
Abstract
In our previous work, a two-plasmid CRISPR/Cas9 system was constructed for genome editing in Corynebacterium glutamicum. To increase the transformation efficiency and simplify the plasmid curing steps, an all-in-one CRISPR/Cas9 system was constructed for efficient genome editing. In addition, to research proteolysis during the production of recombinant proteins and generate a host for enhanced expression of recombinant proteins, the system was used to delete three genes, clpC, porB, and mepA in C. glutamicum CGMCC1.15647, which encoded the Clp protease subunit ClpC, anion selective channel protein B, and metallopeptidase A, respectively. After the evaluation of different plasmids and hosts, small ubiquitin-like modifier-N-terminal pro-brain natriuretic peptide (SUMO-NT-proBNP), an important protein used for the diagnosis of mild heart failure was successfully expressed in the triple mutant ΔclpCΔporBΔmepA, which exhibit threefold higher levels of protein expression compared with the wild-type. In conclusion, we created a simplified CRISPR tool for genome editing in C. glutamicum, provided a method to generate a host for enhanced expression of recombinant proteins and successfully expressed SUMO-NT-proBNP in C. glutamicum. This tool and method will greatly facilitate genetic engineering and metabolic optimization of this important platform organism.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Corynebacterium glutamicum is a non-sporulating Gram-positive species of soil bacterium that has been widely used for the production of amino acids and chemical compounds for several decades [1]. This species is a “generally regarded as a safe” microorganism [2], and large-scale fermentation technology has been established for the efficient production of diverse value-added compounds by these bacteria [3, 4]. However, compared with Escherichia coli, C. glutamicum has some intrinsic disadvantages, such as a much lower transformation efficiency and lower levels of protein expression [5].
Recent technological developments have allowed a deeper exploration into the full potential of C. glutamicum as a production host for value-added chemicals [6]. The recent development of the CRISPR/Cas9 system provides a simple, sequence-specific platform for genome engineering [7]. The CRISPR/Cas9 system has been widely applied in both prokaryotes and eukaryotes, including E. coli [8], Bacillus subtilis [9], Saccharomyces cerevisiae [10], and mammalian cells [11]. Moreover, there are many tries to adapt this system into C. glutamicum. CRISPR interference (CRISPRi) technology was used to manipulate the expression levels of specific genes in C. glutamicum [12, 13]. A genome editing tool based on the CRISPR-Cpf1 system has been developed for C. glutamicum [14]. By harboring CRISPR/Cas9 and RecT, a CRISPR/Cas9-coupled recombineering system was developed for rapid, efficient and scarless knockout of multiple genes in C. glutamicum [15]. In a previous work, we created a two-plasmid CRISPR/Cas9 gene editing system [16]. However, the transformation efficiency of that system was too low for co-transformation, and two-plasmid curing steps were required. Additionally, we did not apply this system to modification of host cells for the expression of practical pharmaceutical proteins. Thus, it is necessary to improve this CRISPR/Cas9 system and extend its application in optimization of C. glutamicum expression system.
Proteolysis can be a serious problem during the production of recombinant proteins because it can lead to lower yields and contamination by partially degraded products. Therefore, modification of protease expression in a host is a widely acknowledged strategy for the production of recombinant protein [17], such as in E. coli BL21 (DE3). Lon and Clp proteases cause most of the damage in the production of recombinant proteins in E. coli, together they account for 70–80% of the degradation of proteins in vivo [18]. Only Clp protease has been found in C. glutamicum, and it consists of two proteolytic subunits. The proteolysis subunit, ClpP, formed by two heptameric rings encloses a large chamber containing 14 proteolytic active sites; the ATPase subunits, ClpC and ClpX, are attached to the proteolytic core and determine substrate specificity, allowing suitable substrates to enter the proteolytic chamber [19, 20]. ClpP is essential and cannot be deleted from the chromosome [21], and therefore, ClpC and ClpX are potential targets for enhancing the production of recombinant protein.
Based on the results of previous research, the deletion of ClpX impaired the growth of C. glutamicum and it therefore cannot be used for host optimization, thus Clp protease subunit ClpC [19] is the only target for protease modification to enhance the production of recombinant protein in C. glutamicum. Additionally, our previous work revealed that changes in the expression level of porB and mepA genes had strong effects on recombinant protein expression [16]. The porB gene encodes an anion selective channel [22] and mepA gene encodes metallopeptidase A, which is involved in cell wall metabolism in C. glutamicum. These two genes are also targets to enhance the production of recombinant protein in C. glutamicum [23]. Therefore, in this study, the two-plasmid CRISPR/Cas9 system was improved to an all-in-one system to increase the transformation efficiency and simplify the plasmid curing steps. In addition, the clpC, porB, and mepA genes were deleted using this all-in-one CRISPR/Cas9 system, and ΔclpCΔporBΔmepA was constructed as a potentially suitable host for the production of recombinant proteins. Finally, N-terminal pro-brain natriuretic peptide (NT-proBNP), which is an important protein used for the diagnosis of preclinical and mild heart failure was attempted to express in C. glutamicum.
Brain natriuretic peptide (BNP) is a cardiac neurohormone that derives from the precursor pre-proBNP, containing 134 amino acids and including a signal peptide of 26 amino acids [24]. The proBNP, produced by cleavage of the signal peptide, is further split into a biologically active BNP and an inactive NT-proBNP [25]. BNP or NT-proBNP measurements are recommended as aids in diagnosis, risk stratification, and therapy monitoring in patients with heart failure (HF) [26]. NT-proBNP was more accurate than BNP in differentiating stage patients and was a better predictor of HF [27]. Thus, production of NT-proBNP in C. glutamicum provides a potential use for C. glutamicum in the production of protein for diagnostic applications.
Materials and methods
Strains, media, and reagents
All bacterial strains and plasmids used in this study are described in Table S1. DH5α was used as a cloning host for plasmid construction and was purchased from TaKaRa (Dalian, China). C. glutamicum ATCC13032 was purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA). C. glutamicum CGMCC1.15647 was used as the host for foreign protein expression and was donated by Huachang Pharmaceutical Co. (Zhangjiagang, China), this strain was deposited in the China General Microbiological Culture Collection Center (CGMCC), strain number 1.15647.
