
Abstract
A novel prolyl endopeptidase gene from Aspergillus oryzae was cloned and expressed in Pichia pastoris. Amino acid sequence analysis of the prolyl endopeptidase from Aspergillus oryzae (AO-PEP) showed that this enzyme belongs to a class serine peptide S28 family. Expression, purification and characterization of AO-PEP were analyzed. The optimum pH and temperature were pH 5.0 and 40 °C, respectively. The enzyme was activated and stabilized by metal ion Ca2+ and inhibited by Zn2+, Mn2+, Al3+, and Cu2+. The K m and k cat values of the purified enzyme for different substrates were evaluated. The results implied that the recombinant AO-PEP possessed higher affinity for the larger substrate. A fed-batch strategy was developed for the high-cell-density fermentation and the enzyme activity reached 1,130 U/l after cultivation in 7 l fermentor. After addition of AO-PEP during the fermentation phase of beer brewing, demonstrated the potential application of AO-PEP in the non-biological stability of beer, which favor further industrial development of this new enzyme in beer stabilization, due to its reducing operational costs, as well as no beer losses unlike regeneration process and beer lost with regenerated polyvinylpolypyrrolidone system.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Maintaining beer, wine, and fruit juices’ quality through the various stages of maturation, distribution and shelf storage remains an extensive challenge [3]. In beer, the formation of haze is a serious quality problem because of the native effect on the quality of the final product. Two forms of haze exist: cold break (chill haze) and age-related haze [23]. Beer contains numerous barley proteins that are modified chemically and proteolytically during the malting and brewing processes, which can influence the final beer haze stability. Researchers found that the most frequent cause of haze in beer, wine, and clear fruit juices resulted from protein–polyphenol interaction [9, 12], and these clear beverages are typically stabilized to delay the onset of protein–polyphenol haze formation. Furthermore, it has been known for many years that the haze-forming activity of a polypeptide depends greatly on its proline content [17]. Of particular interest is the finding that, prolyl endopeptidase (EC3.4.21.26) would effectively hydrolyze haze-active protein in beer haze formation [20].
Prolyl endopeptidases are the enzymes that hydrolyze substrates on the carboxyl site of proline residues located internally in a peptide (or ester), which are widely distributed in plants [19, 29], mammalians [1, 25, 26], bacteria [4, 8, 14, 18, 21, 34–36], and fungi [6, 7, 15, 16, 20, 27, 28]. The roles of these enzymes among various organisms are diverse including the activation of cell-mediated immunity, autoimmune and inflammatory responses [2, 22], and nutrient digestion. From an application point of view, Flavobacterium meningosepticum [36, 37], Xanthomonas sp. [35], Aeromonas hydrophilic [14], and Pseudomonas sp. KU-22 [27], most of them belong to pathogenic bacteria, are obviously not good choices for food processing industry. Several prolyl endopeptidases from bacteria have been characterized and some of their genes have been cloned [5, 10, 11, 13, 14, 31, 32]. Up to now, among the genus Aspergillus, two prolyl endopeptidases from A. niger and A. fumigatus are known [6, 24]; however, only A. niger is the food-grade microorganism.
In this research, we cloned the gene of prolyl endopeptidase (AO-PEP) from another food-grade microorganism, A. oryzae, which is non-toxigenic and non-pathogenic, and demonstrate the AO-PEP encoding a hydrolytic enzyme acting preferentially on proline residues of peptides. The gene of AO-PEP was cloned and expressed in P. pastoris. Then the effect of physical and chemical parameters on the activity of AO-PEP and its application in beer was investigated.
Materials and methods
Strains, vectors, and chemicals
The strain A. oryzae WX2011(also called S1) was preserved in our laboratory (isolated from soil in Wuxi) [16] and maintained on malt extract medium. E. coli strain JM109 and P. pastoris GS115 were used as host cells. Vectors pMD-19T simple (Promega, USA) and pPIC9K (Invitrogen, USA) were used for gene manipulations. Oligonucleotides, restriction enzymes, Taq polymerase, and T4 DNA ligase, were purchased from TaKaRa biotechnology (Dalian, China). The Z-Gly-Pro-pNA substrate was obtained from Bachem (King of Prussia, PA, USA). The standard mini Plasmid Prep Kit and the DNA gel extraction kit were purchased from Omega. DNA sequencing was performed using an ABI377 sequencer (Applied Biosystems, Foster City, CA, USA). All other chemicals were of analytical grade and were commercially available.
