis et al. 2022) and combined with 1541 sequences available in GenBank and published by other authors (Baker and Hoelzel 2013; Biosa et al. 2015; Gentile et al. 2009; Lorenzini et al. 2014; Randi et al. 2004; Royo et al. 2007; see Supplementary Table S1). The study area ranged from Portugal to the European part of Russia (6°35’ W–43°23’ E) and from Greece to Finland (38°44’ N–67°42’ N). All samples were divided into 14 demes according to their spatial distribution (demes 1–14, Fig. 1). Number of samples in each deme varied from 24 (western France, deme 2) to 665 (central-northern Italy, deme 6) (Table 1).

Distribution of roe deer (Capreolus capreolus) samples analysed in this study divided into 14 demes in Europe. Black dots and triangles—individuals belonging to the European mtDNA lineage: grey dots—individuals assigned to the Siberian lineage (see Plis et al 2022). The analysed samples covered the whole modern range of the species in Europe
The sequences were aligned against a reference sequence of European roe deer (GenBank accession number AY625869.1), manually edited in BioEdit v.7.0.5.3 and assigned to haplotypes using Arlequin 3.5.1.3 software (Excoffier and Lischer 2010). All haplotypes were also checked in NCBI Basic Local Alignment Search Tool (BLAST) to confirm their confirm their assignment to C. capreolus or C. pygargus mtDNA lineage. All haplotypes were analysed in MEGA7 (Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets; Kumar et al. 2016) to create a phylogenetic tree that would assign them to one of two roe deer lineages: the European (Cc) or the Siberian roe deer (Cp) (for details see Plis et al. 2022). In further analyses, three sets of data were used: all roe deer samples (N = 3010), the subset composed of individuals assigned to the European (C. capreolus) mtDNA lineage (N = 2744), and the subset composed of individuals assigned to the Siberian (C. pygargus) mtDNA lineage (N = 266). We used Arlequin 3.5.1.3 software to calculate the following genetic diversity indices for each of the demes: number of unique haplotypes (h), number of segregating (polymorphic) sites (S), haplotype diversity (Hd), nucleotide diversity (π). The average number of pairwise nucleotide differences (k) was calculated in DnaSP 6 (Rozas et al. 2017). Additionally we included index B (Levins 1968) to express the diversity of haplotypes, using the formula
where pi is the proportion of samples with haplotype i in a deme. The minimum value of index B is 1, and its upper bound is equal to the number of haplotypes in the sample.
In the next step, nucleotide diversity, average number of pairwise differences, haplotype diversity and index B calculated for each of the demes were extrapolated on the area of whole Europe to reflect the spatial gradient of mtDNA diversity of roe deer in the continent. We used the Multilevel B-Spline Interpolation method implemented in the System for Automated Geoscientific Analyses (SAGA) in QGIS 3.10.14 (QGIS Development Team 2020), which predicts values to unknown points by multivariate interpolation of a known set of scattered points.
The population genetic structure of roe deer in Europe was investigated using two clustering methods. First, we used the spatial analysis of molecular variance, implemented in the software SAMOVA 1.0 (Dupanloup et al. 2002), which calculates genetic structure based on the genetic data and the a priori defined geographical locations representing maximum number of groups (K). We ran SAMOVA for values of K from 2 to 14. To make sure that the results were consistent between runs, we ran the analyses twice for each K-value. Second, we analysed population genetic structure using a Bayesian model executed in a Markov chain Monte Carlo (MCMC) scheme and implemented in the GENELAND, a computer package for landscape genetics (Coulon et al. 2006; Guedj and Guillot 2011; Guillot et al. 2005). GENELAND offers the opportunity to identify genetic clustering and to infer what influence the spatial pattern has on the population structure (Coulon et al. 2006). It is commonly used in studies, where spatial aspect plays an important role. GENELAND attempts to maximize Hardy–Weinberg and linkage equilibrium and uses MCMC to estimate the number of clusters (K). The spatial location of individuals are incorporated to elucidate the influence of geographic spread on e.g., gene flow, history of demographic and spatial changes. The number of clusters in our study was determined by running the MCMC iterations 50 times, allowing K to vary from 1 to 10, with the following parameters: 1 000 000 MCMC iterations, maximum rate of the Poisson process equal to the number of used samples, uncertainty attached to the spatial coordinates fixed at 5 km, maximum number of nuclei in the Poisson–Voronoi set as a triple value of the number of used samples (9030 for all roe deer samples and 8232 for the European lineage subset, respectively). To assess the population structure irrespective of geographical coordinates, and hence provide a more reliable, independent overview of genetic clusters, we performed PCA analyses in the adegenet package (Jombart 2008) and visualized the results with the ggplot2 package (Wickham 2016) in R ver. 4.1.3 software (R core team 2021).
