
Abstract
The acetolactate synthase (als)-deficient mutant of Klebsiella pneumoniae fails to produce 1,3-propanediol (1,3-PD) or 2,3-butanediol (2,3-BD), and is defective in glycerol metabolism. In an effort to recover production of the industrially valuable 1,3-PD, we introduced the Zymomonas mobilis pyruvate decarboxylase (pdc) and aldehyde dehydrogenase (aldB) genes into the als-deficient mutant to activate the conversion of pyruvate to ethanol. Heterologous expression of pdc and aldB efficiently recovered glycerol metabolism in the 2,3-BD synthesis-defective mutant, enhancing the production of 1,3-PD by preventing the accumulation of pyruvate. Production of 1,3-PD in the pdc- and aldB-expressing als-deficient mutant was further enhanced by increasing the aeration rate. This system uses metabolic engineering to produce 1,3-PD while minimizing the generation of 2,3-BD, offering a breakthrough for the industrial production of 1,3-PD from crude glycerol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Copious raw glycerol is formed as the main by-product of biodiesel production, corresponding to as much as 10 % (w/w) of the generated biodiesel [1]. This surplus of raw glycerol poses a significant environmental problem, since it cannot be discharged directly into the environment without any treatment [2]. Numerous researchers have sought to use glycerol as a low-cost feedstock for refinement into industrially valuable materials [3–5], including 1,3-propanediol (1,3-PD). 1,3-PD is a valuable chemical that is mainly polymerized with terephthalates for the synthesis of polymethylene terephthalates, which are used in the manufacturing of textile fiber, film and plastic [6].
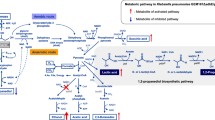
Klebsiella pneumoniae is a typical microbial strain that is capable of producing 1,3-PD from glycerol. The metabolic pathway responsible for microbial conversion has been well studied (Fig. 1) [7]. In K. pneumoniae, glycerol is used as a carbon and energy source for the generation of biomass, resulting in the production of various metabolites, including acetate, ethanol, lactate and 2,3-butanediol (2,3-BD) (Fig. 1). Additionally, a reductive pathway was accompanied to balance intracellular redox level during glycerol assimilation. In the reductive pathway, 1,3-PD is generated from glycerol by the action of reduced nicotinamide adenine dinucleotide (NADH)-dependent oxidoreductase).

The glycerol metabolic pathway in Klebsiella pneumoniae. Pyruvate decarboxylase (Pdc) and aldehyde dehydrogenase (AldB) from Zymomonas mobilis are indicated in dotted line. LdhA lactate dehydrogenase, Als acetolactate synthase, Pta phosphate acetyltransferase
Based on the genetic elucidation of fermentative glycerol metabolism [8], researchers have sought to enhance the production yield of 1,3-PD [9–13]. One efficient strategy for increasing 1,3-PD production is engineering the oxidative pathway to minimize the production of by-products. A successful result was obtained following inactivation of the lactate dehydrogenase (ldhA) gene, which is involved in lactate synthesis and is a major metabolite of the glycerol oxidation pathway [14]. We previously inactivated 2,3-BD production and assessed subsequent glycerol metabolism in K. pneumoniae [14]. This genetic engineering reduced the production level of 1,3-PD, and was thus unsatisfactory for industrial applications. Given that the boiling point of 2,3-BD is similar to that of 1,3-PD [15], potentially hampering the high-purity recovery of 1,3-PD, strategies should be developed to maximize the production of 1,3-PD while minimizing that of 2,3-BD. Here, we successfully used genetic engineering to enhance the production of 1,3-PD in a 2,3-BD-deficient mutant.
Materials and methods
Bacterial strains, plasmids, and growth media
The K. pneumoniae ΔldhA and Δ(ldhA als) mutant, derived from ATCC 200721, was described previously [14]. Escherichia coli DH5α was used for DNA manipulation. The λ Red and FLP recombinases were expressed by helper plasmids pKD46 and pCP20, respectively. The replication of these plasmids is temperature-sensitive, allowing them to be easily eliminated. The pIJ773 vector was used as the source of the apramycin-resistance gene. The pBR322 vector was used to create pBR-aldB-pdc, which encoded the Zymomonas mobilis pyruvate decarboxylase (Pdc) and aldehyde dehydrogenase (AldB) genes. Microbial cells were grown in LB [yeast extract (Difco), 0.5 % (w/v); Bacto-tryptone (Difco), 1.0 % (w/v); and NaCl, 1.0 % (w/v)] or germ medium [16] supplemented with appropriate antibiotics [ampicillin (50 μg mL−1), apramycin (50 μg mL−1), or tetracycline (10 μg mL−1)]. The germ medium contained 20 g m−1 crude glycerol (purity 80 % w/w), 2 g L−1 (NH4)2SO4, 3.4 g L−1 K2HPO4, 1.3 g L−1 KH2PO4, 0.2 g L−1 MgSO4, 0.02 g L−1 CaCl2·2H2O, 1 g L−1 yeast extract, 1 mL Fe solution [5 g L−1 FeSO4·7H2O and 4 mL L−1 HCl (37 %, w/v)], and 1 mL trace element solution [70 mg L−1 ZnCl2, 100 mg L−1 MnCl2·4H2O, 60 mg L−1 H3BO3, 200 mg L−1 CoCl2·4H2O, 20 mg L−1 CuCl2·2H2O, 25 mg L−1 NiCl2·6H2O, 35 mg L−1 Na2MoO4·2H2O, and 4 mL L−1 HCl (37 %, w/v)]. Crude glycerol was obtained from a biodiesel-producing company (GSBio, Yeosu, Korea).