Cloning of the full-length prolyl endopeptidase gene from A. oryzae
Aspergillus oryzae WX2011 was grown in a medium containing 1.0 g of K2HPO4, 0.4 g of KH2PO4, 0.5 g of KCl, 0.5 g of MgSO4·7H2O, 0.01 g of FeSO4·7H2O, 5 g of glucose, and 15 g of collagen (Sigma). Young mycelia were harvested after 48 h grown at 30 °C. Total RNA from A. oryzae WX2011 was isolated using the Trizol reagent exactly as described by the supplier (Sangon Biotech. Shanghai, China), and its purity was evaluated by electrophoresis on 2 % agarose gel.
Reverse transcription was performed by using 2 μg total RNA, 1× Prime Script Buffer, 25 pmol Oligo dT Primer (50 μM), and 50 pmol Random 6 mers (100 μM). Reactions were carried out at 37 °C for 15 min, 85 °C for 5 s, and 4 °C for 10 min. The resulting cDNA from the A. oryzae WX2011 was then used for PCR. To obtain prolyl endopeptidase gene fragment, two degenerate primers were designed (Table 1) according to the conserved sequences of prolyl endopeptidase from Flavobacterium meningosepticum (Genbank: AAA24925.1), Aeromonas hydrophila (Genbank: BAA03105.1), Novosphingobium capsulatum (Genbank: BAA34052.1), Myxococcus xanthus (Genbank: AAD31004.1), especially A. niger (Genbank: CAK45422.1) and Aspergillus fumigatus (Genbank: XP_749261.2).
PCR mixture contained 0.5 μM final concentrations of primers, which contained added sites for the restriction enzymes SnaBI and NotI (underline), respectively, TaKaRa Taq DNA polymerase 1.25 U, 10× PCR buffer (Mg2+ plus) 5 μl, dNTP Mixture (each 2.5 mM) 4 μl, primer up (20 μM) 1 μl, primer down (20 μM) 1 μl, and cDNA 0.5 μg, in a 50 μl volume PCR amplification was performed by incubating the samples at 94 °C for 3 min preheating, followed by 30 cycles at 94 °C for 30 s, 63 °C for 60 s, and at 72 °C for 60 s, with a final extension at 72 °C for 10 min. At the end of amplification, samples were submitted to electrophoresis on 1.5 % agarose gel with a 2,000 bp DNA ladder as a size marker. Primers for full-length amplification (Table 1) were designed according to the complete sequence assembled by the DNAMAN program. Appropriated bands of PCR products were purified, cloned into pMD19T simple vector, sequenced, and then assembled with the known fragment sequences.
DNA sequence analysis and alignments
Sequence similarity searches were carried out using the National Center for Biotechnology Information (NCBI) database and European Molecular Biology Laboratory (EMBL). Sequence analysis, such as putative amino acid sequence, open reading frame (ORF), molecular mass, and theoretical isoelectric point, was inferred with ExPASy. A multiple alignment of prolyl endopeptidase amino acid sequences from various prokaryotic organisms was constructed with the software package DNAMAN, EMBL-EBI Clustal Omega and the ESPript 2.2 network station.
The expression and purification of the prolyl endopeptidase
After SnaBI–NotI digestion in pMD-19T-AO-PEP, the prolyl endopeptidase gene from A. oryzae (short for AO-PEP) was cloned in the pPIC9K vector between the SnaBI (5′ end) and NotI (3′ end) restriction sites to generate the recombinant plasmid pPIC9K-AO-PEP. The resulting plasmid pPIC9K-AO-PEP was transformed into JM109, and then the recombinant E. coli cells were selected on ampicillin containing LB plates and screened by restriction with SnaBI and NotI and PCR using the P5, P6 primers. Plasmid DNAs were purified from the recombinant E. coli DH5α and subjected to DNA sequence analysis to confirm the serine peptidases cDNA fragments.
Pichia pastoris wild-type strain GS115 was used as a host for the expression of the gene encoding prolyl endopeptidase. The expression vector pPIC9K-AO-PEP described above was linearized by digestion with restriction enzyme SalI and introduced into P. pastoris wild-type strain GS115 by electroporation using a Micropulser (Bio-Rad. USA). According to manufacturer recommendations (Invitrogen), the following culture media: minimal dextrose medium (MD), buffered glycerol-complex medium (BMGY), and buffered minimal methanol (BMMY), were prepared for the transformation of P. pastoris, selection of recombinant clones, and expression of serine peptidases. For cultures in liquid BMMY, which contain methanol as an inducer and carbon source, methanol was added every 24 h to a final concentration of 1 % (v/v).