For the genetic groups defined by the SAMOVA and GENELAND programmes, we calculated pairwise FST values (Wright 1965) as a measure of population differentiation based on genetic structure in Arlequin 3.5.1.3 software, and created the heatmap in the R statistic software 4.1.3 with the qqplot2 package. To evaluate possible models of expansion, we performed two neutrality tests in DnaSP: Tajima’s D (Tajima 1989) and Fu’s Fs (Fu 1997). We also used mismatch distribution with the sudden expansion model and goodness-of-fit tests (sum of squared deviation − SSD; Harpending’s raggedness index R) calculated in Arlequin 3.5.1.
Results
Genetic diversity and its spatial pattern in roe deer population across the Europe
Most of the roe deer samples (91%) belonged to the European mtDNA lineage (Table 1). Samples assigned to the Siberian lineage were present in 6 out of 14 demes, mainly in Central and Eastern Europe (demes 8, 9, 11, 12, 13 and 14; Fig. 1). Among all analysed sequences, we identified 327 haplotypes with 95 polymorphic sites. The highest number of roe deer haplotypes (87) was detected in Central Europe (deme 10). The lowest number of haplotypes, and polymorphic sites were found in southern England (deme 3; Table 1). The highest number of haplotypes belonging to the Siberian lineage occurred in deme 13 in Eastern Europe, covering the territory of Belarus, Lithuania, Latvia, and the European part of Russia. In demes where both Siberian and European lineages occurred, the number of polymorphic sites was higher than in those where only the European lineage occurred (Table 1).
Haplotype diversity was high among all roe deer samples (Hd = 0.98), while the nucleotide diversity was moderate (π = 0.015; Table 1. Values for the Siberian lineage were lower than those for the European one (Hd = 0.83 vs. 0.98; π = 0.010 vs. 0.011, respectively; Table 1). The highest haplotype diversity among all roe deer individuals inhabiting Europe was detected in the central part of the continent: from north-western Poland through the Czech Republic, and Austria to Slovenia and the northern Balkan peninsula to south-western Ukraine (demes 9 and 10, Hd = 0.98), while the lowest value was found in the northern Great Britain (deme 4, Hd = 0.66). The similar spatial pattern of highest and lowest values was observed for the European lineage subset (Table 1). The haplotype diversity of the Siberian lineage ranged from 0.83 (deme 14) to 0.60 (deme 9).
Nucleotide diversity in all roe deer peaked in the eastern parts of the continent (demes 13 and 14, π = 0.024 and 0.025, respectively; Table 1, Fig. 2). In the European lineage subset, the highest values were found in Central Europe (demes 9, 10, and 12; Fig. 2). For both datasets, the lowest values were detected in southern Italy (deme 7, π = 0.004).

Pan-European spatial pattern of the molecular diversity indices of roe deer (Capreolus capreolus), calculated for the whole dataset (including individuals belonging to both the European and the Siberian mtDNA lineages) and individuals belonging to the European mtDNA lineage only. Numbers show values for the 14 demes (see Fig. 1 and Table 1)
The average number of pairwise differences among all roe deer was 9.05, while in the European and the Siberian lineages the difference values were 6.04 and 5.80, respectively (Table 1). Among all roe deer the highest value of this parameter was recorded in Eastern Europe (deme 14, k = 15.29; Fig. 2). In both subsets, the European and the Siberian lineage, the highest values were found in South-Eastern Europe (southern Ukraine, Romania, southern Hungary, Serbia, and Croatia; deme 9, k = 6.57 and k = 10.21 respectively; Table 1, Fig. 2). The lowest number of pairwise differences occurred in southern Italy (deme 7, k = 2.15, Table 1, Fig. 2). In the Siberian lineage, the lowest value was found in North-Eastern Europe (deme 13, k = 2.95).
Index B of diversity for all individuals and for the European lineage subset reached the highest value in the central part of roe deer range (demes 9, 10 and 12; Fig. 2). The lowest values of the index B for both datasets were recorded in northern Great Britain (deme 4, B = 2.87, Table 1, Fig. 2). Interestingly, in the Siberian lineage subset, the areas with the lowest (B = 2.25) and highest values (B = 5.03) values were located close to each other (demes 9 and 14, respectively) (Table 1).
