
Abstract
To improve the thermostability of the lipase (r27RCL) from Rhizopus chinensis through rational design, a newly introduced buried disulfide bond F223C/G247C was proved to be beneficial to thermostability. Interestingly, F223C/G247C was also found to improve the alkali tolerance of the lipase. Subsequently, six other thermostabilizing mutations from our previous work were integrated into the mutant F223C/G247C, leading to a thermo-alkali-stable mutant m32. Compared to the wild-type lipase, the associative effect of the beneficial mutations showed significant improvements on the thermostability of m32, with a 74.7-fold increase in half-life at 60 °C, a 21.2 °C higher \(T_{50}^{30}\) value and a 10 °C elevation in optimum temperature. The mutated m32 was also found stable at pH 9.0–10.0. Furthermore, the molecular dynamics simulations of m32 indicated that its rigidity was enhanced due to the decreased solvent-accessible surface area, a newly formed salt bridge, and the increased ΔΔG values.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lipases (triacylglycerol lipase EC.3.1.1.3) are a class of enzymes that can catalyze the long-chain triglyceride hydrolysis, ester synthesis, and ester exchange reactions. Owing to their high catalytic efficiency and environmental friendliness, lipases have found important applications in various industries including food, oil, pharmaceuticals, paper, leather, detergent, and cosmetics [1]. Rhizopus sp. is one of the most important lipase producers [2]. The high sn-1, 3-positional specificity, enantioselectivity, and activity in non-aqueous media of Rhizopus lipases are the most desirable characters for the industrial applications [2]. However, most Rhizopus lipases are mesophilic and exhibit low thermostability [3]. Thus, endeavors have always been pursued to improve their thermostability, for even slight improvement can save huge costs during the manufacturing processes [4, 5].
The thermostability of the enzyme is mainly affected by salt bridge, hydrogen bond, hydrophobic stacking, and disulfide bond. Among these, disulfide bond is one of the most important interactions to stabilize the enzyme structure [6], which can reduce the conformational entropy of the unfolded state of protein, and provide an increase in the stabilization of the folded protein conformation [7, 8]. It has been reported that each native disulfide bond provided an increase of 2.3–5.2 kcal/mol for the stability of a protein [9]. Most of the stabilizing disulfide bond mutations were bridged between longer loop lengths (between 25 and 75 residues) and involved in the regions with higher flexibility (as assessed by the B-factor of the residues) [10, 11]. Disulfide by Design 2.0 (DbD2) is a web-based program to calculate the χ3 torsion angle and angle of all residue pairs, and assessed for the potential formation of a disulfide bond, assuming that the residues were mutated to cysteine. This approach has proved very powerful and successful in improving thermostability of industrial enzymes [12].
In our previous studies, the lipase (r27RCL) from Rhizopus chinensis CCTCC M201021 was highly expressed in Pichia pastrois [2, 13, 14], and great efforts have been put to improve its thermostability. First, directed evolution (two rounds error-prone PCR and two rounds DNA shuffling) was applied to engineer r27RCL, and a variant called S4-3 containing five mutations with 23 times longer half-life at 65 °C was obtained [15]. Subsequently, a surface disulfide bond was introduced to the lipase [16]. Thereafter, the predicted thermal unstable points were mutated and generated a mutant with a 41.7-fold increase in half-life at 60 °C and a 15.8 °C increase in \(T_{50}^{30}\) [17].
Herein, our goal was to further improve the thermostability of r27RCL. The potential buried disulfide bonds formed in r27RCL were selected and evaluated through the DbD2 program, together with the combination of thermostabilizing mutations reported in our previous studies, for better enhancement of the lipase thermostability. Moreover, molecular dynamic (MD) simulations were conducted to explore the mechanism of increased protein stability upon mutations. The root-mean-square deviation (RMSD), the root-mean-square fluctuation (RMSF), the radius of gyration (Rg) and the solvent-accessible surface area (SASA), as well as the secondary structure change of mutant, and the formation of salt bridges and H-bonds of r27RCL and m32 were analyzed at 313 K, while the ΔΔG values of r27RCL and m32 were determined at 313 K and 343 K.
Materials and methods
Plasmid, strains, cultures, and reagents
The restriction enzymes (Sal I, Dpn I), PrimeSTAR GXL DNA polymerase, Taq DNA polymerase, plasmid miniprep kit, and agarose gel DNA purification were supplied by Takara (Dalian, China). The expression plasmid pPIC9K and protein expression host strain P. pastoris GS115 (His− Mut+) were purchased from Invitrogen Company. Escherichia coli JM109 was used for plasmid amplification, and HPLC-purified DNA primers were purchased from Sangon Biotech (Shanghai, China). Recombinant plasmid pPIC9K-r27RCL was constructed by Yu et al.[8]. Yeast growth medium YPD/BMGY, yeast screening medium MD, and lipase induction medium BMMY were prepared according to “P. pastoris expression Kit” (Pichia Multi-Copy Expression Kit, version A, Invitrogen BV, The Netherlands). All chemicals were of analytical grade or higher quality and purchased from Sigma.