After the induction for 96 h, the entire medium was harvested by centrifugation at 12,000 rpm for 20 min. Solid ammonium sulfate was added to the supernatant to 60 % saturation at 4 °C. The precipitate was collected and dissolved in citrate/disodium phosphate buffer (pH 5.0) and dialyzed overnight against the same buffer. After dialysis, the enzyme solution was filtered. The enzyme solution was then injected into an AKTA purifier (GE Healthcare) through ion-exchange (HiTrap DEAE FF, Amersham Biosciences). Buffer A (equilibration buffer) contained 20 mM phosphate buffer (pH 5.0). Buffer B (elution buffer) contained 20 mM phosphate buffer (pH 5.0) and 1 M NaCl. The flow rate was 1.0 ml/min. Buffer A was used to absorb the target enzyme to the column. A linear elution was done by ramping buffer B from 0 to 100 % after the unbound proteins were eluted. The fractions containing activity were collected for activity assays and SDS-PAGE analysis.
High-cell-density fermentation at laboratory scale
The recombinant strain with AO-PEP activity was grown in a laboratory fermentor vessel of 7 l total volume (New Brunswick Scientific, USA) to scale up expression. Dissolved oxygen (DO) in fermentation culture was monitored by using the DO sensor (Inpro 6800 Series O2 Sensors 12 mm, Mettler Toledo, Switzerland). pH was monitored by pH sensor (405-DPAS-SC-K8S/325, Mettler Toledo, Switzerland). The general control and operation were run according to Pichia protocols and a recommendation by the fermentor manufacturer. 1 ml of frozen cell stock was transferred to 50 ml of shaking flask containing 10 ml of YPD medium, and incubated with shaking at 30 °C overnight. Then the 1-ml culture was transferred to six 250 ml shaking flasks, respectively, containing 50 ml of YPD medium, and incubated with shaking at 30 °C for 20–24 h when the culture reached a OD600 nm of between 10 and 12. Then the seed cultures were inoculated into a 7 l fermentor containing 3 l fermentation medium consisted of basic medium [CaSO4 0.93 g/l, 85 % H3PO4 28.70 ml/l, MgSO4·7H2O 14.90 g/l, K2SO4 18.20 g/l, KOH 4.13 g/l, with 4 % (W/V) glycerol for growth period] and 4 ml/l trace salt solution (CuSO4·5H2O 6.0 g/l, KI 0.08 g/l, MnSO4·H2O 3.0 g/l, Na2MoO4·2H2O 0.2 g/l, H3BO3 0.02 g/l, ZnSO4·7H2O 42.2 g/l, FeSO4·7H2O 65.0 g/l, CoC12·6H2O 0.5 g/l, Biotin 0.2 g/l, H2SO4 5.0 ml/l). To control excessive foaming, 3–5 drops of undiluted antifoam was added to the fermentor through a 0.45-µm syringe filter. The culture was started with a batch fermentation step using glycerol as the sole carbon source, then a methanol fed-batch phase steps were followed and methanol was used as an inducer of the AOX promoter. The initial cultivation was continued until the glycerol was consumed out (about 24 h, OD600 = 30), and the methanol feeding is started and maintained approximately 0.1 % concentration throughout the methanol induction phase. This methanol induction phase lasted for about 96 h at 28 °C. The cell density was measured every 24 h by measuring absorbance at 600 nm. The three important index of dissolved oxygen, pH value, and feeding rate were all monitored online and coupled, and controlled automatically by a computer preset threshold of 30 % higher of dissolved oxygen and stable pH value of 5.5, so that the viability of the culture and accumulation of enzyme protein were kept normal.
Enzymes activity
Prolyl endopeptidase activity was fluorimetrically assayed by monitoring cleavage of a synthetic fluorogenic peptide, Z-Gly-Pro-pNA [20]. Briefly, the reaction mixture consisted of 80 µl of 0.1 M citrate/disodium phosphate buffer (pH 5.0), 10 µl of purified enzyme solution, and of 10 µl of Z-Gly-Pro-pNA(250 μM, dissolved in 40 % 1, 4-dioxane). Other assay condition including fluorescence measurement was as described previously [15, 20]. The protein concentration was measured with the Bradford method, with bovine albumin (Sangon Biotech, Shanghai, China) as the standard.
Computer-aided modeling of the tertiary structure
The theoretical structures of prolyl endopeptidase were obtained by homology modeling with the Swiss Model server.