Analysis of the spatial patterns of genetic diversity revealed that both all roe deer and the European lineage subset reached the highest values of almost all diversity indices in the central part of the continent (demes 9, 10, and 12; Fig. 2). The only exceptions were nucleotide diversity and the average number of pairwise differences in the set of all individuals, which were low in Western Europe and increased from central Poland towards the east (Fig. 2).
Genetic population structure and expansion processes
For roe deer inhabiting Europe, two genetic populations were identified with SAMOVA: SP1, comprising individuals from northern, western, central and southern Europe (Fig. 3), and SP2 in the eastern part of the continent (Fig. 3). The fixation index (FST = 0.361) between these two populations was significant (p < 0.001). When the Siberian lineage of roe deer was excluded, SAMOVA indicated three genetic populations (S1–S3, Figs. 3 and S1), although suggestions for 11, 12 and 13 populations were almost as likely (see Figs. S1and S2). The range of S1 covered almost all of Europe, except for individuals from southern Italy, which formed the second population S2, and individuals from Bulgaria and Greece, which constituted the third population S3. All roe deer samples that belonged to SP2, when the whole dataset was analysed, were now included in one population S1. The highest value of pairwise FST was between S2 and S3 (0.640), while the values between each of these populations and S1 were lower by about half (S1–S2: 0.302; S1–S3: 0.301; p < 0.001 in all cases).

Genetic populations of roe deer (Capreolus capreolus) in Europe, indicated by the SAMOVA (upper panel) and GENELAND (lower panel) analysis of the whole dataset (including individuals belonging to both the European and the Siberian mtDNA lineages), left-side panels), and the European lineage only (right-side panels)
PCA analyses grouped the roe deer samples by their phylogenetic origin. Most of the samples were located in one panmixed population with a wide range of variation between individuals (Fig. S3). It contained individuals belonging to the Central and Western clades (see Plis et al. 2022). Two clusters, which were located on the edges of the dataset, corresponded to the Eastern clade and thesubspecies C.c.italicus (haplogroup C7 in the Central clade; Plis et al. 2022).
GENELAND indicated a more complicated genetic structure and spatial distribution of roe deer populations defined by mtDNA determination. The whole dataset was divided into 16 or 15 genetic populations, while the European lineage subset was divided into 15 or 16 populations (Figs. 3 and S4). Both divisions were characterized by a high level of admixture between samples as well as low FST values between central and northern European populations (populations 9, 10, 11, 12) and the rest of populations (Table 2, Fig. S5). Distinct genetic populations were identified in the Iberian Peninsula, the British Isles, in Fennoscandia, Western and Eastern Europe, the Apennine Peninsula, the Balkans, the Black Sea region, and the Caucasus region. Genetic populations identified in the Iberian Peninsula (GP1, G1), Great Britain (GP3, G3) and Fennoscandia (GP14, G13) were separated in the same way in both analyses (including the whole data set and only the subset of European lineage) (Figs. 3 and S5). Similar separation of genetic populations in the southern parts of Italy (GP6, G6) and Greece (GP7, G7) was also indicated by the SAMOVA analyses (compare Fig. 3, 3S1–S3). This was supported by the high and statistically significant values of FST between each of them and the rest of the GENELAND populations (Table 2, Fig. S5). Interestingly, GENELAND analyses of both datasets (the whole dataset and the European lineage subset only) revealed three geographically discontinuous populations (Fig. 3). One of them (GP14 or G13; Fig. 3) was divided between western France and Fennoscandia. The second (GP2 or G2) and third (GP4 or G4; Fig. 3) one were divided between Great Britain and the continental Western Europe.
The goodness-of-fit tests, which compared expansion model with the observed mismatch distribution among individuals, showed evidence for demographic expansion in 7 out of 15 populations and spatial expansion in all populations of the European lineage defined by GENELAND. v Fu’s FS tests confirmed the recent expansion (p < 0.05) but Tajima’s D did not (all p values > 0.05). Only three populations (2, 6 and 11) showed strong unimodal distributions of the pairwise differences, which indicated one main expansion event in each case. All other populations had multimodal distributions, which indicate multiple expansion events (data not shown).