E. coli was cultured in Luria–Bertani (LB) medium (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl) at 37 °C with shaking at 200 rpm. C. glutamicum was cultured in LBB medium (LB supplemented with brain–heart infusion: 10 g/L tryptone, 5 g/L yeast extract, 10 g/L brain–heart infusion broth, and 10 g/L NaCl) at 30 °C with shaking at 200 rpm. LBHIS medium (LB supplemented with brain–heart infusion and sorbitol: 5 g/L tryptone, 2.5 g/L yeast extract, 18.5 g/L brain–heart infusion broth, 91 g/L sorbitol, and 5 g/L NaCl) was used for obtaining transformants of C. glutamicum. When needed, chloramphenicol was added to a final concentration of 30 µg/mL for E. coli and 10 µg/mL for C. glutamicum.
Plasmid DNA was extracted using an AxyPrep Plasmid Miniprep kit (Axygen, Union City, CA, USA). DNA fragments from polymerase chain reactions (PCRs) and restriction enzyme digestions were purified using the AxyPrep Gel Extraction kit (Axygen). Genomic DNA was extracted using a Bacterial Genomic DNA Extraction kit (Tiangen, Beijing, China). All kits were used according to the manufacturer’s instructions. Taq polymerase, T4 DNA ligase, and In-Fusion HD Cloning Plus kits were purchased from Takara (Dalian, China). Restriction endonucleases were purchased from Thermo Scientific (San Jose, CA, USA). High-sig ECL western blotting reagents were purchased from Tanon (Shanghai, China). Substrate primers were purchased from Genweiz (Suzhou, China). All of the primers used in this study are listed in Table S2.
Construction of the gene editing plasmid
The all-in-one CRISPR/Cas9 plasmid consists of the Cas9 module, the synthetic guide RNA (sgRNA) module, and the ori module. In our previous work the Cas9 expression plasmid, pFSC, was constructed. The Cas9 module was generated using the primers casF and casR to amplify the sequence containing the cat gene, the lacIq gene, the Ptac promoter, the cas9 gene, and the rrnB terminator from pFSC. Plasmid psgRNA was constructed as follows: the synthetic sgRNA scaffold sequence produced by Genweiz (Suzhou, China) (GGATCCTACAATGGAAATACAGATTGGGGATGAGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTGCGGCCGC) was designed to contain the AjuI, BamHI, and NotI sites for use in the assembly of sgRNA, and the resulting sgRNA scaffold was subcloned into pECXK99. The sgRNA module was then generated using the primers sgRNAF and sgRNAR to amplify the sequence containing the Ptrc promoter, sgRNA scaffold, and T1 and T2 terminators from psgRNA. Plasmid p109 was constructed as follows: pDTW109 was digested by SmaI, the fragment containing rep and oriE, which are replicons of C. glutamicum and E. coli, respectively, was purified, and the resulting ligated fragment was named p109. The ori module was then generated using primers 109F and 109R to amplify the sequence containing rep and oriE. Each module contained a 15-bp overlap with the other two modules. The all-in-one CRISPR/Cas9 plasmid pFSTC was constructed via assembling the Cas9 module, sgRNA module, and ori module using an In-Fusion HD Cloning Plus kit. AflII, BstBI, SnaBI, and SwaI sites were included at the end of the sgRNA module for assembling the homologous repair template (Fig. 1a).

Schematic illustration of the all-in-one CRISPR/Cas9 system in C. glutamicum. a Construction of the all-in-one CRISPR/Cas9 vector pFSTC. The cas9 module was amplified via PCR using the vector pFSC (blue). The sgRNA module was amplified via PCR using the vector psgRNA (violet). The ori module was amplified via PCR using the vector p109 (brown). The three fragments were assembled to construct the all-in-one CRISPR/Cas9 vector pFSTC. The BamHI and NotI sites were included for assembling the sgRNA, and the AflII, BstBI, SnaBI, and SwaI sites were included for assembling the HD repair template. b Construction of the vector for gene editing. The sgRNA was assembled into pFSTC to generate the pFSTC-sgRNA vector; the HDarm was assembled into pFSTC-sgRNA to generate the pFSTC-sgRNA-HDarm vector
The gene knockout plasmid contained both the sgRNA and the homolog-directed repair arm (HDarm) (Fig. 1b). The sgRNA was designed using the online-based sgRNA guide sequence designer CRISPy-web [28] (http://crispy.secondarymetabolites.org/). All candidates were searched using NCBI Blast against the NCBI C. glutamicum reference genome to identify sgRNA off-target sites that might produce off-target effects (https://blast.ncbi.nlm.nih.gov/Blast.cgi, Reference Sequence: NC_003450.3). The N20 sequence was selected for use in the design of the sgRNA based on the specific protospacer-adjacent motif site found using this website. The sgRNA was amplified using two primers, sgRNAF (5ʹ-CGCGGATCCN20GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCC-3ʹ) and sgRNAR (5ʹ-ATAAGAATGCGGCCGCAAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTT-3ʹ). After amplification, the sgRNA was ligated into the BamHI and NotI cloning sites of the all-in-one plasmid to construct the plasmid pFSTC-sgRNA. The HDarm containing the left and right repair arms, which are the regions upstream and downstream, respectively, of the target locus, was amplified from C. glutamicum genomic DNA. The left arm was amplified by primers LF and LR, and the right arm was amplified by primers RF and RR. The outer two primers (LF and RR) contained 15-bp overhang regions of the assembled site of plasmid pFSTC and the inner two primers (FR and RF) contained 15-bp overhang regions for the other repair arm. The repair template was finally assembled into the plasmid pFSTC-sgRNA at the AflII, BstBI, SnaBI, or SwaI sites using an In-Fusion HD Cloning Plus kit. After sequence validation, an appropriate plasmid was selected for gene editing.
Gene editing
Transformation of C. glutamicum was performed according to a previous publication [4]. After electroporation, the cells were incubated at 25 °C for 2 h, spread onto LBHIS agar containing chloramphenicol (10 µg/mL) and IPTG (0.01 mM), and then incubated for 24 h at 25 °C. Transformants were confirmed by PCR using primers targeted to sequences outside of the homologous repair template. The transformation efficiency was calculated as total number of colony-forming unit (CFU) generated per µg plasmid DNA [29].