The characterization of the recombinant AO-PEP
To estimate the optimal pH of AO-PEP activity, the purified protein was incubated in citrate/disodium phosphate buffer (pH 2.2–8.0), Tris–HCl (pH 9.0–10.5), and glycine/NaOH (pH 11–12). The pH stability of prolyl endopeptidase activity was determined at pH ranging from 2.2 to 12 at 37 °C for 30 min. After incubation, the prolyl endopeptidase activity was measured at 37 °C. Thermal dependence of AO-PEP activity was determined incubating the reaction mixture (enzyme in 0.1 M citrate–phosphate buffer, optimal pH) at temperatures between 25 and 80 °C. To evaluate thermal stability, 50 μl of the enzyme solution and 500 μl of 0.1 M citrate–phosphate buffer (optimal pH) were incubated for 30 min at different temperatures (25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, and 80 °C). Then, the activity was determined according to the enzyme activity test at optimal pH and temperature.
The metal ion (Ca2+, Na+, K+, Mg2+, Mn2+, Fe2+, Zn2+, and Al3+) was added to enzyme and incubated for 30 min followed by enzyme assay under the standard condition, respectively. Enzymatic activities were expressed as relative values (%) and the sample without any metal ion or reagent was taken as control (100 %).
Enzyme kinetics
The Michaelis–Menten constant (K m), maximal velocity (V max), and k cat of the purified enzyme were determined using Z-Gly-Pro-pNA, Ala-Pro-pNA, Ala-Ala-Pro-pNA, and Z-Ala-Ala-Ala-Pro-pNA as substrates in the range of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 mM under the optimal assay conditions in the standard method. The kinetic data were calculated from Lineweaver–Burk plots using Michaelis–Menten equation using the software SkanIt RE for MS 2.4.2.
Effect on the improvement of non-biological stability of beer by prolyl endopeptidase
Comparisons of beer stabilized by Kieselguhr filter, Silica gel filtration, and enzymolysis were made. Testing was carried out on a range of beer types with different kinds of treatment. Haze testing included 0–6 weeks, and 60 °C forced hazes. Most of the early assessment on the degree of stabilization achieved was based upon forced haze data. This allowed for relatively quick responses to any issues, which arose during the early stages of the trial. The analyses carried out during the trials were as follows:
Haze measurement. Turbidity analyses were carried out with the use of a Hach 2100N Turbidimeter (90º) (Hach, Loveland, CO, USA) at 0 °C, and measured in EBC formazin units (EBC).
Results and discussion
Cloning and sequence analysis of the prolyl endopeptidase gene
A 1,016-bp fragment containing the A. oryzae prolyl endopeptidase gene was amplified with degenerate primers. The downstream fragment of the prolyl endopeptidase fragment was amplified by PCR with specific primers and arbitrary degenerate primers, while the upstream fragment was amplified with a gene-specific primer and a degenerate primer, as described in “Materials and methods”. The full-length arbitrary degenerate primers gene sequence consisted of an ORF of 1,740 bp, starting with ATG and terminating with TAA, and encoded a putative polypeptide of 581 amino acid residues. The calculated molecular mass was 64.8 kDa, and the theoretical isoelectric point was 5.7. The amino acid sequence from prolyl endopeptidase gene of A. oryzae was compared with those of other organisms. It exhibited 27 and 25 % identity to prolyl endopeptidase of Aspergillus niger (Genbank: CAK45422.1) and Aspergillus fumigatus Af293 (Genbank: XP_749261.2) (Fig. 1), while 31, 28, 26 and 42 % identity to prolyl endopeptidase of Flavobacterium meningosepticum (Genbank: AAA24925.1), Aeromonas hydrophila (Genbank: BAA03105.1), Novosphingobium capsulatum (Genbank: BAA34052.1), and Myxococcus xanthus (Genbank: AAD31004.1), respectively. Though the amino acid identity from different sources is very low, the consensus sequence Gly-X-Ser-X-Gly was observed among them. In this study, a prolyl endopeptidase gene from A. oryzae was successfully over expressed in P. pastoris for the first time.

Multiple sequence alignment of the active site consensus motif of A. oryzae with a predicted prolyl endopeptidase from A. niger (Genbank: AX458699) and A. fumigatus (Genbank: EDP53789.1). Conserved regions were in the box and active sites were denoted by asterisks
Proline-specific endopeptidases (PEPs) are a unique class of serine proteases with considerable therapeutic potential for the treatment of celiac sprue and beer stability. However, very little is known about their structure and mechanism, therefore, it is necessary to analyze its structure, which may provide a foundation for further optimization of the useful features of PEPs.