Discussion
Hot and cold spots of roe deer genetic diversity in Europe
Most of the European continent is inhabited by roe deer belonging to the European mtDNA lineage, while the eastern part of the continent, harbours populations showing the evidence of introgression of C. pygargus genes into the C. capreolus mtDNA genome (Plis et al. 2022; this study). The overall haplotype diversity was high while the nucleotide diversity of roe deer in Europe was moderate in comparison to other ungulate species from the Palearctic zone (Fig. S6). Individuals possessing the mtDNA of the Siberian roe deer co-occur with those of the European lineage in Eastern Finland, Lithuania, Estonia, Belarus, Ukraine, Russia, eastern regions of Poland, Slovakia, Romania, and Hungary. The proportion of individuals with Siberian haplotypes in roe deer population amounted to 80% in Western Russia (Zvychainaya et al. 2011). In consequence, the nucleotide diversity and the average number of pairwise differences in roe deer demes were highest in the eastern part of the continent. High nucleotide diversity estimates in that region indicate that the C. pygargus mtDNA haplotype is rather divergent from C. capreolus mtDNA.
Introgression between C. pygargus and C. capreolus has restored genetic diversity (i.e., haplotype diversity) to some extent, but apparently only marginally, because the populations at either end of the contact zone were depleted and could exchange a limited number of haplotypes, only. However, the highest values of the haplotype diversity and index B of diversity were recorded in Central Europe and its vicinity (Poland, Ukraine, Slovakia, Czech Republic). This is not only due to the presence of individuals with the Siberian mtDNA introgression, but mainly to the overlapping distribution of the three main mtDNA clades (central, western, and eastern) of European roe deer in this region (Plis et al. 2022). This is a novel finding, as previous studies suggested the Balkan and the Iberian Peninsulas as the regions with the highest mtDNA genetic diversity of European roe deer (Randi et al. 2004). We assume that Central Europe has been colonized by roe deer from different refugia where the species survived the LGM. A similar genetic pattern was recently reported in a phylogeographic study on wild boar (Sus scrofa) in Central and Eastern Europe (Niedziałkowska et al. 2021). As palaeontological and phylogeographic studies indicate, refugia for large ungulates were not only located in the southernmost part of the continent, but also in the Carpathian region and the around the Black Sea (Doan et al. 2021; Plis et al. 2022; Sommer and Nadachowski 2006).
In contrast, the remnant populations of the species occurring in the geographically isolated southernmost areas of Italy, Greece and the northern parts of Great Britain had very low values of the diversity indices. Low values of these parameters were also found in Fennoscandia and the peripheral parts of Eastern Europe. The situation in Fennoscandia can be explained by the founder effect, as roe deer recovered there after a significant bottleneck (Randi et al. 2004). In the case of Eastern Europe, the low values of the genetic indices might be due to the low densities of the species and the strong dominance of the one (Siberian) mtDNA lineage.
Spatial genetic structure of roe deer population in Europe
At a macrogeographic level, previous studies did not define any structuring among roe deer in Europe, except for the subspecies C. c. italicus in southern Italy (Mucci et al. 2012; Vernesi et al. 2002). Moreover, Tsaparis et al. (2019) pointed out that roe deer in southern Greece are significantly different from the mainland populations, while Barros et al. (2020) revealed distinctiveness of the westernmost parts of Spain. In the Iberian Peninsula, there were signs of admixture from the Central parts of Europe, while the Italian and Greek populations are most probably endemic ones that did not spread from their LGM refugia. Similar endemic populations in the southernmost areas of the continent were found in wolves (e.g. Stronen et al. 2013), red deer (Doan et al. 2021) and wild boar (Niedziałkowska et al. 2021; Veličković et al. 2015).
The results of our analyses (performed in SAMOVA software) were similar for both datasets. First, we noted the basic division into two clusters/groups: one dominated by specimens of the Siberian mtDNA lineage and another belonging to the European lineage. When only the specimens of the European lineage were considered, the two smaller clusters (southern Italian and southern Greek) were separated from the third, large roe deer cluster covering most of the Europe.
Division obtained in PCA analyses allowed to assess population structure irrespective of geographical localizations of the populations. It revealed the basic phylogenetic structuring of the roe deer population (see Plis et al. 2022). As could be expected, most of the individuals in the SAMOVA population S2 and S3 fell into their respective phylogenetic clusters in PCA plot: S2 from southern Italy into PCA cluster grouping C.c.italicus, and S3 from the southern Balkans and Greece (the region inhabited almost exclusively by the Eastern clade)—into PCA cluster containing Easten clade roe deer.
Further analyses in GENELAND confirmed the distinctiveness of the "Siberian", Italian, and Greek populations, and revealed also a higher resolution of the genetic structure of roe deer in Europe. First, we found more such local ("endemic") genetic populations, which have limited ranges and, more importantly, a high proportion of only one or two genetic haplogroups (comp. Plis et al. 2022), which are hardly found elsewhere (see Fig. 3 lower panel: population G15 along northern shore of the Black Sea; G14 in Lithuania, Belarus, and western Russia; G1 in the Iberian Peninsula and G3 in Great Britain). Second, we recorded several highly diverse populations in Central Europe, resulting from post-glacial recolonization by roe deer, with different proportions of individuals belonging to the three main mtDNA clades (Western, Central and Eastern). Finally, the GENELAND analysis identified some relocation events that gave rise to new populations (see below).