Plasmid curing and multigene modification
After gene editing, plasmid curing must be performed before the next round of gene editing. To achieve this, the mutant strains were inoculated into 5 mL of LBB medium and incubated for 16 h at 37 °C. The cells were then streaked onto LBB plates in the absence of antibiotics and cultured overnight at 30 °C. Colonies were confirmed by streaking onto LBB plates containing chloramphenicol and by PCR amplification of the cas9 gene. Colonies in which the plasmid was successfully eliminated failed to grow on LBB plates containing chloramphenicol. These colonies were used in the next round of genome editing.
GFP expression and detection
To evaluate recombinant protein expression in porB-, mepA-, and clpC-disrupted strains, GFP was chosen as a model protein using plasmid pXMJ19-EGFP [16]. The expression of GFP was detected via a microplate reader, fluorescence microscopy, and 12% (w/v) sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) according to previously reported methods.
NT-proBNP expression and protein preparation
In this study, three NT-proBNP expression plasmids were constructed as follows. The NT-proBNP gene and small ubiquitin-like modifier (SUMO) gene were codon-optimized for C. glutamicum (GenBank accession number: MG766217) using the Genweiz Sequence synthetic program. SUMO, a fusion partner, has shown the function of enhancing the expression and solubility of target protein [30]. The sequence of SUMO gene was based on S. cerevisiae Smt3p (Protein id: XP_018224028.1) and the DNA size was 294 bp. The human NT-proBNP gene (Protein id: NP_002512.1) was codon-optimized for its expression in C. glutamicum and the DNA size was 231 bp. An SD sequence (AAAGGAGGACAACTA) and His tag were added in front of the SUMO gene. BEP5 promoter was amplified from pE-BEP5 [31] via PCR using the primers BEP5F and BEP5R. The NT-proBNP gene fragment was then introduced into pXMJ19 at the SmaI site using an In-Fusion HD Cloning Plus kit. The lacIq gene of pXMJ19 was removed as described previously. After DNA sequencing and PCR identification, this plasmid was transformed into C. glutamicum.
The bacterial colonies containing the NT-proBNP expression plasmid were cultured in a 24-well plate and then diluted 1:10 into 2 mL of fresh medium and grown at 30 °C for 16 h. In the study, all samples were taken with three independent biological replicates. After the cultivation, the optical density of each samples was measured at 600 nm (OD600) by a UV–Vis spectrophotometer (Lengguang, Shanghai, China) and 1 mL of the cells were harvested by centrifugation at 12,000g for 10 min. After washing with phosphate buffered saline (PBS; NaCl 137 mmol/L, KCl 2.7 mmol/L; Na2HPO4 4.3 mmol/L; KH2PO4 1.4 mmol/L, pH 7.4) three times the cells were resuspended in 1 mL PBS and disrupted by sonication on ice. Ultrasonic disruption of cells was carried out with an ultrasonic processor at 25% amplitude and pulse mode (2 s on/2 s off) for 10 min (Sonics Vibra Cell VCX800, Newtown, USA). After sonication, the dispersions were centrifuged at 12,000g for 10 min and then the supernatant was used for analysis the expression of NT-proBNP.
NT-proBNP detection methods
The expression of NT-proBNP was determined by 15% (w/v) SDS-PAGE, western blotting, and immunofluorescence assays. After SDS-PAGE, all protein bands were electrophoretically transferred onto a polyvinyl difluoride membrane using a Bio-Rad transblot device (Bio-Rad, Hercules, CA, USA) for 15 min. The membrane was incubated in 5% nonfat milk powder for 1 h to block nonspecific binding sites. It was then incubated for 1 h with a monoclonal horseradish peroxidase (HRP)-conjugated anti-His6 antibody (Sanying, Wuhan, China) (1:10,000) for the immunodetection of His-tagged protein. The membrane was washed and the antibody was detected using a Tanon High-sig ECL Western Blotting Substrate.
For NT-proBNP quantification, the sandwich two-step immunofluorescence assay was performed as described previously [32]. Capture antibodies were biotinylated, whereas detection antibodies were labeled with horseradish peroxidase (HRP). Proteins were quantified through HRP-catalyzed luminol-H2O2-p-iodophenol (PIP) chemiluminescent (CL) system (Autobio, Zhengzhou, China). Fluorescence was measured on a LUmo Luminometer (Autobio). Recombinant NT-proBNP (Roche, Rotkreuz, Switzerland) was used as a calibrator and 5 different concentrations of calibrator (200 pg/mL, 1000 pg/mL, 5000 pg/mL, 15,000 pg/mL and 48,000 pg/mL) were measured. The concentration of SUMO-NT-proBNP in the disruption supernatant was measured and the amount of NT-proBNP was expressed as mg per g of dry cell weight (DCW). The DCW was calculated from a curve relating the OD600 to DCW, an OD600 of 1.0 represented 0.23 g dry weight per liter.
Fed-batch cultivation and purification of SUMO-NT-proBNP
The fed-batch cultivations were carried out using C. glutamicum wild-type harboring p19BEP5-SUMOBNP and ΔclpCΔporBΔmepA harboring p19BEP5-SUMOBNP to achieve large-scale production of SUMO-NT-proBNP. A seed culture was inoculated into 200 mL of LBB medium in a 500 mL baffled flask and cultivated at 30 °C for 24 h with shaking at 200 rpm. Then the seed was inoculated into 2 L of fresh LBB (30 g/L glucose) medium in a 5 L bioreactor (Applikon EZ-control). Throughout the cultivation, the temperature was maintained at 30 °C. The dissolved oxygen concentration was controlled at 30% (v/v) by automatically varying the agitation speed from 400 to 1000 rpm and supplying pure oxygen through a solenoid valve. The pH was maintained at 7.0 with ammonia solution. To prevent glucose starvation, a glucose solution (300 g/L) was added into the cultures when the glucose concentration dropped lower than 5% (w/v). Cell growth was monitored by measuring the OD600. Glucose concentrations in the culture medium were monitored by a glucose assay kit (Sigma, St. Louis, Missouri, USA). The expression of SUMO-NT-proBNP was determined by SDS-PAGE and immunofluorescence assays.
The SUMO-NT-proBNP purification method was performed as follows. The cells were harvested by centrifugation at 4000g for 10 min at 4 °C, washed twice in PBS and then disrupted by sonication on ice. Finally, SUMO-NT-proBNP was purified using an AKTA purifier system (GE Healthcare, Uppsala, Sweden) with a HisTrap HP affinity column. The purity and quantity of SUMO-NT-proBNP was determined by SDS-PAGE analysis.