A web-based tool for protein structure homology modeling called Swiss-Model and a software named Discovery Studio were used for predicting the structure of AO-PEP (Fig. 2). This revealed the active site of prolyl endopeptidase from A. oryzae was located in Ser206-Asp479-His528, based on the template 3n2z of the structure of human prolylcarboxypeptidase with sequence identity of 21.3 %, while prolyl endopeptidase from A. niger was located in Ser179-Asp458-His491, based on the same template 3n2z with sequence identity of 17.6 %.

Structural models of prolyl endopeptidase (AO-PEP) from A. oryzae. The α helices and β sheets are shown in red and blue, respectively. a The conserved region and active sites were marked. b The structural model of AO-PEP was constructed using the crystal structure of human prolylcarboxypeptidase (3n2z) as a template. The catalytic residues are shown by the Corey-Pauling-Koltun (CPK) representation scheme
Prolyl endopeptidase has been classified as a serine protease, and the catalytic triad residues for enzymes from different sources have already been identified. According to amino acid sequence analysis, Ser206, Asp479, and His528 are expected to be the catalytic residues of A. oryzae prolyl endopeptidase (Fig. 2). The 3D modeling first revealed the active site was located in Ser206-Asp479-His528, based on the template 3n2z with sequence identity of 21 %. Therefore, the result indicated that the prolyl endopeptidase from A. oryzae has the same active site Ser-Asp-His with human prolylcarboxypeptidase belongs to S28 protease family [30, 33]. The catalytic mechanism of this specific prolyl endopeptidase might be very interesting for further investigation.
Expression and purification of the recombinant prolyl endopeptidase
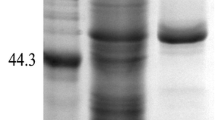
The full-length gene encoding prolyl endopeptidase was expressed in P. pastoris, and the expression was induced by methanol at 28 °C. When compared to the sample without induction, only the induced cells containing the recombinant vector secreted an extra 64 kDa protein. The recombinant prolyl endopeptidase was purified as described in “Materials and methods” (Fig. 3). The purified recombinant enzyme migrated as a single band on SDS-PAGE, similar to the calculated MW of 64.8 kDa.

The SDS-PAGE of the purified recombinant prolyl endopeptidase from A. oryzae. M protein marker, 1 recombinant prolyl endopeptidase
Characterization of the recombinant prolyl endopeptidase
Using Z-Gly-Pro-pNA as a substrate, the optimal pH and stability of recombinant prolyl endopeptidase were determined. According to the pH profile, the enzyme showed the highest activity at pH 5.0, and over 60 % of the maximum activity was retained between pH 3.0 and 8.0. However, the optimal pH and stability of the purified native prolyl endopeptidase from A. oryzae are pH 4.0–5.0 and pH 3.0–6.0, respectively [35]. Within the pH range of 3.5–7.0, it retained more than 80 % of the initial activity after incubation at 37 °C for 30 min (Fig. 4a). The recombinant prolyl endopeptidase showed the maximal activity at 40 °C (Fig. 4b), and it showed higher stability below 40 °C, with over 80 % activity after incubation for 30 min. But the enzyme was not stable at 65 and 70 °C, decreasing to 40 and 32 % maximum activity, respectively; and the activity was almost lost after incubation of 30 min at 80 °C (Fig. 4b). The requirement was satisfied to different extents by Ca2+, Na+, K+, Mg2+, Mn2+, Fe2+, Zn2+, Cu2+, and Al3+ (Table 2). Only Ca2+ showed activating effect on enzyme activity, while Na+, K+, and Mg2+ showed moderate effects. Mg2+ was better than Fe2+ under the same condition (1 mM), but the largest negative effect was observed by adding Zn2+, Mn2+, Cu2+, and Al3+. It is reasonable to speculate that Ca2+ was prevalent physiological activator for prolyl endopeptidase.

a The effect of pH on the recombinant prolyl endopeptidase activity and stability. The activity was determined under conditions at pH 2.2–12.0 and 37 °C. The pH stability was determined by incubating enzyme in citrate/disodium phosphate buffer (pH 2.2–8.0), Tris–HCl (pH 9.0–10.5), and glycine/NaOH (pH 11–12) for 30 min and assay at optimum temperature, respectively. b The effect of temperature on recombinant prolyl endopeptidase activity and stability. The activity for optimum temperature was determined under conditions at 25–80 °C and optimum pH. The effect of temperature on enzyme stability was determined by incubating the enzyme for 30 min at temperatures 25–80 °C. All the experiments were conducted in triplicate
The substrate specificity of prolyl endopeptidase was investigated by the release of the intramolecularly quenched fluorescence after hydrolysis of synthetic peptides containing the pNA group.