Production of r27RCL variants by rational design of interior disulfide bond
The crystal structure of r27RCL (PDB: 4L3W) at 2.2 Å resolution [18] was resolved and used DbD2 online (https://cptweb.cpt.wayne.edu/DbD2/index.php) [12] to predict its potential disulfide bonds under the calculated condition with bond energy (χ3 angle) around + 97 (± 30). Within all candidates, those disulfide bonds located within 3–5 Å distance from the molecular surface, but not within the range of 6–8 Å from the central amino acid S172 of the lipase catalytic triad (S172-H284-D231), were our research targets. PyMOL software was used to analyze the location of the mutations, and those with obvious irrational structures were discarded. If different substitutions were predicted to form disulfide bonds with a certain amino acid, the one with a larger B-factor value was selected.
Construction of the variants
Mutations were performed by whole-plasmid PCR using pPIC9K-r27RCL as the template and the primers have been listed in Table S1. The 25 μL reaction mixture included 1/5 volume of PrimeSTAR GLX polymerase buffer, 25 ng template, 125 ng of each primer, 100 μM of each dNTP, and 1 U of PrimeSTAR GLX polymerase. PCR conditions were as follows: 95 °C for 3 min; 95 °C for 20 s, 54–59 °C for 1 min, 72 °C for 11 min, 20 cycles in total, and then 72 °C for 10 min. PCR products were digested by 10 U Dpn I at 37 °C for 30 min to eliminate the template plasmid. The digested product was transformed into E. coli JM109 competent cells for nick repair and plasmid amplification. After sequencing, Sal I-linearized mutated plasmids were transformed to P. pastoris GS115 competent cells by electroporation, and then incubated in a selective MD plate at 28 °C for 48 h. The positive variants were detected through PCR amplification of genomic DNA with the primers of 5′AOX/3′AOX.
Expression and purification of the lipase variants
The positive variants were inoculated into 25 mL BMGY medium and incubated at 28 ℃ to reach an OD600 of 2–6. Then, the cells were centrifuged and re-suspended in 100 mL BMMY medium, with supplemental addition of 1% (v/v) methanol to the medium every 24 h to induce expression. After 84 h induction, the medium was centrifuged at 7000 × g for 30 min and the histidine-tagged mutant lipase in the supernatant was subjected to the 0.22 μm syringe filters and the Nickle-affinity column (Ni Sepharose 6 Fast Flow) chromatography connected to an ÄKTA purifier (GE Co., Ltd). Subsequently, the purified lipase was concentrated by ultrafiltration through a 10-kDa membrane (Millipore, USA) and analyzed by SDS-PAGE. The purified lipase was desalinized and dissolved in storage buffer (150 mM NaCl, 25 mM Tris–HCl, pH 8.0, 10% glycerol), followed by storing at − 20 °C for further use within one month.
Property characterization of lipase
Lipase activity assay was carried on emulsified p-nitrophenyl butyrate (pNPB) [19] at the optimum temperature and pH, while the product was detected at 410 nm using a UV–Vis spectrophotometer. One unit of enzyme was defined as the amount of enzyme releasing 1 μmol of p-nitrophenol per minute under the assay conditions. The optimal temperature was determined by measuring the enzyme activity at pH 8.0 under various temperatures from 30 °C to 60 °C using pNPB as the substrate. The optimum reaction pH was calculated at pH values from 5.0 to 10.0 at the optimum temperature. For pH stability, the lipase was incubated in different 50 mM buffers, including phosphate buffer (pH 6.0–7.5), Tris–HCl buffer (pH 8.0–9.0), carbonate buffer (pH 9.5–10.0), and sodium carbonate sodium hydroxide buffer (pH 10.5–11.0) at room temperature for 60 min, and then the residual activity was measured and represented as a percentage of the initial activity. To determine the thermostability of each variant, the purified lipases were incubated at 25–70 °C for 30 min, followed by cooling in the ice bath for 20 min and equilibration at room temperature for 5 min. Then, their residual activities were measured and represented as a percentage of the initial activity. Thermostability, represented as \(T_{50}^{30}\) values, was defined as the temperature at which the enzyme remained 50% of its activity after 30 min of heat treatment. Then, the half-lives of variants were detected by calculating the thermal denaturation time at which lipase lost 50% catalytic activity at 60 °C, 65 °C or 70 °C. The Circular Dichroism (CD) spectra were recorded at wavelengths from 190 to 260 nm at a speed of 60 nm per min, using Xe lamp (100 W) and 1 mm path length quartz cuvette, while the spectrum of a buffer blank was subtracted. To construct the melting curves and determine the melting temperature (Tm) of mutants, samples were heated at 1 °C per min from 20 °C to 80 °C and the CD ellipticity signal at 220 nm, which showed the maximal change with temperature, was monitored. The lipase concentration used in the CD analysis was 0.4 ± 0.05 mg/mL. Kinetic parameters of variants toward pNPB (10–100 mM) hydrolysis reaction were determined at their optimum conditions, and non-linear regression software was used for the calculation of kinetic parameters [20].
Generation of a thermostable combinatorial variant m32
The combinatorial variant m32 contained mutations of S85C/Q145C/F223C/G247C/S142A/D217V/Q239F/S250Y was generated by the recombination of beneficial mutations including S85C/Q145C (by DbD2) [16] and S142A/D217V/Q239F/S250Y (by FoldX) [17]. Their primers have been shown in Table S2. After expression in P. pastoris, the enzyme properties of the purified m32 were characterized.