Construction of the gene overexpression strains
The gene overexpression plasmids were each constructed using the pXMJ19 plasmid as the backbone. First, porB and mepA were amplified from C. glutamicum genomic DNA via PCR using the primers porbgeneF-porbgeneR and mepageneF-mepageneR, respectively. The blunt-ended products were assembled into a linearized pXMJ19 fragment at the SmaI site using an In-Fusion HD Cloning Plus kit. The lacIq gene of pXMJ19 was removed to create a constitutive expression vector. A map of these plasmids is shown in Fig. S1. The porB overexpression strain OporB and mepA overexpression strain OmepA were constructed by transforming the porB and mepA gene overexpression plasmids into wild-type C. glutamicum, respectively.
Assessment of the effects of porB and mepA deletions on the lysozyme and NaCl sensitivity of C. glutamicum
The lysozyme sensitivities of C. glutamicum ΔporB, ΔmepA, ΔporBΔmepA, porB overexpression strain OporB, and mepA overexpression strain OmepA were each evaluated with a growth assay in LBB liquid medium with lysozyme using a previously described method [33]. For the NaCl sensitivity analysis, each strain was cultured in 5 L of liquid medium and subjected to fivefold serial dilutions to the same OD, after which 10 µL of each dilution were spotted onto LBB plates containing 1% (control) or 10% NaCl. The plates were incubated at 30 °C for 16 h.
Results
Construction of the all-in-one CRISPR/Cas9 system
In the previous work, a two-plasmid CRISPR/Cas9 system was constructed for genome editing [16], here, an all-in-one system was constructed to increase the transformation efficiency and simplify the plasmid curing steps. The all-in-one CRISPR/Cas9 system contained three parts: the cas9 module, the sgRNA module, and the ori module (Fig. 1a). The cas9 module was amplified from pFSC. In this module, Cas9 was under the control of an IPTG-inducible Ptac promoter and rrnb terminator, and cat gene expression was driven by a psiGA1 promoter to make the organism resistant to chloramphenicol. The psgRNA was constructed by subcloning the sgRNA scaffold into pECXK99, and the sgRNA module was then amplified from psgRNA. In this module, the sgRNA was driven by a Ptrc promoter and terminated by the T1 and T2 terminators. The AjuI, BamHI, and NotI sites were included in the sgRNA module for use in the assembly of sgRNA. Additionally, AflII, BstBI, SnaBI, and SwaI sites were included at the end of the sgRNA module for use in the assembly of the HDarm. The ori module was amplified from p109, which was constructed by deletion of the cat gene in pDTW-109. To allow the all-in-one plasmid to shuttle between E. coli and C. glutamicum, rep and oriE were included in the module, which are replicons of C. glutamicum and E. coli, respectively. The all-in-one CRISPR/Cas9 vector pFSTC was finally constructed by assembling the cas9 module, sgRNA module, and ori module into a single plasmid. The resulting vector was validated by digestion, PCR, and sequence analysis (Fig. S2).
Validation of the improved CRISPR/Cas9 system by disruption of the porB gene
To test the all-in-one system, the porB gene was knocked out. A sgRNA containing a 20-bp spacer was designed based on the porB gene sequence, and BLAST searches against the C. glutamicum genomic sequence were performed to ensure that the sgRNA did not have any predicted off-target effects. The sgRNA was constructed and assembled into the all-in-one plasmid pFSTC to generate plasmid pFSTC-porBsgRNA. The HDarm was used in this system to provide the repair template for the homolog-directed repair DNA repair pathway. The HDarm was constructed by assembling the left arm and the right arm regions upstream and downstream, respectively, of the porB gene. The porB deletion plasmid pFSTC-porB was constructed by assembling the HDarm into the plasmid pFSTC-porbsgRNA (Fig. 1b). After pFSTC-porB was transformed into C. glutamicum, PCR and sequence analysis were used to validate the deletions. The system was tested in both C. glutamicum ATCC13032 and C. glutamicum CGMCC1.15647, and the resulting efficiencies were as high as 100% (Fig. 2a, b). Furthermore, the new system has a higher transformation efficiency (1.5*102 CFU µg−1 DNA) compared to the two-plasmid system (0.4*102 CFU µg−1 DNA) (Fig. 2c) and easier plasmid curing step (Fig. 2d). These results demonstrated that this new all-in-one CRISPR/Cas9 system can be used in C. glutamicum and has high gene editing efficiency.

Evaluation of the all-in-one CRISPR/Cas9 system. a The CRISPR/Cas9 system mediated disruption of the porB gene in C. glutamicum ATCC 13032. b The CRISPR/Cas9 system mediated disruption of the porB gene in C. glutamicum CGMCC1.15647. Lane M is marker DL10000; Lane ck shows the PCR product using wild-type strain genomic DNA as the template; Lane 1–10 are PCR products using the genomic DNA of transformations as the template. The primers using for PCR are targeted to sequences outside of the homologous repair template. The 2.3 and 2 kb bands represent the wild-type and the porB gelation genotype, respectively. The editing efficiency was calculated as the ratio of deletion and wild-type. c Transformation efficiency of the all-in-one CRISPR/Cas9 system (left) and the two-plasmid CRISPR/Cas9 system (right). The transformation efficiency was calculated as CFU per µg plasmid DNA. d The pFSTC plasmid curing. Four individual colonies obtained after the curing steps were streaked onto the LBB agar plates in the absence (left) or presence (right) of chloramphenicol (Cm) to test the effectiveness of curing
Construction of porB, mepA, and clpC deletion mutations
To enhance the ability of C. glutamicum to express heterologous protein, two other genes, mepA and clpC, were selected for targeting via genome modification. The knockout plasmids pFSTC-mepA and pFSTC-clpC were constructed using the method described above. After eliminating the porB deletion plasmid from the porB-deleted strain, the plasmid for mepA deletion was transformed into this strain. This produced a strain with a double mutation of porB and mepA, namely ΔporBΔmepA. Last, using the same method, we obtained ΔclpCΔporBΔmepA, a strain with mutations of all three target genes, clpC, porB, and mepA (Fig. S3).