Kinetic parameters of AO-PEP on different substrates were determined at 40 °C and pH 4.0. Prolyl endopeptidase from A. oryzae had no catalyzing activity toward Ala-Pro-pNA, but had the capacity to hydrolyze Z-Gly-Pro-pNA, Ala-Ala-Pro-pNA, and Z-Ala-Ala-Ala-Pro-pNA. The K m, k cat, and k cat/K m of Z-Gly-Pro-pNA were determined to be 0.37 mM, 105.7 S−1, and 285.7 S−1 mM−1, respectively, in comparison to Z-Ala-Ala-Ala-Pro-pNA (K m = 0.56 mM, k cat = 93 S−1, k cat/K m = 166.1 S−1 mM−1), and Ala-Ala-Pro-pNA (K m = 0.28 mM, k cat = 139.4 S−1, k cat/K m = 496.4 S−1 mM−1).
The activity of AO-PEP in this study was relatively high, which made it a promising candidate for industrial application. Yet the K m value was also high, which indicated that the affinity between the substrate and the enzyme was weak. Therefore, future studies would be focused on lowering the K m value of this recombinant prolyl endopeptidase by protein engineering.
High-cell-density fermentation
To investigate the expression of AO-PEP in an larger scale, the batch high-cell-density fermentation was performed in a 7 l fermentor. During the initial glycerol-batch phase, the biomass (dry cell weight, DCW) increased exponentially to 34 gDCW/l and the initial glycerol content was depleted within 24 h. Then induction-batch phase began, the biomass increased linearly up and reached 116.9 gDCW/l at the end of the methanol induction-batch phase (Fig. 5). The activity of the culture supernatant was determined every 12 h during induction. As shown in Fig. 5, the highest prolyl endoprotease activity (1,130 U/l) appeared to be at 84 h and the expression level was fourfold higher than that in flask.

Constitutive expression of the prolyl endoprotease from A. oryzae in P. pastoris during fed-batch fermentation. The prolyl endoprotease clone was grown in a 7 l fermentor at fixed pH 5.5 and 30 °C. The fed-batch phase started at 24 h using glycerol as the carbon source for growth. Culture samples (10 ml) were collected at 12 h intervals and centrifuged. The prolyl endoprotease activity was assayed in the culture supernatant (extracellular activity) and the biomass (DCW dry cell weight )
Effect on the improvement of non-biological stability of beer by prolyl endopeptidase
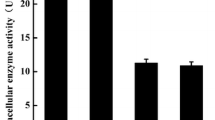
The researcher showed that non-biological stability of beer caused mainly due to the beer contained polyphenols and proteins caused by polymerization [17]. Therefore, in the beer brewing process, it is necessary to remove the protein-polyphenol in beer. The most common type of problem with beer clarity is haze. Typically the haze that causes most home brewers problems is the chill haze (which is temporary). In our experiments, after addition of AO-PEP during the fermentation phase of beer brewing(test3), the beer turbidity became the lowest, compared to that of the test1 and test2 (Fig. 6a), while the beer turbidity had little change during storage (Fig. 6b). The results showed that the recombinant prolyl endopeptidase from A. oryzae could effectively reduce the beer turbidity and improve non-biological stability of beer.

Effect on the improvement of non-biological stability of beer by prolyl endopeptidase. a Comparisons of beer stabilized by Kieselguhr filter (test1), Kieselguhr filter plus Silica gel filtration (test2), and Kieselguhr filter plus Silica gel filtration plus recombinant AO-PEP (test3) were made. b Beer turbidity changes during storage. 6 + 1 heat haze: the samples were incubated for 6 days at 60 °C in the thermostat and then for 1 days at 4 °C. After that, the turbidities of samples were immediately tested. Turbidity analyses were carried out with the use of a Hach 2100N Turbidimeter (90°) (Hach, Loveland, CO, USA) at 0 °C, and measured in EBC formazin units (EBC). Chill haze: the result of haze-producing proteins that reside in the beer. Turbidity analyses were carried out with the use of a Hach 2100N Turbidimeter (90 o) (Hach, Loveland, CO, USA) at 0 °C, and measured in EBC formazin units (EBC)
On the other hand, we found that the most frequent cause of haze in beer, wine, and clear fruit juices resulted from protein–polyphenol interaction, and these clear beverages are typically stabilized to delay the onset of protein–polyphenol haze formation [17]. Furthermore, it has been known for many years that the haze-forming activity of a polypeptide depends greatly on its proline content. This is undesirable as it can have a negative effect on the shelf life of beer, wine, and fruit juices. One way to reduce this effect is to remove these polypeptides using silica gel, or the polyphenol component with PVPP (polyvinylpolypyrrolidone). However, there are a number of disadvantages, including limited absorbing capacity of silica gel, the high capital costs of PVPP regeneration, and the inherent lowering of natural antioxidant potential of beer. The recent research found that prolyl endopeptidase would effectively hydrolyze haze-active protein in beer haze formation. In our study, the recombinant prolyl endopeptidase from A. oryzae had a significant effective on improving the non-biological stability of beer and fulfilling industrial requirements.