Interestingly, the results of our analysis at the continental scale are consistent with those of studies conducted at a regional scale. Buzan et al. (2020) found that roe deer in Slovenia are divided into three genetic populations inhabiting separate areas in the north-eastern, central, and south-western parts of the country. This division, supported by microsatellite analysis, was explained by the presence of natural barriers (mountain ridges, coastal areas) as well as anthropogenic barriers (urban areas, highways). In this area, we identified the same genetic pattern (populations G4, G5 and G10; see Fig. 3) and showed similarities between the Slovenian populations and roe deer inhabiting the neighbouring countries. Moreover, our GENELAND analyses confirmed the division of the Italian roe deer into a northern and southern population (G5, G6), already described by Gentile et al. (2009) and Mucci et al. (2012). In addition, we detected three populations in British roe deer, which is in line with previous findings by Baker and Hoelzel (2013). Introduced populations in southern Great Britain (G2 and G4) were separated from the population inhabiting central and northern areas (G3). The latter one is a remnant of the autochthonous Scottish population that survived the bottleneck and recolonised its former range (Baker and Hoelzel 2013). In addition, we were able to show genetic connections between the introduced populations in Great Britain and roe deer inhabiting their ancestral regions in Germany, where they were translocated from (Baker and Hoelzel 2013). Similarly, the roe deer in Finland, which originated from natural spread and specimens translocated from Sweden, also formed a genetic unit with its source population (see Fig. 3). However, it must be kept in mind that for a complete picture of the genetic population structure of roe deer in Europe, a large spatial gap in data in some areas (see e.g., in France and the Benelux countries) needs to be filled.
The discrepancy between the genetic structures revealed by the PCA, SAMOVA and GENELAND analyses can be explained by the different approaches of the programmes. PCA is not taking into account the geographical location of samples, SAMOVA considers a priori defined populations (in our study according to their geographical location), while GENELAND infers the number of populations by integrating geographical and genetic information from each of the analysed individuals separately. The last approach is better when individuals are genetically structured as a cline (Jombart et al. 2008), and such a model fits the roe deer population in Europe, where the frequencies of different mtDNA lineages, clades and haplogroups have gradually changed across the continent (comp. Plis et al. 2022).
The performed neutrality as well as demographic and spatial expansion tests gave contradictory results, so they were not conclusive. Probably this is an effect of panmixia of roe deer from different source populations and overlap of various demographic processes, which took place in the studied populations in the past.
Conclusions
The roe deer population in Europe is genetically very diverse, with the genetic hotspot occurring in the central parts of the continent. The genetic diversity of the species and its structure has been strongly shaped by the processes of introgression of the Siberian roe deer mtDNA into the population of the European roe deer, which affected mostly the eastern part of the continent. The high level of genetic diversity among European roe deer (C. capreolus) populations in Central Europe is the result of the recolonization of the continent by roe deer from different geographical regions in the post-glacial period. The low diversity of the peripheral populations is the result of a significant bottleneck in the past (e.g., at the British Isles) or isolation in the former southernmost LGM refugia. By combining genetic and geographical information, we were able to define the detailed internal structuring of the European roe deer populations, which had largely been shaped by natural processes in the species historical demography. High population numbers, wide distribution of the species, relatively rare translocation events and in many areas moderate interest in trophy hunting, have meant that roe deer have not been seriously affected by human-induced changes in their genetic composition compared to for example red deer (Niedziałkowska et al. 2011). However, the role of natural processes versus translocations in the introgression of the Siberian mtDNA genes to the European roe deer populations needs further studies.
Data availability
The data that support the findings of this study will be openly available in the public data repository and complete mtDNA sequences will be deposited in GenBank after the manuscript is accepted for publication.