Molecular dynamic simulation
The protein sequence of mutant m32 was uploaded to the Swiss-Model protein automated modeling program for three-dimensional structure generation based on the crystal structure of 4L3W [21]. All simulations were performed with GROMACS 2019.3 simulation software (www.gromacs.org) [22], using the Gromos 54A7forcefeld [23]. Each system was solvated with explicit solvent SPC/E water molecules in a cubic box which left a 1.2 nm distance around the solute. Several Na+ cations were added to neutralize the systems. Each simulation was performed for 100 ns at 313 K using a time step of 2 fs with periodic boundary conditions. The steepest descent method was used to minimize the energy. Long-range electrostatic effects were handled using PME and the system was weakly coupled to an external bath using Berendsen’s method [24, 25]. H-bonds were constrained with LINCS [26]. GROMACS built-in tools and VMD 1.9.3 [27] were used to analyze the RMSD, the RMSF, the Rg and the SASA, the secondary structure changes, the intermolecular interactions (salt bridge and H-bond), and the ΔΔG of lipase structure during MD trajectory. Structure visualizations were performed using VMD.
Results and discussion
Rational design of buried disulfide bonds of the lipase
R. chinensis lipase r27RCL contains 296 amino acids, forming nine α-helices and eight β-sheets in the secondary structure. There naturally existed seven cysteines in its protein sequence form three disulfide bonds (C56–C295, C67–C70, and C262–C271) in the structure, which play an important role in the correct folding and structural stabilization of r27RCL.
Based on algorithms created for disulfide identification in protein fold recognition method, DbD2 has been successfully applied for disulfide bond prediction [12]. For r27RCL, twenty potential disulfide bonds were found to meet the conformational criteria by this prediction method. It is well known that heat denaturation of protein begins from the enzyme surface [28, 29]. In our previous study, we designed some potential disulfide bonds on the surface of r27RCL and screened out S85C-Q145C for the improvement of the lipase thermostability without the damage of lipase catalytic activity [16]. However, the mutations buried in the lipase structure have higher possibilities to affect the properties of the lipase of different aspects, such as thermostability, catalytic ability, stereoselectivity, substrate specificity, and the tolerance to severe pH conditions. Therefore, we focused on the potential buried disulfide bond inside of the enzyme structure in the current study. On one hand, we continued to seek the mutation sites capable of stabilizing lipase. On the other hand, we intended to investigate whether the formation of the buried disulfide bonds affects other properties of lipase. Thus, the predicted disulfide bond situated within 3–5 Å depth from the lipase molecular surface and more than 6–8 Å away from the catalytic triads was the target. According to this rule, we selected five pairs of disulfide bond mutations from a total of 20 potential disulfide bonds predicted by DbD2 for further research (Table 1 and Fig. 1).

Three-dimensional structures prior (a) and after (b) mutation of all evaluated buried disulfide bonds in the lipase molecular. The catalytic triad (S172-H284-D231) are shown in green; The short α-helix, called the “lid” region, is shown in yellow; The five evaluated disulfide bonds were shown in red (A133C/F212C), in orange (V216C/P246C), in blue (F223C/G247C), in pink (G182C/I199C), and in violet (A36C/V259C)
Preliminary screening of the variants for improved thermostability
The mutants introduced with additional disulfide bonds were subjected to heat treatment at 55 °C for 30 min as a preliminary screening. The mutated lipases were diluted to 0.125 ± 0.025 μg/mL and subjected to the enzymatic activity assay. The residual activity of r27RCL was defined as 100%, and the residual activities of different mutants were compared with that of r27RCL (Table 1). Among the five mutants, two (m17/18 and m19/20) exhibited increased residual activity, whereas the left three (m15/16, m21/22, and m23/24) showed lower residual activities than that of r27RCL after thermal inactivation. The specific activities of m15/16, m21/22, and m23/24 were also decreased after mutation. Meanwhile, m19/20 showed a very slight improvement in thermostability but a 37.4% decline in the specific activity. Thus, we selected m17/18 as the target for further research, which retained 90.8% catalytic activity and a 1.78-fold increase in residual activity after heat treatment. The SDS-PAGE of the purified r27RCL and m17/18 has been shown in Fig. S1.
The thermostability and enzymatic property of m17/18
The effects of temperature on the activity and the stability of the variant m17/18 were determined. As shown in Fig. 2a, r27RCL and m17/18 both exhibited their maximal activities at 40 °C. The wild-type r27RCL lost half of its activity when the temperature was elevated to higher than 45 °C, whereas the mutant m17/18 maintained > 60% activity at 50 °C. Their thermostabilities were examined after 30 min heat incubation at temperature ranges from 25 °C to 70 °C (Fig. 2b). For r27RCL, it maintained > 50% activity below 45 °C, sharply declined at 50 °C, with quick and complete inactivation. However, m17/18 retained > 60% and 30% activities, respectively, after heat incubations at 50 °C and 55 °C for 30 min, but it was completely inactivated when the temperature was elevated to 60 °C.