Conclusions
The present study reports the gene cloning, purification, characterization, and expression of prolyl endopeptidase from A. oryzae and its potential application to improve the non-biological stability of beer. The AO-PEP showed low identity from other known prolyl endopeptidase in its amino acid sequence, which is grouped into family S28. The relatively broad pH and temperature made it to be a potential application in food industry. However, the expression of prolyl endopeptidase including our recombinant AO-PEP was not very high to application in the industry directly. Therefore, in the future, we focus on the strategy in improving the expression and characterization of AO-PEP with either protein engineering or on engineering the protein expression host. Furthermore, we are interested in the AO-PEP and will do some research on its catalytic mechanism and specificity.
References
Andrews PC, Minth CD, Dixon JE (1982) Immunochemical characterization of a proline endopeptidase from rat brain. Its relationship to proline endopeptidase from other tissues and from other species. J Biol Chem 257:5861–5865
Arentz-Hansen H, McAdam SN, Molberg O, Fleckenstein B, Lundin KEA (2002) Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology 123:803–809
Aron PM, Shellhammer TH (2010) A discussion of polyphenols in beer physical and flavour stability. J Inst Brew 116:369–380
Capiralla H, Hiroi T, Hirokawa T, Maeda S (2002) Purification and characterization of a hydrophobic amino acid -specific endopeptidase from Halobacterium halobium S9 with potential application in debittering of protein hydrolysates. Process Biochem 38:571–579
Chevallier S, Goeltz P, Thibault P, Banville D, Gagnon J (1992) Characterization of a prolyl endopeptidase from Flavobacterium meningosepticum. Complete sequence and localization of the active-site serine. J Biol Chem 267:8192–8199
Edens L, Dekker P, Van der Hoeven R, Deen F, De Roos A, Floris R (2005) Extracellular prolyl endoprotease from Aspergillus niger and its use in the debittering of protein hydrolysates. J Agric Food Chem 53:7950–7957
Esparza Y, Huaiquil A, Neira L, Leyton A, Rubilar M, Salazar L, Shene C (2011) Optimization of process conditions for the production of a prolyl endopeptidase by Aspergillus niger ATCC 11414 in solid state fermentation. Food Sci Biotechnol 20:1323–1330
Gass J, Ehren J, Strohmeier G, Isaacs I, Khosla C (2005) Fermentation, purification, formulation, and pharmacological evaluation of a prolyl endopeptidase from Myxococcus xanthus: implications for Celiac Sprue therapy. Biotechnol Bioeng 92:674–684
Goertges S (1982) Problematik der Eiweissstabilisierung (Problems with protein stabilization in winemaking). Weinwirtschaft 118:931–935
Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen J, Ronning CM et al (2006) Evolution of sensory complexity recorded in a myxobacterial genome. Proc Natl Acad Sci USA 103:15200–15205
Goptar IA, Lysogorskaya EN, Filippova IY, Vinokurov KS, Zhuzhikov DP, Elpidina EN (2005) Prolyl endopeptidases from the midgut of the yellow mealworm Tenebrio molitor. FEBS J 272:154–155
Hough JSBD, Stevens R, Young TW (1982) Malting and brewing science, 2nd edn. Chapman & Hall, London, 2
Kabashima T, Fujii M, Meng Y, Ito K, Yoshimoto T (1998) Prolyl endopeptidase from Sphingomonas capsulata: isolation and characterization of the enzyme and nucleotide sequence of the gene. Arch Biochem Biophys 358:141–148
Kanatani A, Yoshimoto T, Kitazono A, Kokubo T, Tsuru D (1993) Prolyl endopeptidase from Aeromonas hydrophila: cloning, sequencing, and expression of the enzyme gene, and characterization of the expressed enzyme. J Biochem 113:790–796
Kang C, Yu XW, Xu Y (2013) Gene cloning and enzymatic characterization of an endoprotease Endo-Pro-Aspergillus niger. J Ind Microbiol Biotechnol 40:855–864
Kang C, Yu XW, Xu Y (2014) Purification and characterization of a prolyl endopeptidase isolated from Aspergillus oryzae. J Ind Microbiol Biotechnol 41:49–55
Siebert KJ (1999) Effects of protein-polyphenol interactions on beverage haze, stabilization, and analysis. J Agric Food Chem 47:353–362