References
Andersen R, Duncan P, Linnell JDC (1998) The European Roe Deer: the biology of success. Scandinavian University Press
Baker KH, Hoelzel AR (2013) Evolution of population genetic structure of the British roe deer by natural and anthropogenic processes (Capreolus capreolus). Ecol Evol 3(1):89–102. https://doi.org/10.1002/ece3.430
Baker KH, Hoelzel AR (2014) Influence of Holocene environmental change and anthropogenic impact on the diversity and distribution of roe deer. Heredity 112(6):607–615. https://doi.org/10.1038/hdy.2013.142
Barros T, Ferreira E, Rocha RG, Brotas G, Carranza J, Fonseca C, Torres RT (2020) The multiple origins of roe deer populations in western Iberia and their relevance for conservation. Animals 10:2419. https://doi.org/10.3390/ani10122419
Biosa D, Scandura M, Tagliavini J, Luccarini S, Mattioli L, Apollonio M (2015) Patterns of genetic admixture between roe deer of different origin in Central Italy. J Mammal 96(4):827–838. https://doi.org/10.1093/jmammal/gyv098
Buzan E, Gerič GU, Potušek PS, Flajšman FK, Pokorny B (2020) First insights into the population genetic structure and heterozygosity-heterozygosityfitness relationship in roe deer inhabitingdi the area between the Alps and Dinaric Mountains. Animals 10(12):2276. https://doi.org/10.3390/ani10122276
Buzan E, Potušek S, Duniš L, Pokorny B (2022) Neutral and selective processes shape MHC diversity in roe deer in Slovenia. Animals 12(6):723. https://doi.org/10.3390/ani12060723
Caballero A (1994) Developments in the prediction of effective population size. Heredity 73:657–679. https://doi.org/10.1038/hdy.1994.174
Clark AG (2001) Population Genetics. In: Brenner S, Miller JH (eds) Encyclopedia of Genetics. Academic Press, pp 1513–1519
Clark PU, Dyke AS, Shakun JD, Carlson AE, Clark J, Wohlfarth B, Mitrovica JX, Hostetler SW, McCabe AM (2009) The Last glacial maximum. Science 325(5941):710–714. https://doi.org/10.1126/science.1172873
Coulon A, Guillot G, Cosson J-F, Angibault JMA, Aulagnier S, Cargnelutti B, Galan M, Hewison AJM (2006) Genetic structure is influenced by landscape features: empirical evidence from a roe deer population. Mol Ecol 15(6):1669–1679. https://doi.org/10.1111/j.1365-294X.2006.02861.x
Danilkin A (1996) Behavioural Ecology of Siberian and European Roe Deer. Springer Netherlands. https://www.springer.com/gp/book/9780412638800
Danilkin A (2014) Roe Deer (Biological Bases of Resource Management). Management KMK Scientific Press, Moscow
Doan K, Niedziałkowska M, Stefaniak K, Sykut M, Jędrzejewska B, Ratajczak-Skrzatek U, Piotrowska N, Ridush B, Zachos FE, Popović D, Baca M, Mackiewicz P, Kosintsev P, Makowiecki D, Charniauski M, Boeskorov G, Bondarev AA, Danila G, Kusak J, Stanković A (2021) Phylogenetics and phylogeography of red deer mtDNA lineages during the last 50 000 years in Eurasia. Zool J Linn Soc-Lond. https://doi.org/10.1093/zoolinnean/zlab025
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11(12):2571–2581. https://doi.org/10.1046/j.1365-294x.2002.01650.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10(3):564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Gentile G, Vernesi C, Vicario S, Pecchioli E, Caccone A, Bertorelle G, Sbordoni V (2009) Mitochondrial DNA variation in roe deer (Capreolus capreolus) from Italy: evidence of admixture in one of the last C. c. italicus pure populations from central-southern Italy. Ital J Zool 76(1):16–27. https://doi.org/10.1080/11250000802018725
Guedj B, Guillot G (2011) Estimating the location and shape of hybrid zones. Mol Ecol Res 11(6):1119–1123. https://doi.org/10.1111/j.1755-0998.2011.03045.x
Guillot G, Mortier F, Estoup A (2005) Geneland: a computer package for landscape genetics. Mol Ecol Notes 5(3):712–715. https://doi.org/10.1111/j.1471-8286.2005.01031.x
Hague MTJ, Routman EJ (2016) Does population size affect genetic diversity? A test with sympatric lizard species. Heredity 116(1):92–98. https://doi.org/10.1038/hdy.2015.76
Helle P (1996) Metsäkauris [Roe deer]. In: Lindén H, Hario M, Wikman M (eds), Riistan jäljille, pp 100–102. Finnish Game and Fisheries Research Institute, Edita, Helsinki. (In Finnish with English summary)