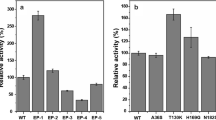
GFP was chosen as a model protein to evaluate the protein expression abilities of the porB-, mepA-, and clpC-deleted strains (ΔporB, ΔmepA, ΔclpC, ΔporBΔmepA, and ΔclpCΔporBΔmepA, respectively). The plasmid used in these experiments was pXMJ19-EGFP. The GFP expression levels were determined using a microplate reader, SDS-PAGE, and fluorescence microscopy analysis. Compared with the wild-type, the ΔporB, ΔmepA, and ΔclpC strains showed higher GFP fluorescence intensities of 39.2, 28.8, and 40.6%, respectively. The double mutant, ΔporBΔmepA, had an increased GFP fluorescence intensity of about 73.1%, and the triple mutant, ΔclpCΔporBΔmepA, showed the highest expression of GFP, about 92.8% higher than that of the wild-type (Fig. 3). The results indicated that the ΔclpCΔporBΔmepA strain had an enhanced ability to express recombinant protein.

GFP expression in the clpC-, porB-, and mepA-deleted C. glutamicum strains. a Fluorescence intensities normalized against a culture of OD600 were used to indicate the expression of GFP in various C. glutamicum strains. b SDS-PAGE analysis of GFP expression in various C. glutamicum strains. Lane M, Protein Molecular Weight Marker number 26610 (Thermo Scientific). c The expression of GFP was determined by fluorescence microscopy with an exposure time of 1 s. CK: negative control of the wild-type strain containing pXMJ19 without the gfp gene. 1: WT-GFP is a positive control of the wild-type strain containing pXMJ19-EGFP; 2: ΔporB-GFP is the porB-deleted mutant with plasmid pXMJ19-EGFP; 3: ΔmepA-GFP is the mepA-deleted mutant with plasmid pXMJ19-EGFP; 4: ΔclpC-GFP is the clpC-deleted mutant with plasmid pXMJ19-EGFP; 5: ΔporBΔmepA-GFP is the porB and mepA-deleted mutant containing pXMJ19-EGFP; 6: ΔclpCΔporBΔmepA-GFP is the clpC, porB, and mepA-deleted mutant containing pXMJ19-EGFP
Enhanced expression of SUMO-NT-proBNP in C. glutamicum
To expand the applications of C. glutamicum for the production of diagnostic proteins, we attempted to express NT-proBNP in C. glutamicum. To examine the effects of different promoters and SUMO on NT-proBNP expression in C. glutamicum, three plasmids were constructed to find a suitable NT-proBNP expression vector (Fig. 4a). The correctly constructed plasmids were selected for use in the transformation of C. glutamicum strains. SDS-PAGE, western blotting, and immunofluorescence assays were used to detect NT-proBNP expression. The results showed that NT-proBNP cannot be expressed in the strain harboring p19C-BNP (Fig. 4b, c). While production yield of SUMO-NT-proBNP in C. glutamicum harboring p19C-SUMOBNP and p19BEP5-SUMOBNP were 32.96 mg/g DCW and 39.92 mg/g DCW, respectively (Table 1). Thus, the p19BEP5-SUMOBNP with BEP5 promoter and SUMO was the suitable NT-proBNP expression vector.

SUMO-NT-proBNP expression in C. glutamicum. a Schematic illustration of plasmids to express NT-proBNP in C. glutamicum strains. b SDS-PAGE and c western blot analysis of SUMO-NT-proBNP expression in C. glutamicum wild-type strain containing various plasmids. M, Protein Molecular Weight Marker number 26632 (Thermo Scientific). CK: negative control of the wild-type strain containing pXMJ19 without the NT-proBNP gene. 1: the wild-type strain containing p19C-BNP; 2: the wild-type strain containing p19C-SUMOBNP; 3: the wild-type strain containing p19BEP5-SUMOBNP. d SDS-PAGE, e Western blot and f Immunofluorescence analysis of SUMO-NT-proBNP expression in various C. glutamicum strains. CK: negative control of the wild-type strain containing pXMJ19 without the NT-proBNP gene. 1: WT-SUMO-NT-proBNP is a positive control of the wild-type strain containing p19BEP5-SUMOBNP; 2: ΔporB-SUMO-NT-proBNP is the porB-deleted mutant with plasmid p19BEP5-SUMOBNP; 3: ΔmepA-SUMO-NT-proBNP is the mepA-deleted mutant with plasmid p19BEP5-SUMOBNP; 4: ΔclpC-SUMO-NT-proBNP is the clpC-deleted mutant with plasmid p19BEP5-SUMOBNP; 5: ΔporBΔmepA-SUMO-NT-proBNP is the porB- and mepA-deleted mutant containing p19BEP5-SUMOBNP; 6: ΔclpCΔporBΔmepA-SUMO-NT-proBNP is the clpC-, porB-, and mepA-deleted mutant containing p19BEP5-SUMOBNP
To evaluate the SUMO-NT-proBNP expression abilities of the different mutant strains, the p19BEP5-SUMOBNP was selected for use in the transformation of the different C. glutamicum strains. The results showed that the ΔporB and ΔmepA strains had levels of SUMO-NT-proBNP expression that were 72.42 mg/g DCW and 60.06 mg/g DCW, respectively. Additionally, the ΔclpC and ΔporBΔmepA strains had SUMO-NT-proBNP expression levels that were 65.89 mg/g DCW and 97.24 mg/g DCW, respectively. Notably, ΔclpCΔporBΔmepA showed the highest level of SUMO-NT-proBNP expression with 118.80 mg/g DCW, about threefold higher than that of the wild-type (Fig. 4d–f, Table 1). Therefore, ΔclpCΔporBΔmepA was chosen as the suitable host.
Production of SUMO-NT-proBNP in a fed-batch bioreactor
To achieve large-scale production of SUMO-NT-proBNP, fed-batch cultivations were carried out in a lab-scale (5 L) bioreactor system. C. glutamicum wild-type harboring p19BEP5-SUMOBNP continued to grow up to an OD600 of 43.76 for 24 h, after which the cell density decreased. The cell specific growth rate in the exponential growth phase was 0.204 h−1. The SUMO-NT-proBNP band first appeared at 12 h, then its concentration gradually increased, with the maximum yield of SUMO-NT-proBNP (11.5 mg/g DCW) being obtained at 24 h (Fig. 5a, b). In the fed-batch cultivation of ΔclpCΔporBΔmepA producing SUMO-NT-proBNP, ΔclpCΔporBΔmepA harboring p19BEP5-SUMOBNP grew up to an OD600 of 47.30 for 28 h with a specific growth rate of 0.197 h−1 in the exponential growth phase. SUMO-NT-proBNP was first produced at 12 h, then its concentration gradually increased, with the maximum yield of SUMO-NT-proBNP (29.96 mg/g DCW) being obtained at 28 h (Fig. 5c, d). In the fed-batch cultivation the cell density was higher, but the yield per unit was lower, compared to that in the 24-well plate. Thus, the fermentation process needs to be optimized in further study.