Effect of temperature and pH on the activity and the stability of r27RCL, m17/18, and m32. a The optimum temperature. The activity of r27RCL was determined at 30–65 °C and pH 8.0, and the activity of m17/18 and m32 was determined at 30–65 °C and pH 9.0 using pNPB as the substrate; b The temperature stabilities of r27RCL, m17/18 and m32 were determined at 40 °C/pH 8.0, 40 °C/pH 9.0, and 50 °C/pH 9.0, respectively, after incubation for 30 min at temperatures of 25 to 70 °C; c The optimum pH. The activity of r27RCL and m17/18 was determined at pH 6.0–10.0 and 40 °C, and the activity of m32 was determined at pH 6.0–10.0 and 50 °C; d The pH stabilities of r27RCL, m17/18 and m32 were determined at 40 °C/pH 8.0, 40 °C/pH 9.0, and 50 °C/pH 9.0, respectively, after being incubated in pH 6.0 − 11.0 50 mM buffers for 60 min. Error bars indicated the standard deviation of three biological replicates
As shown in Table 2, the half-life of the mutant m17/18 at 60 °C was enhanced by 19.1-fold (16.2 min), compared to the wild type (0.85 min), and represented a great increase in \(T_{50}^{30}\) of 8.5 °C over the wild type. The Tm value of m17/18 was 3.9 °C higher than that of r27RCL (Table 2, Fig. S2b).
Thereafter, the effects of pH on the activity and the stability of m17/18 were determined. Both r27RCL and m17/18 were alkaline lipases, and r27RCL had an optimum pH of 8.0 (Fig. 2c), while mutant m17/18 lifted its optimum pH value to 9.0. Moreover, m17/18 had a broader catalytic range (under which condition > 50% catalytic ability remained) from pH 7.0 to 10.0 compared to that of r27RCL (pH 7.5 to 9.5). At pH 10.0, the wild type was completely inactivated, whereas m17/18 retained > 30% catalytic activity. As shown in Fig. 2d, m17/18 represented remarkably enhanced alkali tolerance, with > 80% residual activity retained after incubation at pH 10.0 for 30 min. These results indicated that the buried disulfide bond exerted great impacts on both thermostability and alkali tolerance.
Kinetic parameters were further measured for r27RCL and m17/18 from their initial reaction rates. The kcat/Km value of m17/18 was 3.53-fold lower than that of r27RCL (Table 3). This result revealed that m17/18 had a weaker affinity to the substrate of pNPB and a slower reaction rate compared to that of r27RCL. The distance between F223C/G247C to the catalytic triads was about 9 Å; however, it still affected the substrate affinity and the kcat value, improving the thermostability at the expense of catalytic efficiency of the lipase.
As demonstrated in this study, the newly added disulfide bond of F223C/G247C was buried within the lipase molecule and displayed dramatic impacts both on thermostability and alkali tolerance. The disulfide bond F223C-G247C situates within 3–5 Å deep from the surface, not very deep in the lipase molecule, and about 8 Å away from the lipase triad. The result was in agreement with the notion that the buried disulfide bonds were more likely to be located fairly close to the protein surface with a depth of 3–5 Å [11]. The disulfide bond F223C-G247C situates at the loop connecting the 9th β-sheet and the loop between the 10th β-sheet and the 4th η-random coil [18]. The crosslinking between two loops through the disulfide bond F223C-G247C contributed to the increased thermostability by reducing the conformational entropy of the unfolded state [8]. Another possible explanation was that the backbone atoms in the regions containing the respective mutated sites moved closer, which greatly increased the overall packing of lipase [30, 31]. Similar effects were observed in the case of disulfide bond mutants of Bacillus xylanase [32] and barnase [33]. Besides thermostability, F223C/Q247C also led to better alkali tolerance. The increased overall packing and rigidity of the lipase structure might also contribute to the better alkali tolerance of the mutant. Among five predicted disulfide bond mutations, four (A36C/V259C, A133C/F212C, G182C/I199C, and V216C/P246C) showed destabilization effects on the lipase. The disulfide bond prediction accuracy is also low in other reports. For example, only four out of 16 lytic polysaccharide monooxygenase variants with additional disulfide bonds predicted by MODiP and DbD displayed improved thermostability [34]. The rest four disulfide bond mutations showed destabilization effects of lipase mainly due to the atypical stereochemical features or the loss of existing interactions. A good stereochemistry of the introduced disulfide was indeed essential since the atypical stereochemical features can lead to higher strain energy, which can offset the stabilizing effect of the disulfide cross-linking [35]. Moreover, the loss of existing interactions after substituting with cysteine, and the introduction of unfavorable steric contacts can also have a destabilizing effect [36].
Generation of a thermostable variant by combinatorial mutagenesis and the property analysis
To further enhance the thermostability of the lipase, we integrated six other beneficial mutations, including a pair of surface disulfide bond sites (S85C/Q145C) [16] and four mutations (S142A/D217V/Q239F/S250Y) [17] to m17/18 (F223C/G247C), and generated a mutant named m32 (S85C/Q145C/F223C/G24C/S142A/S250Y/Q239F/D217V). In our previous work, we reported that the addition of a disulfide bond S85C/Q145C on the surface of r27RCL was very efficient in stabilizing the lipase structure at higher temperatures, neither compromising catalytic ability nor changing the pH optimum and pH stability of the lipase [16]. Four beneficial single-point mutations (S142A/D217V/Q239F/S250Y) in r27RCL were screened out through a rational design approach of FoldX. By combining these four mutations, the variant exhibited a 5 °C increase in Topt and 15.8 °C elevation in \(T_{50}^{30}\) compared to that of r27RCL, without compromising its enzyme activity [17].