Jombart T, Devillard S, Dufour A-B, Pontier D (2008) Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity 101:92–103
Kashinina NV, Danilkin AA, Zvychaynaya EY, Kholodova MV, Kiryakulov VM (2018) On the gene pool of roe deer (Capreolus) of Eastern Europe: analysis of the Cyt b gene sequence variability. Russ J Genetvariability 54(7):825–831. https://doi.org/10.1134/S1022795418070049
Kimura M (1955) Solution of a process of random genetic drift with a continuous model. Proc Nat Acad Sci 41:144–150. https://doi.org/10.1073/pnas.41.3.144
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for Bigger Datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1093/molbev/msw054
Levins R (1968) Evolution in changing environments. Princeton University Press. https://press.princeton.edu/books/paperback/9780691080628/evolution-in-changing-environments
Lorenzini R, Garofalo L, Qin X, Voloshina I, Lovari S (2014) Global phylogeography of the genus Capreolus (Artiodactyla: Cervidae), a Palaearctic meso-mammal. Zool J Linn Soc-Lond 170(1):209–221. https://doi.org/10.1111/zoj.12091
Lovari S, Herrero J, Masseti M, Ambarli H, Lorenzini R, Giannatos G (2016) Capreolus capreolus. The IUCN Red List of Threatened Species 2016. IUCN Red List of Threatened Species. https://doi.org/10.2305/IUCN.UK.2016-1.RLTS.T42395A22161386.en
Markov G, Zvychaynaya E, Danilkin A, Kholodova M, Sugar L (2016) Genetic diversity and phylogeography of roe deer (Capreolus capreolus L.) in different biogeographical regions in Europe. CR Acad Bulg Sci 69(5):579–584
Matosiuk M, Borkowska A, Świsłocka M, Mirski P, Borowski Z, Krysiuk K, Danilkin AA, Zvychaynaya EY, Saveljev AP, Ratkiewicz M (2014) Unexpected population genetic structure of European roe deer in Poland: An invasion of the mtDNA genome from Siberian roe deer. Mol Ecol 23(10):2559–2572. https://doi.org/10.1111/mec.12745
Mucci N, Mattucci F, Randi E (2012) Conservation of threatened local gene pools: landscape genetics of the Italian roe deer (Capreolus c. italicus) populations. Evol Ecol Res 14(7):897–920
Niedziałkowska M, Tarnowska E, Ligmanowska J, Jędrzejewska B, Podgórski T, Radziszewska A, Ratajczyk I, Kusza S, Bunevich AN, Danila G, Shkvyria M, Grzybowski T, Woźniak M (2021) Clear phylogeographic pattern and genetic structure of wild boar Sus scrofa population in Central and Eastern Europe. Sci Rep 11(1):9680. https://doi.org/10.1038/s41598-021-88991-1
Olano-Marin J, Plis K, Sönnichsen L, Borowik T, Niedziałkowska M, Jędrzejewska B (2014) Weak population structures in European roe deer (Capreolus capreolus) and evidence of introgressive hybridization with Siberian roe deer (C. pygargus) in Northeastern Poland. PLoS ONE 9(10):e109147. https://doi.org/10.1371/journal.pone.0109147
Plis K, Niedziałkowska M, Borowik T, Lang J, Heddergott M, Tiainen J, Bunevich A, Šprem N, Paule L, Danilkin A, Kholodova M, Zvychaynaya E, Kashinina N, Pokorny B, Flajšman K, Paulauskas A, Djan M, Ristić Z, Novák L, Jędrzejewska B (2022) Pan-European phylogeography of the European roe deer (Capreolus capreolus). Ecol Evol. https://doi.org/10.1002/ece3.8931
Pulliainen E (1980) Occurrence and spread of the roe deer (Capreolus capreolus L.) in eastern Fennoscandia since 1970. Memo Soc Fauna Flora Fenn 56:28–32
QGIS Development Team (2020) QGIS Geographic Information System. Open Source Geospatial Foundation. http://qgis.org
R Core Team (2021) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Randi E, Alves PC, Carranza J, Milošević-Zlatanović S, Sfougaris A, Mucci N (2004) Phylogeography of roe deer (Capreolus capreolus) populations: the effects of historical genetic subdivisions and recent nonequilibrium dynamics. Mol Ecol 13(10):3071–3083. https://doi.org/10.1111/j.1365-294X.2004.02279.x
Royo LJ, Pajares G, Álvarez I, Fernández I, Goyache F (2007) Genetic variability and differentiation in Spanish roe deer (Capreolus capreolus): A phylogeographic reassessment within the European framework. Mol Phylogenet Evol 42(1):47–61. https://doi.org/10.1016/j.ympev.2006.05.020