Deletion of the pta gene
For construction of the pta deletion mutant (Supplementary Fig. 1), the 300-bp DNA sequences located upstream and downstream of pta were PCR amplified using oligonucleotides P1 (5′-acccgcaataattcgagctg-3′) and P2 (5′-gagcgctgtaccgctttgtaGTTAACgcgataggtttaaagacgctcag-3′; italics indicate an HpaI site) for the upstream region, and P3 (5′-ctgagcgtctttaaacctatcgcGTTAACtacaaagcggtacagcgctc-3′; italics indicate an HpaI site) and P4 (5′-agagagcgagcgcgaataaa-3′) for the downstream region. The PCR products were annealed via overlapping regions of the P2 and P3 primers, amplified as a single fragment using primers P1/P4, and cloned into the pGEM-T Easy vector. The resulting plasmid was digested with HpaI and ligated with an apramycin-resistance gene [aac(3)IV] obtained from pIJ773 by digestion with EcoRI and HindIII and treatment with the Klenow fragment. The resultant plasmid, designated pT-pta-Apra, was used as a template for PCR amplification of the deletion cassette, which was introduced into K. pneumoniae Δ(ldhA als) by electroporation [17] to induce homologous recombination. Correct integration of the DNA fragment was confirmed by Southern hybridization using the upstream regions of pta and aac(3)IV to probe DraI-digested chromosomal DNA.
Construction of recombinant strain expressing pdc and adh gene
The 1.8-kb open reading frame (orf) of pdc was amplified from chromosomal DNA of Z. mobilis ZM4 using the following primers: Ppdc-F (5′-TCTAGAATGAGTTATACTGTCGGTACCTATTTAGC-3′; italicized bases indicate an XbaI site) and Ppdc-R (5′-CTCGAG CTGCAGCTAGAGGAGCTTGTTAACAGGCTTAC-3′; italicized and underlined letters indicate an XhoI and a PstI site, respectively). A DNA fragment including a 1.15-kb aldB segment was amplified from chromosomal DNA of Z. mobilis ZM4 using the following primers: PaldB-F (5′-AGATCTATGGCTTCTTCAACTTTTTATATTCC-3′; italicized letters indicate a BglII site) and PaldB-R (5′-CTCGAG TCTAGATTAGAAAGCGCTCAGGAAGAGTT-3′; italicized and underlined letters indicate XhoI and XbaI sites, respectively). The lacZ promoter sequence (P lacZ -aldB) was amplified using specific primers; these were PlacZ-aldB-F (5′-GAATTCAGCGGGCAGTGAGCGCAA-3′; italicized letters indicate an EcoRI site) and PlacZ-aldB-R (5′-CTCAGA AGATCTAGCTGTTTCCTGTGTGAAATTG-3´; italicized and underlined letters indicate XhoI and BglII sites, respectively). Amplified DNA fragments were cloned into the pGEM TEasy (Promega) vector, followed by nucleotide sequencing to confirm the absence of any sequence error. A BglII–XhoI fragment including the aldB gene was inserted between equivalent restriction sites downstream of a lacZ promoter sequence. An XbaI–XhoI fragment, including the pdc gene, was next inserted between corresponding sites of pGEM-P lacZ -aldB to create pGEM-P lacZ -aldB-pdc. Finally, an EcoRI–PstI fragment including P lacZ -aldB-pdc was inserted between the corresponding sites of pBR322 to create pBR-aldB-pdc. The final plasmids was transformed into K. pneumoniae Δ(ldhA als) by electroporation.
Preparation of cell-free extracts and enzyme activity assays
Cells were grown in culture and shaken overnight at 100 rpm in 250-mL flasks containing 50 mL of glycerol media, until they reached the stationary phase. The cells were then harvested by centrifugation (13,000 rpm, 4 °C, 10 min), and each cell pellet was washed twice with cold potassium phosphate buffer (50 mM, pH 7.0) and re- suspended in potassium phosphate buffer (50 mM, pH 7.0). The samples were sonicated in an ice bath for 90 cycles (each cycle = 3 s at 200 W followed by a 5-s pause). Cell debris was removed by centrifugation (13,000 rpm, 4 °C, 20 min), and the enzyme activities of pyruvate decarboxylase (Pdc) and aldehyde dehydrogenase (AldB) were measured as described by Postma et al. [18]. One unit of enzyme activity was defined as the amount of enzyme that consumed 1 µmol of substrate per min. Protein concentrations were determined by a protein assay kit (Bio-Rad), with BSA used as a standard. All activity measurements were performed in triplicate.