First, m32 was expressed in P. pastoris and purified. The SDS-PAGE of the purified m32 has been shown in Fig. S1. The specific activity of m32 was comparable to that of m17/18, reaching 598 U/mg (Table 1). As shown in Fig. 2a, m32 exhibited a 10 ℃ higher Topt value (50 ℃) compared to that of r27RCL (40 ℃). Subsequently, m32 also displayed significantly increased thermostability from 25 ℃ to 70 ℃ (Fig. 2b). It retained nearly 90% activity after heat incubation at 50 ℃ for 30 min, whereas the wild type had already completely inactivated. At higher temperatures, m32 still showed great thermo-tolerance and retained about 85%, 80%, 60%, and 30% activity after 30 min heat incubation at 55 ℃, 60 ℃, 65 ℃, and 70 ℃, respectively.
The effects of pH on the activity and stability of m32 were subsequently measured. The pHopt of m32 was lifted from 8.0 of the wild type to 9.0, which was identical to that of m17/18 (Fig. 2c). As mentioned above, m17/18 processed broader catalytic range (from pH 7.0 to 10.0), while m32 remained in a similar catalytic range from pH 7.5 to 9.0 with r27RCL, indicating that the introduction of other beneficial mutations narrowed the catalytic range of m17/18 (Fig. 2c). However, m32 possessed a greater alkali tolerance than m17/18. m32 remained > 70% activity after incubation for 30 min from pH 7.0 to 10.0. r27RCL was most stable at pH 8.0; however, m32 preferred pH 9.0–10.0 (Fig. 2d). The pHopt and pH stability of m32 were identical to that of m17/18, revealing that the property of m32 was greatly affected by the newly introduced disulfide bond of F223C/G247C.
As shown in Fig. 3, the thermal inactivation of m32 and r27RCL was characterized by incubation at 60 °C, 65 °C, and 70 °C. The curves of r27RCL at each temperature were much steeper than those of m32. r27RCL lost 100% enzyme activity after heat treatment at 60 °C and 65 °C for 10 min; however, mutant m32 retained > 50% and 40% residual activity after undergoing thermal inactivation at 60–65 °C for 60 min (Fig. 3a, b). At 70 °C, r27RCL was denatured very fast and lost 100% of its activity after 2.5 min, whereas, m32 retained half of its activity after 7.5 min, and even 10% after 20 min heat treatment (Fig. 3c). Table 2 showed that m32 exhibited a significant increase (74.7-fold) of its half-life (63.5 min) at 60 °C and a 21.2 °C higher \(T_{50}^{30}\) value (66.2 °C), compared to those of r27RCL. At 65 °C and 70 °C, the half-lives of m32 reached 56.5 min and 7.5 min, respectively. The Tm value of m32 was 11.5 °C higher than that of r27RCL (Table 2, Fig. S2b). The significantly enhanced thermostability of m32 proved the beneficial associative effect of the combination of multiple beneficial mutations.

The thermostability of r27RCL and m32 after heat-treatment at 60 ℃ (a), 65 (b), and 70 ℃ (c) for different times. The residual activity of r27RCL was measured at 40 ℃ and pH 8.0, and the residual activity of m32 was measured at 50 ℃ and pH 9.0. Error bars indicated the standard deviation of three biological replicates
As for the kinetic properties, the mutant m32 exhibited 2.22-fold lower kcat/Km value than that of r27RCL (Table 3). The mutation of F223C/G247C can probably be the major cause of the decrease of catalytic efficiency of m32, since other six mutations in m32 were located more than 13 Å away from the catalytic triad with minimum effects on the catalytic efficiency. In addition, the newly added six mutations brought a slight increase of catalytic efficiency of m32, compared with m17/18.
The associative effect of eight mutations
The integrated strategy in this study, including FoldX, MD, and disulfide bond design to generate four beneficial single-point mutations and two pairs of disulfide bonds, appeared to be a very efficient and universal way to further enhance the thermostability of the lipase without laborious work.
The obtained final mutant m32 showed greatly improved thermostability and alkali tolerance, indicating the significant associative effect of these beneficial mutations. Table 4 summarized the enzymatic properties of m32 and its individual mutation, demonstrating that the desirable lipase property of m32 was inherited from these mutations. In summary, m32 was presented as a thermos-alkali-stable mutant, and these desirable properties endowed it with great potential in industrial applications.
Elucidation of thermostability mechanism by protein modeling and B-factor analysis
Based on the crystal structure of the wild-type (PDB: 4L3W), the protein sequence of m32 was uploaded to the Swiss-Model protein automated modeling program for three-dimensional structure generation. As shown in Fig. 4, except F223C/G247C, the other six substitutions were all located on or close to the lipase surface and far from the catalytic triad. Mutation F223C/G247C was buried inside of the lipase and about 9 Å away from the catalytic triad.

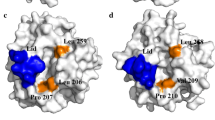
Substitutions in m32. The catalytic triad residues (S172-H284-D231) are shown in green. The short α-helix, called the “lid” region, is shown in yellow. S142A, S250Y, Q239F, and D217V were shown in red. The disulfide bonds of S85C-Q145C and F223C-Q247C were shown in blue and orange, respectively
The disulfide bond S85C-Q145C situates at the flexible surface regions between the 2nd β-sheet and the 4th α-helix, which is more likely to generate beneficial mutants as the relax of the protein structure can happen without incurring a significant energetic penalty [16]. As for the wild type, residues 85, 142, and 145 were all hydrophilic residues. After mutation (Fig. 5), the hydrophobic interactions between 85C, 142A, and 145C and the surrounding hydrophobic residues of F139, L140, L86, L87, A176, A178, L179, L180, and A181 were strengthened, along with the introduced disulfide bonds, resulted in tighter packing of these hydrophobic regions.