Fed-batch cultivation of C. glutamicum for the production of SUMO-NT-proBNP. a Fed-batch cultivation of the wild-type strain containing p19BEP5-SUMOBNP. Time profiles of cell growth (filled square), glucose concentration (filled circle), and SUMO-NT-proBNP concentration (filled triangle). b SDS-PAGE analysis of the wild-type strain containing p19BEP5-SUMOBNP. Lane M, molecular weight size markers. Lanes 1 to 10, samples at 0, 4, 8, 12, 16, 20, 24, 28, 32, and 36 h, respectively. c Fed-batch cultivation of ΔclpCΔporBΔmepA containing p19BEP5-SUMOBNP. Time profiles of cell growth (filled square), glucose concentration (filled circle), and SUMO-NT-proBNP concentration (filled triangle). d SDS-PAGE analysis of ΔclpCΔporBΔmepA containing p19BEP5-SUMOBNP. Lane M, molecular weight size markers. Lanes 1 to 10, samples at 0, 4, 8, 12, 16, 20, 24, 28, 32, and 36 h, respectively. e SDS-PAGE analysis of purified SUMO-NT-proBNP produced by ΔclpCΔporBΔmepA. Lane 1: purified SUMO-NT-proBNP; Lane M: molecular weight size markers
The SUMO-NT-proBNP expressed by ΔclpCΔporBΔmepA was purified using an AKTA purifier system. SUMO-NT-proBNP was successfully purified with high purity (> 90%) after this simple affinity purification step (Fig. 5e).
Effect of porB and mepA mutations on the lysozyme and NaCl sensitivity of C. glutamicum
Both porB and mepA are relatives of cell wall proteins; thus, mutations in these proteins may affect the response of the strain to environmental stress. To investigate the mechanism for the enhanced protein expression ability in the porB and mepA deletion mutants, we performed lysozyme and NaCl sensitivity experiments in ΔporB, ΔmepA, ΔporBΔmepA, the porB overexpression strain OporB, and the mepA overexpression strain OmepA. All strains exhibited similar growth rates under control conditions (Fig. 6a), whereas the porB overexpression strain OporB and the mepA overexpression strain OmepA showed impaired growth when 25 µg/mL of lysozyme was added to the growing cultures (Fig. 6b). For the NaCl sensitivity analysis, 10 μL dilutions of each strain were spotted onto LBB plates containing 10% NaCl. The porB overexpression strain OporB and mepA overexpression strain OmepA showed impaired growth on LBB plates containing 10% NaCl compared with their growth on LBB plates containing 1% NaCl (Fig. 6c). The results show that overexpression of porB and mepA increased the sensitivity of the strains to NaCl and lysozymes. A representative photo of the porB and mepA overexpression strains also demonstrates that the mepA overexpression strain had elongated colony morphology (Fig. 6d).

Lysozyme and NaCl sensitivity of C. glutamicum strains. aC. glutamicum strains were incubated in LBB medium at 30 °C, and their growth was monitored by measuring the OD600 of the cultures at various time points. b Lysozyme (final concentration, 25 µg/mL) was added to cultures of various C. glutamicum strains. Growth was monitored as described in a). cC. glutamicum strains were grown in LBB plates containing 1% NaCl (top) or 10% NaCl (bottom). 1: WT; 2, ΔporB; 3, ΔmepA; 4, OporB; 5, OmepA, and 6, ΔporBΔmepA. d Micrographs of different C. glutamicum strains. Abbreviations: 1: WT is the wild-type strain of C. glutamicum; 2: ΔporB is the porB-deleted mutant; 3: ΔmepA is the mepA-deleted mutant; 4: OporB is the porB gene overexpression strain; 5: OmepA is the mepA gene overexpression strain; 6: ΔporBΔmepA is the porB- and mepA-deleted mutant
Discussion
The CRISPR/Cas9 system has been proven to be a useful tool for gene editing [34]. Previously, our group constructed a two-plasmid CRISPR/Cas9 system for gene deletion, gene insertion, and point mutation. However, when using this system, we found that the transformation efficiency was low owing to the need to co-transform two-plasmids and perform two plasmid curing steps. Here, the CRISPR/Cas9 system was improved by generating an all-in-one system, which has a high editing efficiency in both C. glutamicum ATCC13032 and C. glutamicum CGMCC1.15647, as well as the advantages of higher transformation efficiency and easier plasmid curing (Fig. 2). Therefore, the new system generated here is simpler and more useful than the previous CRISPR/Cas9 system.
C. glutamicum has been considered as an alternative host cell for recombinant protein expression for a few years [2]. However, the relatively low expression efficiency of C. glutamicum for some proteins remains a key challenge in fulfilling its potential [5]. Previous omics analysis revealed that the porB and mepA genes undergo substantial changes under different dissolved oxygen conditions [35], further investigation demonstrated that the deletion of each of these genes was helpful for enhancing the GFP expression level in C. glutamicum [16]. ClpC and ClpX are potential targets for the modification of protease expression in C. glutamicum to enhance its production of recombinant protein [21]. However, deletion of ClpX impaired the growth of C. glutamicum so cannot be used for host optimization, thus ClpC is the only target for modification of protease expression to enhance the production of recombinant protein in C. glutamicum. Therefore, it was speculated that deleting these genes might be a viable strategy for host modification to enhance the recombinant protein expression ability of C. glutamicum. In this study, the improved all-in-one CRISPR/Cas9 system was used to edit the C. glutamicum genome and to generate clpC, porB, and mepA deletion mutants.