Sommer RS, Nadachowski A (2006) Glacial refugia of mammals in Europe: Evidence from fossil records. Mammal Rev 36(4):251–265. https://doi.org/10.1111/j.1365-2907.2006.00093.x
Sommer RS, Fahlke JM, Schmölcke U, Benecke N, Zachos FE (2009) Quaternary history of the European roe deer Capreolus capreolus. Mammal Rev 39(1):1–16. https://doi.org/10.1111/j.1365-2907.2008.00137.x
Steinbach P, Heddergott M, Weigand H, Weigand AM, Wilwert E, Stubbe M, Helm B, Campbell RE, Stubbe A, Frantz AC (2018) Rare migrants suffice to maintain high genetic diversity in an introduced island population of roe deer (Capreolus capreolus): evidence from molecular data and simulations. Mamm Biol 88(1):64–71. https://doi.org/10.1016/j.mambio.2017.11.009
Stronen AV, Jędrzejewska B, Pertoldi C, Demontis D, Randi E, Niedziałkowska M, Pilot M, Sidorovich VE, Dykyy I, Kusak J, Tsingarska E, Kojola I, Karamanlidis AA, Ornicans A, Lobkov VA, Dumenko V, Czarnomska SD (2013) North-south differentiation and a region of high diversity in European wolves (Canis lupus). PLoS ONE 8(10):e76454. https://doi.org/10.1371/journal.pone.0076454
Thulin C-G (2006) Microsatellite investigation of roe deer (Capreolus capreolus) in Scandinavia reveals genetic differentiation of a Baltic Sea Island population. European J Wildl Res 52(4):228–235. https://doi.org/10.1007/s10344-006-0047-1
Tsaparis D, Sotiropoulos K, Legakis A, Kotoulas G, Kasapidis P (2019) New phylogeographic insights support the distinctiveness and conservation value of the little-known Greek roe deer populations. Mamm Biol 96(1):23–27. https://doi.org/10.1016/j.mambio.2019.03.010
Veličković N, Djan M, Ferreira E, Stergar M, Obreht D, Maletić V, Fonseca C (2015) From north to south and back: The role of the Balkans and other southern peninsulas in the recolonization of Europe by wild boar. J Biogeogr 42(4):716–728. https://doi.org/10.1111/jbi.12458
Vernesi C, Pecchioli E, Caramelli D, Tiedemann R, Randi E, Bertorelle G (2002) The genetic structure of natural and reintroduced roe deer (Capreolus capreolus) populations in the Alps and central Italy, with reference to the mitochondrial DNA phylogeography of Europe. Mol Ecol 11(8):1285–1297. https://doi.org/10.1046/j.1365-294X.2002.01534.x
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer-Verlag, New York
Woodruff DS (2001) Populations, species, and conservation genetics. Encyclopedia of Biodiversity, pp 811–829
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159. https://doi.org/10.1093/genetics/16.2.97
Wright S (1965) The interpretationinterpretation of population structurepopulation structure by F-statisticsstatistics with special regardspecial regard to systemssystems of matingmating. Evol 1:395–420
Zvychainaya EY, Danilkin AA, Kholodova MV, Sipko TP, Berber AP (2011) Analysis of the variability of the control region and cytochrome b gene of mtDNA of Capreolus pygargus Pall. Biology Bull 38(5):434–439. https://doi.org/10.1134/S1062359011050189
Zvychaynaya EY, Volokh AM, Kholodova MV, Danilkin AA (2013) Mitochondrial DNA polymorphism of the European roe deerdeer, Capreolus capreolus (Artiodactyla, Cervidae), from the South-west of Ukraine. Vestn Zool 47(5):27–32. https://doi.org/10.2478/vzoo-2013-0044
Acknowledgements
The study was financed by the National Science Centre in Poland under the project No. 2013/11/B/NZ8/00884 for BJ. The paper forms part of the PhD thesis of KP. The research was supported by the European Commission’s project BIOGEAST No. PIRSES-GA-2009-247652. The authors thank the Polish Hunting Association, J. Žák, K. Turek, M. Mati, V. Sidarovich, I. Dykiy, D. Vishnevsky, M. Kolesnikov and V. Lobkov, as well as numerous hunters and hunting associations across Europe for their help in collecting samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Handling editor: Laura Iacolina.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Plis, K., Niedziałkowska, M., Borowik, T. et al. Mitochondrial DNA diversity and the population genetic structure of contemporary roe deer (Capreolus capreolus) in Europe. Mamm Biol 102, 1743–1754 (2022). https://doi.org/10.1007/s42991-022-00274-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42991-022-00274-y