The hydrophobic interactions after mutation of S142A, S85C, and Q145C in m32. a The hydrophilic residues before mutation are shown in yellow; b The hydrophobic residues are shown in blue
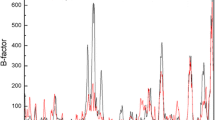
The B-factor is commonly used to represent residue flexibility and is considered as a reliable parameter to select appropriate residues for single-point mutation [37] or disulfide bond mutations [34], and the residues with higher B-factors are more likely to generate stabilizing mutations. Herein, we visualized the B-factor distribution of r27RCL (Fig. S3), and the top 20% most flexible residues (with B-factor > 35) are shown in red. Among these eight mutations, only two of them (residues 239 and 145) were within the 20% flexible residues, and residue 142 was very close to the highly flexible residue of 141 (with B-factor > 35). This result suggested that B-factor can be used as a partial assistant in selecting hot-spot, meanwhile other factors should also be considered [38]. Since the B-factor was measured based on the crystal state of the protein, under which temperature, the protein was far from its physiological state [39], thus, B-factor was not able to represent the dynamics of protein residues in solution [40]. Besides, the crystal packing also affected the dynamic information extracted from the B-factor [40].
Elucidation of thermostability mechanism by MD simulations
To explore the effect of mutations on the structural stability of m32, the values of RMSD, RMSF, Rg, and SASA of m32 and r27RCL were calculated from 0 to 100 ns at 313 K. RMSD is an important parameter for estimating the thermal fluctuation of the protein conformation, which reflects the unfolding enthalpy and entropy change of enzyme [41]. The RMSF represents the fluctuation of an atom relative to its average position, and higher RMSF values mean increasing instability of the conformation of residues/regions. Rg is related to the structural stability of a protein, and the smaller value of Rg marked a more stable protein structure. SASA value represents the aggregation-related properties of a protein, and smaller SASA values meant a tighter packed structure of a protein. As shown in Fig. 6a, after the sudden increase in the first 20 ns at the beginning, the average RMSD values were around 0.3 nm. Moreover, the values of r27RCL from 80 to100 ns suddenly increased dramatically and reached 0.45 nm. The sudden increase on RMSD value of r27RCL after 80 ns might result from the flexibility of the lid domain. Q145 is located in an alpha helix connected with the lid domain in yellow (Fig. 5). When the disulfide bonds between S85C and Q145C were constructed, the flexibility of the lid domain might be constrained, leading to slightly lowered RMSD value of m32 than that of r27RCL, and there is no sudden change of RMSD value for m32 at the end of MD simulation. The Rg and SASA values of r27RCL and m32 at 313 K were evaluated to further explore the structural stability of r27RCL and m32 (Fig. 6b, c). The overall Rg values of m32 were slightly lower than that of r27RCL during 5 to100 ns. The average Rg of r27RCL stayed around 1.86 nm, and the average Rg value of m32 was 1.83 nm (Fig. 6b). The average SASA of r27RCL was 140 nm2, while the SASA value of m32 decreased to 133 nm2 (Fig. 6c). The decreased Rg and SASA values of m32 indicated more tightly packed structure of m32, according to the 100 ns simulation [42].

The comparison of MD simulation results of r27RCL and m32 at 313 K. a RMSD, b Rg, c SASA
The RMSF values of r27RCL and m32 at 313 K were compared in Fig. S4. The residues 85, 142, and 145 displayed much lower RMSF values after mutation, while no obvious changes were detected for the RMSF values of the rest five residues 217, 239, 223, 247, and 250 after mutations. However, the overall RMSF values of m32 were much lower than that of the wild type, especially in some highly flexible areas, including residues of S23-E32, T82-N91, R108-F122, E254-A257, and L285-G293. The overall narrowed fluctuations of m32 proved its remarkably enhanced thermostability than that of r27RCL.
To further understand the molecular basis of the conformational changes in m32, we analyzed the differences of the following parameters between r27RCL and m32, including secondary structure, intermolecular interactions (salt bridges and H-bonds), and ΔΔG. The α-helix, β-sheet, turn, and the coil of r27RCL and m32 during 100 ns MD simulations at 313 K were evaluated (Fig. S5). Stable secondary structures represent the stability of a protein to a certain extent. The residue numbers included in α-helix, β-sheet, turn structures were generally stable along the 100 ns MD simulations, with a little change exhibited. The difference of the conformational change between m32 and r27RCL could also be proved by the altered spectrum of m32 at wavelengths from 190 to 260 nm (Fig. S2a).
In Fig. S6, at the first 50 ns, the numbers of internal hydrogen bonds of m32 were slightly lower than those of r27RCL. However, from 80 to 100 nm both numbers are relatively similar and stable during the MD simulation.
Salt bridges are very important in stabilizing protein structure, as the buried salt bridges or those located in hydrophobic core contributed > 4 kcal/mol [43]. However, mutations in protein might destroy or introduce a new salt bridge. Herein, we evaluated the possible noncovalent interactions for salt bridge formation (the distance between two expected atoms was < 3.2 Å) in r27RCL and m32 [44]. Besides distance, we also considered the stability of salt bridges, for which distance was continuously kept within 3.2 Å. From the 100 ns trajectories shown in Fig. 7, in addition to the common three existing salt bridges (Asp231:His284, Glu292:His284, and Asp283:His228) in both r27RCL and m32 structures, m32 formed an extra salt bridge (Glu292:His171), compared to r27RCL. As shown in Fig. 7a, the distance between residues Glu292 and His171 in r27RCL was > 3.2 Å for most of the time, which was too weak to form a salt bridge. However, the distance between residues Glu292 and His171 in m32 was successively < 3.2 Å since 40 ns (Fig. 7b), indicating the formation of a new salt bridge between these two residues. This newly formed salt bridge between Glu292-His171 might be a major contribution to the thermostability of m32.