NT-proBNP has been used for many years as a screening marker for heart disease. In this study, we attempted to use C. glutamicum as a host for NT-proBNP expression; it would expand the applications of C. glutamicum for the production of diagnostic proteins. However, we found that NT-proBNP cannot be expressed without a fragment of SUMO. SUMO is a fusion partner that has been shown to enhance the expression and solubility of protein [30]. After adding the SUMO gene in front of the ATG of the NT-proBNP gene, SUMO-NT-proBNP was successfully expressed in C. glutamicum. The isoelectric point of NT-proBNP is 10.95, compared with 4.66 for SUMO and 6.59 for the SUMO–NT-proBNP fusion protein. It was speculated that an isoelectric point near to neutral contributes to the expression of SUMO–NT-proBNP. After evaluation of various plasmids and hosts, SUMO–NT-proBNP was successfully expressed in C. glutamicum, with 118.80 mg/g DCW of soluble protein achieved, to the best of our knowledge, this is the first report on the production of SUMO–NT-proBNP in C. glutamicum, and these results provide another potential use for C. glutamicum in the production of protein for diagnostic applications.
Compared with the wild-type C. glutamicum, we obtained a twofold increase in the GFP expression and a threefold increase in the SUMO-NT-proBNP expression by the ΔporBΔmepAΔclpC mutant. ClpC is a protease in C. glutamicum [21], and the clpC deletion may reduce the degradation of recombinant protein [19]. As in E. coli, using the protease Clp mutation can improve the cytoplasmic protein ZZT2 accumulation [36]. Mutant strains lacking the ability to express proteases such as OmpT, DegP and protease III, have an increased availability of recombinant protein [37]. MepA and PorB are cell wall-related proteins, thus mepA and porB deletion mutants may affect the integrity of the cell surface structure [23, 38]. A double mutation of cell wall proteins CSpB and PBP1a increased secretion of the antibody Fab fragment by C. glutamicum [39]. The cell wall gene deletion mutants (hsps82Δ, ato2Δ, and ssa3Δ) exhibited more efficient in proton efflux and lower in membrane permeability, and those physiological changes are likely to contribute to enhanced acetic acid tolerance of S. cerevisiae [40]. The results of lysozyme and NaCl sensitivity tests support this idea. The porB and mepA overexpression strains were more sensitive to environmental stresses (Fig. 6), and therefore, we suspect that the mepA and porB deletion mutants reduced the death rate of these strains in response to stress. However, we did not find obviously improved tolerance to Nacl and lysozyme of the deletions compared with the wild type. The reason why deletions of the mepA and porB genes are able to enhance protein expression requires further study.
In conclusion, we have developed a rapid and efficient gene editing tool for C. glutamicum, and this tool increased the transformation efficiency and simplified the plasmid curing steps. We used this new all-in-one CRISPR/Cas9 system to delete the clpC, porB, and mepA genes in C. glutamicum, and construct ΔclpCΔporBΔmepA, which had significantly improved expression levels of GFP compared with the wild-type. Moreover, this study is the first to successfully express SUMO-NT-proBNP in C. glutamicum, which, like GFP was expressed in higher levels in ΔclpCΔporBΔmepA compared with the wild-type strain. The method used here for the modification of the C. glutamicum host could also be generally applicable to other microorganisms.
Abbreviations
- sgRNA:
-
Single-guide RNA
- HDarm:
-
Homolog-directed repair arm
- GFP:
-
Green fluorescent protein
- scFv:
-
Single-chain variable fragment
- SUMO:
-
Small ubiquitin-related modifier
- NT-proBNP:
-
N-terminal pro-brain natriuretic peptide
References
Becker J, Zelder O, Hafner S, Schroder H, Wittmann C (2011) From zero to hero-design-based systems metabolic engineering of Corynebacterium glutamicum for l-lysine production. Metab Eng 13:159–168. https://doi.org/10.1016/j.ymben.2011.01.003
Woo HM, Park JB (2014) Recent progress in development of synthetic biology platforms and metabolic engineering of Corynebacterium glutamicum. J Biotechnol 180:43–51. https://doi.org/10.1016/j.jbiotec.2014.03.003
Becker J, Wittmann C (2012) Bio-based production of chemicals, materials and fuels -Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol 23:631–640. https://doi.org/10.1016/j.copbio.2011.11.012
Park SH, Kim HU, Kim TY, Park JS, Kim SS, Lee SY (2014) Metabolic engineering of Corynebacterium glutamicum for l-arginine production. Nat Commun 5:4618. https://doi.org/10.1038/ncomms5618
Liu X, Yang Y, Zhang W, Sun Y, Peng F, Jeffrey L, Harvey L, McNeil B, Bai Z (2016) Expression of recombinant protein using Corynebacterium glutamicum: progress, challenges and applications. Crit Rev Biotechnol 36:652–664
Heider SA, Wendisch VF (2015) Engineering microbial cell factories: metabolic engineering of Corynebacterium glutamicum with a focus on non-natural products. Biotechnol J 10:1170–1184
Jacobi AM, Rettig GR, Turk R, Collingwood MA, Zeiner SA, Quadros RM, Harms DW, Bonthuis PJ, Gregg C, Ohtsuka M, Gurumurthy CB, Behlke MA (2017) Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods 121–122:16–28. https://doi.org/10.1016/j.ymeth.2017.03.021
Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S (2015) Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol 81:2506–2514. https://doi.org/10.1128/AEM.04023-14
Jakočiūnas T, Jensen MK, Keasling JD (2016) CRISPR/Cas9 advances engineering of microbial cell factories. Metab Eng 34:44–59
DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41:4336–4343. https://doi.org/10.1093/nar/gkt135
Wang T, Wei JJ, Sabatini DM, Lander ES (2014) Genetic screens in human cells using the CRISPR-Cas9 system. Science 343:80–84
Cleto S, Jensen JV, Wendisch VF, Lu TK (2016) Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi). ACS Synth Biol 5:375–385
Park J, Shin H, Lee S-M, Um Y, Woo HM (2018) RNA-guided single/double gene repressions in Corynebacterium glutamicum using an efficient CRISPR interference and its application to industrial strain. Microb Cell Fact 17:4