Kubota K, Tanokura M, Takahashi K (2005) Purification and characterization of a novel prolyl endopeptidase from Aspergillus niger. Proc Jpn Acad B Phys 81:447–453
Kuwabara T (1992) Characterization of a prolyl endopeptidase from spinach thylakoids. FEBS Lett 300:127–130
Lopez M, Edens L (2005) Effective prevention of chill-haze in beer using an acid proline-specific endoprotease from Aspergillus niger. J Agric Food Chem 53:7944–7949
Mary Booth WJD, Fhaoláin IN, Jennings PV, O’Cuinn G (1990) Proline-specific peptidases of Streptococcus cremoris AM2. J Dairy Res 57:79–88
Mentlein R (1988) Proline residues in the maturation and degradation of peptide hormones and neuropeptides. FEBS Lett 234:251–256
Nadzeyka A, Altenhofen U, Zahn H (1979) The significance of beer proteins in relationship to cold break and age-related haze formation. Brauwissenschaft 32:167–172
Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS et al (2005) Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438:1151–1156
O’Leary RM, Gallagher SP, O’Connor B (1996) Purification and characterization of a novel membrane-bound form of prolyl endopeptidase from bovine brain. Int J Biochem Cell Biol 28:441–449
O’Leary RM, O’Connor B (1995) Identification and localisation of a synaptosomal membrane prolyl endopeptidase from bovine brain. Eur J Biochem 227:277–283
Oyama H, Aoki H, Amano M, Mizuki E, Yoshimoto T, Tsuru D, Murao S (1997) Purification and characterization of a prolyl endopeptidase from Pseudomonas sp. KU-22. J Ferment Bioeng 84:538–542
Riggle HM, Fisher MA (2009) Purification of a prolyl endopeptidase from Aspergillus oryzae and evaluation of its ability to digest gluten. Abstracts of Papers of the American Chemical Society 237
Sattar AK, Yamamoto N, Yoshimoto T, Tsuru D (1990) Purification and characterization of an extracellular prolyl endopeptidase from Agaricus bisporus. J Biochem 107:256–261
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucl Acid Res 31:3381–3385
Shan L, Marti T, Sollid LM, Gray GM, Khosla C (2004) Comparative biochemical analysis of three bacterial prolyl endopeptidases: implications for coeliac sprue. Biochem J 383:311–318
Shen GX, Shi JP (1999) Cloning and nucleotide sequencing of prolyl endopeptidase gene from Aeromonas punctata subsp. punctata. Acta Biochim Biophys Sin 31:567–571
Soisson SM, Patel SB, Abeywickrema PD, Byrne NJ, Diehl RE et al (2010) Structural definition and substrate specificity of the S28 protease family: the crystal structure of human prolylcarboxypeptidase. BMC Struct Biol 10:16
Sridhar VR, Hughes JE, Welker DL, Broadbent JR, Steele JL (2005) Identification of endopeptidase genes from the genomic sequence of Lactobacillus helveticus CNRZ32 and the role of these genes in hydrolysis of model bitter peptides. Appl Environ Microb 71:3025–3032
Szwajcer-Dey E, Rasmussen J, Meldal M, Breddam K (1992) Proline-specific endopeptidases from microbial sources: isolation of an enzyme from a Xanthomonas sp. J Bacteriol 174:2454–2459
Yoshimoto T, Walter R, Tsuru D (1980) Proline-specific endopeptidase from Flavobacterium. Purification and properties. J Biol Chem 255:4786–4792
Yoshimoto T, Kanatani A, Shimoda T, Inaoka T, Kokubo T, Tsuru D (1991) Prolyl endopeptidase from Flavobacterium meningosepticum: cloning and sequencing of the enzyme gene. J Biochem 110:873–878
Acknowledgments
Financial support from the National High Technology Research and Development Program of China (863 Program) (No. 2012AA022207), the National Key Basic Research and Development Program of China (973 Program) (No. 2011CB710800), the High-end Foreign Experts Recruitment Program (GDW20123200113) and the 111 Project (111-2-06) are greatly appreciated.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Kang, C., Yu, XW. & Xu, Y. Cloning and expression of a novel prolyl endopeptidase from Aspergillus oryzae and its application in beer stabilization. J Ind Microbiol Biotechnol 42, 263–272 (2015). https://doi.org/10.1007/s10295-014-1571-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1571-8