The presence of salt bridges in r27RCL (a) and m32 (b) during 100 ns MD simulations at 313 K
Herein, we analyzed the variations in ΔΔG during 100 ns of MD simulations for m32 and r27RCL at 313 K and 343 K (Fig. 8). The changes in ΔG between the folded and unfolded states were calculated every 10 ns within the 100 ns of MD simulation at both 313 K and 343 K. The changes in ΔΔG for r27RCL and m32 revealed the tendency of m32 for increased stability both at 313 K and 343 K, while no such obvious tendency was found for r27RCL.

FoldX results for r27RCL and m32. The ΔΔG changes for the two lipase conformations along 100 ns MD simulations at 313 K (a) and 343 K (b)
Conclusion
In this study, we generated a mutant m32 with simultaneously improved thermostability and alkali tolerance by rational design, including engineering of a buried disulfide bond and the combination of previously reported beneficial mutations. The introduction of a disulfide bond F223C/G247C within 3–5 Å away from the lipase surface exhibited enhanced thermostability with a 19.1-fold increase in half-life at 60 °C and an 8.5 °C higher \(T_{50}^{30}\) value compared to that of r27RCL. Most importantly, F223C/G247C simultaneously improved the alkali tolerance of lipase. Thereafter, we combined four beneficial mutations and a disulfide bond on the lipase surface to F223C/G247C. The associative effect of the beneficial mutations was so significant that the combinatorial m32 accumulated the favorable properties of all individual mutations, and exhibited highly increased thermostability and alkali tolerance. MD results indicated that the increased overall packing and rigidity of the lipase structure contributed to the higher thermostability of the mutant, which might be also the reason for its better alkali tolerance. However, the mechanism of alkali tolerance of m32 needs to be further explored in our future study through crystal structure analysis.
References
Hasan F, Shah AA, Hameed A (2006) Industrial application of microbial lipase. Enzyme Microb Tech 39:235–251
Yu X, Xu Y, Xiao R (2016) Lipases from the genus Rhizopus: characteristics, expression, protein engineering and application. Prog Lipid Res 64:57–68
Kamiya N, Ogawa T, Nagamune T (2001) Enhancement of apparent thermostability of lipase from Rhizopus sp. by the treatment with a microbial transglutaminase. Biotechnol Lett 23:1629–1632
Ariaeenejad S, Maleki M, Hosseini E, Kavousi K, Moosavi-Movahedi AA, Salekdeh GH (2019) Mining of camel rumen metagenome to identify novel alkali-thermostable xylanase capable of enhancing the recalcitrant lignocellulosic biomass conversion. Bioresource Technol 281:343–350
Chirstopher LP, Zambare VP, Zambare A, Kumar H, Malek L (2015) A thermos-alkaline lipase from a new thermophile Geobacillus thermodenitrificans AV-5 with potential application in biodiesel production. J Chem Technol Biotechnol 90:2007–2016
Flory PJ (1956) Theory of elastic mechanisms in fibrous proteins. J Am Chem Soc 78:5222–5235
Masazumi M, Giovanni S, Brian WM (1989) Substantial increase of protein stability by multiple disulfide bonds. Nature 342:291–293
Pace CN, Grimsley GR, Thomson JA, Barnett BJ (1988) Conformational stability and activity of ribonuclease T1 with zero, one, and two intact disulfide bonds. J Biol Chem 263:11820–11825
Tidor B, Karplus M (1993) The contribution of cross-links to protein stability: a normal mode analysis of the configurational entropy of the native state. Proteins 15:71–79
Melnik BS, Povarnitsyna TV, Glukhov AS, Melnik TN, Uversky VN, Sarma RH (2012) SS-stabilizing proteins rationally: intrinsic disorder-based design of stabilizing disulfide bridges in GFP. J Biomol Struct Dyn 29:815–824
Dani VS, Ramakrishnan C, Varadarajan R (2003) MODIP revisited: re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng 16:187–193
Craig DB, Dombkowski AA (2013) Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics 14:346–352
Xu Y, Wang D, Mu XQ, Zhao GA, Zhang KC (2002) Biosynthesis of ethyl esters of short-chain fatty acids using whole-cell lipase from Rhizopus Chinesis CCTCCM201021 in non-aqueous phase. J Mol Catal B-Enzym 18:29–37
Yu X, Wang L, Xu Y (2009) Rhizopus chinensis lipase: Gene cloning, expression in Pichia pastoris and properties. J Mol Catal B-Enzym 57:304–311
Yu XW, Wang R, Zhang M, Xu Y, Xiao R (2012) Enhanced thermostability of a Rhizopus chinensis lipase by in vivo recombination in Pichia pastoris. Microb Cell Fact 11:102–112
Wang R, Yu XW, Xu Y (2018) Rationale design of disulfide bond in Rhizopus chinensis lipase to improve thermostability. Microbiol China 11:2311–2319
Wang R, Wang S, Xu Y, Yu XW (2020) Enhancing the thermostability of Rhizopus chinensis lipase by rational design and MD simulations. Int J Biol Macromol 160:1189–1200