Jiang Y, Qian F, Yang J, Liu Y, Dong F, Xu C, Sun B, Chen B, Xu X, Li Y (2017) CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun 8:15179
Cho JS, Choi KR, Prabowo CPS, Shin JH, Yang D, Jang J, Lee SY (2017) CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab Eng 42:157–167
Peng F, Wang X, Sun Y, Dong G, Yang Y, Liu X, Bai Z (2017) Efficient gene editing in Corynebacterium glutamicum using the CRISPR/Cas9 system. Microb Cell Fact 16:201. https://doi.org/10.1186/s12934-017-0814-6
Ahmad M, Hirz M, Pichler H, Schwab H (2014) Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol 98:5301–5317
Zhang J, Ye F, Lan L, Jiang H, Luo C, Yang C-G (2011) Structural switching of Staphylococcus aureus Clp protease a key to understanding protease dynamics. J Biol Chem 286:37590–37601
Trentini DB, Suskiewicz MJ, Heuck A, Kurzbauer R, Deszcz L, Mechtler K, Clausen T (2016) Arginine phosphorylation marks proteins for degradation by a Clp protease. Nature 539:48
Wang F, Mei Z, Qi Y, Yan C, Hu Q, Wang J, Shi Y (2011) Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature 471:331–335. https://doi.org/10.1038/nature09780
Engels S, Schweitzer JE, Ludwig C, Bott M, Schaffer S (2004) clpC and clpP1P2 gene expression in Corynebacterium glutamicum is controlled by a regulatory network involving the transcriptional regulators ClgR and HspR as well as the ECF sigma factor σH. Mol Microbiol 52:285–302
Ziegler K, Benz R, Schulz GE (2008) A putative α-helical porin from Corynebacterium glutamicum. J Mol Biol 379:482–491
Bott M, Brocker M (2012) Two-component signal transduction in Corynebacterium glutamicum and other corynebacteria: on the way towards stimuli and targets. Appl Microbiol Biotechnol 94:1131–1150
Rodseth RN, Biccard BM, Le Manach Y, Sessler DI, Buse GAL, Thabane L, Schutt RC, Bolliger D, Cagini L, Cardinale D (2014) The prognostic value of pre-operative and post-operative B-type natriuretic peptides in patients undergoing noncardiac surgery: B-type natriuretic peptide and N-terminal fragment of pro-B-type natriuretic peptide: a systematic review and individual patient data meta-analysis. J Am Coll Cardiol 63:170–180
Mueller T, Gegenhuber A, Poelz W, Haltmayer M (2005) Diagnostic accuracy of B type natriuretic peptide and amino terminal proBNP in the emergency diagnosis of heart failure. Heart 91:606–612. https://doi.org/10.1136/hrt.2004.037762
Maisel AS, Krishnaswamy P, Nowak RM, McCord J, Hollander JE, Duc P, Omland T, Storrow AB, Abraham WT, Wu AH (2002) Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 347:161–167
Emdin M, Passino C, Prontera C, Fontana M, Poletti R, Gabutti A, Mammini C, Giannoni A, Zyw L, Zucchelli G, Clerico A (2007) Comparison of brain natriuretic peptide (BNP) and amino-terminal ProBNP for early diagnosis of heart failure. Clin Chem 53:1289–1297. https://doi.org/10.1373/clinchem.2006.080234
Blin K, Pedersen LE, Weber T, Lee SY (2016) CRISPy-web: an online resource to design sgRNAs for CRISPR applications. Synth Syst Biotechnol 1:118–121
Yan M-Y, Yan H-Q, Ren G-X, Zhao J-P, Guo X-P, Sun Y-C (2017) CRISPR-Cas12a-assisted recombineering in bacteria. Appl Environ Microbiol 83:e00947-00917
Maffitt M, Auldridge M, Sen S, Floyd S, Krerowicz A, Uphoff M, Thompson J, Mead D, Steinmetz E (2015) Rapid screening for protein solubility and expression. Nat Methods 12(6):586. https://doi.org/10.1038/nmeth.f.382
Zhao Z, Liu X, Zhang W, Yang Y, Dai X, Bai Z (2016) Construction of genetic parts from the Corynebacterium glutamicum genome with high expression activities. Biotechnol Lett 38:2119–2126
Seferian KR, Tamm NN, Semenov AG, Tolstaya AA, Koshkina EV, Krasnoselsky MI, Postnikov AB, Serebryanaya DV, Apple FS, Murakami MM (2008) Immunodetection of glycosylated NT-proBNP circulating in human blood. Clin Chem 54:866–873
Choi JW, Yim SS, Lee SH, Kang TJ, Park SJ, Jeong KJ (2015) Enhanced production of gamma-aminobutyrate (GABA) in recombinant Corynebacterium glutamicum by expressing glutamate decarboxylase active in expanded pH range. Microb Cell Fact 14:21
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87
Liu X, Yang S, Wang F, Dai X, Yang Y, Bai Z (2017) Comparative analysis of the Corynebacterium glutamicum transcriptome in response to changes in dissolved oxygen levels. J Ind Microbiol Biotechnol 44:181–195. https://doi.org/10.1007/s10295-016-1854-3
Rozkov A (2001) Control of proteolysis of recombinant proteins in Escherichia coli. Bioteknologi
Selas Castiñeiras T, Williams SG, Hitchcock AG, Smith DC (2018) E. coli strain engineering for the production of advanced biopharmaceutical products. FEMS Microbiol Lett 365:fny162
Costa-Riu N, Maier E, Burkovski A, Krämer R, Lottspeich F, Benz R (2003) Identification of an anion-specific channel in the cell wall of the Gram-positive bacterium Corynebacterium glutamicum. Mol Microbiol 50:1295–1308
Matsuda Y, Itaya H, Kitahara Y, Theresia NM, Kutukova EA, Yomantas YAV, Date M, Kikuchi Y, Wachi M (2014) Double mutation of cell wall proteins CspB and PBP1a increases secretion of the antibody Fab fragment from Corynebacterium glutamicum. Microb Cell Fact 13:56
Lee Y, Nasution O, Choi E, Choi I-G, Kim W, Choi W (2015) Transcriptome analysis of acetic-acid-treated yeast cells identifies a large set of genes whose overexpression or deletion enhances acetic acid tolerance. Appl Microbiol Biotechnol 99:6391–6403
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21808082), the Natural Science Foundation of Jiangsu Province (BK20150148), the 111 Project (111-2-06) and national first-class discipline program of Light Industry Technology and Engineering (LITE2018-24).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Peng, F., Liu, X., Wang, X. et al. Triple deletion of clpC, porB, and mepA enhances production of small ubiquitin-like modifier-N-terminal pro-brain natriuretic peptide in Corynebacterium glutamicum. J Ind Microbiol Biotechnol 46, 67–79 (2019). https://doi.org/10.1007/s10295-018-2091-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-018-2091-8