Zhang M, Yu X, Swapna GV, Xiao R, Xu Y (2018) G. Montelione, Backbone and Ile-δ1, Leu, Val methyl chemical shift assignments for Rhizopus microsporus var. chinensis lipase. Bio NMR Assigm 12:63–68
Lee D, Koh Y, Kim K, Kim B, Choi H, Kim D, Suhartono MT, Pyun Y (1999) Isolation and characterization of a thermophilic lipase from Bacillus thermoleovorans ID-1. FEMS Microbiol Lett 179:393–400
Zorn H, Li QX (2017) Trends in food enzymology. J Agric Food Chem 65:4–5
Waterhouse A, Bertoni M, Bienert S et al (2018) SWISS-MODEL: homology modeling of protein structures and complexes. Nucleic Acids Res 46:W296–W303
Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX1 2:19–25
Schmid N, Elchenberger AP, Choutko A, Riniker S, Winger M, Mark AE, van Gunsteren WF (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J 40:843–856
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N⋅log (N) method for Ewald sums in large systems. J Chem Phys B 98:98–110
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1998) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38
Eijsink VGH, Bjørk A, Gåseidnes S, Sirevåg R, Synstad B, van den Burg B, Vriend G (2004) Rational engineering of enzyme stability. J Biotechnol 113:105–120
Johannes TW, Woodyer RD, Zhao HM (2005) Directed evolution of a thermostable phosphite dehydrogenase for NAD(P)H regeneration. Appl Environ Microb 71:5728–5734
Pjura PE, Matsumura M, Wozniak JA, Matthews BW (1990) Structure of a thermostable disulfide-bridge mutant of phage T4 lysozyme shows that an engineered cross-link in a flexible region does not increase the rigidity of the folded protein. Biochemistry 29:2592–2598
Jacobson RH, Matsumura M, Faber HR, Matthews BW (1992) Structure of a stabilizing disulfide bridge mutant that closes the active-site cleft of T4 lysozyme. Protein Sci 1:46–57
Wakarchuk WW, Sung WL, Campbell RL, Cunningham A, Watson DC, Yaguchi M (1994) Thermostabilization of the Bacillus circulans xylanase by the introduction of disulfide bonds. Protein Eng 7:1379–1386
Clarke J, Henrick K, Fersht AR (1995) Disulfide mutants of barnase, I: changes in stability and structure assessed by biophysical methods and X-ray crystallography. J Mol Biol 253:493–504
Tanghe M, Danneels B, Last M, Beerens K, Stals I, Desmet T (2017) Disulfide bridges as essential elements for the thermostability of lytic polysaccharide monooxygenase LPMO10C from Streptomyces coelicolor. Protein Eng Des Sel 30:401–408
Katz BA, Kossiakoff A (1986) The crystallographically determined structures of atypical strained disulfides engineered into subtilisin. J Biol Chem 261:15480–15485
Mitchinson C, Wells JA (1989) Protein engineering of disulfide bonds in subtilisin BPN’: enhanced stabili zation through the introduction of two cysteines to form a disulfide bond. Biochemistry 28:4807–4815
Reetz MT, Carballeira JD, Vogel A (2006) Iterative saturation mutagenesis on the basis of B-factors as a strategy for increasing protein thermostability. Angew Chem Int Edit 45:7745–7751
Zhang X, Yang G, Zhang Y, Xie Y, Withers SG, Feng Y (2016) A general and efficient strategy for generating the stable enzymes. Sci Rep 6:33797
Teilum K, Olsen JG, Kragelund BB (2009) Functional aspects of protein flexibility. Cell Mol Life Sci 66:2231–2247
Pezeshgi Modarres H, Dorokhov BD, Popov VO, Ravin NV, Skryabin KG, Dal Peraro M (2015) Understanding and engineering thermostability in DNA Ligase from Thermococcus sp 1519. Biochemistry 54:3076–3085
Liu Z, Lemmonds S, Huang J, Tyagi M, Hong L, Jain N (2018) Entropic contribution to enhanced thermal stability in the thermostable P450 CYP119. PNAS 115:E10049–E10058
Badieyan S, Bevan DR, Zhang C (2012) Study and design of stability in GH5 cellulases. Biotech Bioeng 109:31–44
Anderson DE, Becktel WJ, Dahlquist FW (1990) pH-induced denaturation of proteins: a single salt bridge contributes 3–5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry 29:2403–2408
Donald JE, Kulp DW, DeGrade WF (2011) Salt bridge: geometrically specific, designable interactions. Proteins 79:898–915
Acknowledgements
Financial support from the National Natural Science Foundation of China (31671799), the Six Talent Peaks Project in Jiangsu Province (NY-010), the High-end Foreign Experts Recruitment Program (GDT20153200044), the National First-Class Discipline Program of Light Industry Technology and Engineering (LITE2018-09), and the 111 Project (111-2-06) are greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, R., Wang, S., Xu, Y. et al. Engineering of a thermo-alkali-stable lipase from Rhizopus chinensis by rational design of a buried disulfide bond and combinatorial mutagenesis. J Ind Microbiol Biotechnol 47, 1019–1030 (2020). https://doi.org/10.1007/s10295-020-02324-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-020-02324-1