Fermentation by K. pneumoniae strains
For fermentations, seed cells were prepared in 1-L flasks containing 200 mL of germ medium. The flasks were incubated at 37 °C for 12 h, and then the culture [10 % (v/v)] was inoculated into the growth vessel. Fed-batch fermentations were conducted in a 5-L stirred-vessel system (Kobiotech Co. Ltd.) containing 2 L of germ medium; all fermentation experiments were conducted at 37 °C with stirring at 200 rpm. Unless stated otherwise, the pH was maintained at pH 6.5 ± 0.2 and the aeration rate was 2.0 vvm. The crude glycerol was controlled between 30 and 80 g L−1 in subsequent fed-batch fermentations. All data are given as the average from three independent experiments.
Metabolite analysis
The levels of residual glycerol 1,3-PD, ethanol, acetate, lactate, succinate, and 2,3-BD were determined using an HPLC apparatus equipped with a refractive index detector and an organic acid analysis column (300 × 78 mm; Aminex HPX-87H; Bio-Rad). The mobile phase was 5 mM H2SO4 and the flow rate was 0.6 mL min−1. The column and cell temperatures were 65 and 45 °C, respectively. The biomass concentration was determined by measurement of optical density at 600 nm (OD600).
Results
Effect of pta gene deletion in the als-deficient K. pneumoniae mutant
We previously showed that inactivation of the acetolactate synthase (Als) gene reduces 2,3-BD production and subsequent 1,3-PD production in K. pneumoniae [14]. Here, we found that glycerol assimilation and cell growth were also decreased in the mutant (Fig. 2), and confirmed the low-level production of 1,3-PD (Table 1). We also observed a remarkable increase in the acetate level of the als-deficient mutant (Table 1), prompting us to speculate that there may be an association between acetate accumulation and the observed defect in glycerol metabolism. To test this hypothesis, we constructed and characterized a phosphate acetyltransferase (pta)-deficient mutant. As expected, inactivation of pta in the als-deficient mutant decreased the acetate level (Table 1). However, the metabolic defect of the als mutation was not recovered (Fig. 2; Table 1). These results indicate that the metabolic defects in the als-negative mutant are not directly caused by the observed increase in the acetate level.

Glycerol consumption rate (a) and cell growth (b) of K. pneumoniae mutants. Symbols: closed circles, K. pneumoniae ΔldhA; open circles, K. pneumoniae Δ(ldhA als); closed triangles, K. pneumoniae Δ(ldhA als pta); and open triangles, K. pneumoniae Δ(ldhA als) harboring pBR-aldB-pdc
Effect of expression of the Z. mobilis pdc and aldB genes in the als-deficient K. pneumoniae mutant
To evaluate whether the metabolic defects in the als-deficient mutant were related to accumulation of the metabolic intermediate, pyruvate, we expressed the pyruvate decarboxylase (Pdc) and aldehyde dehydrogenase (AldB) genes from Z. mobilis in the mutant strain to stimulate conversion of pyruvate into ethanol (Fig. 1). Following this genetic engineering, we detected higher activities of Pdc and AldB in the recombinant strain compared to the parent strain (Fig. 3), indicating that our genetic engineering was successful.

Effect of IPTG concentration on the enzyme activity. Symbols: closed bar crude cell lysate from K. pneumoniae Δ(ldhA als), and open bar crude cell lysate from Δ(ldhA als)/pBR-aldB-pdc. Pdc pyruvate decarboxylase, AldB aldehyde dehydrogenase
As shown in Fig. 2, heterologous expression of the pdc and aldB genes recovered the glycerol metabolism and cell growth of the als-negative mutant to levels similar to that of the corresponding als-positive strain (ΔldhA). The production of 1,3-PD was enhanced in the engineered strain, whereas 2,3-BD production remained entirely blocked (Table 1). Ethanol production was strikingly accelerated in the engineered strain, whereas acetate production was inhibited due to the stimulation of conversion of pyruvate to ethanol.
Fed-batch fermentation of the pBR-aldB-pdc-transfected Δ(ldhA als) mutant in a bioreactor
Following fed-batch fermentation, we observed similar 1,3-PD production levels in the als-positive ΔldhA mutant and the Δ(ldhA als)/pBR-aldB-pdc mutant (Fig. 4; Table 2). The 2,3-BD level was lower in the Δ(ldhA als) mutant, as was the molar ratio of 2,3-BD to 1,3-PD. In contrast to our earlier batch-type cultivation, where 2,3-BD was not detected, we did detect 2,3-BD at different levels in the fed-batch-grown mutants.

Fermentation of K. pneumoniae mutant strains fed with crude biodiesel-industry-derived glycerol at an aeration rate of 2 vvm. a K. pneumoniae ΔldhA mutant, b K. pneumoniae Δ(ldhA als) mutant, and c K. pneumoniae Δ(ldhA als) harboring pBR-aldB-pdc
A large amount of acetate accumulated in the culture broth of the als-negative mutant during fed-batch fermentation, due to the decreased conversion of pyruvate to 2,3-BD (Fig. 5). Notably however, the accumulation of acetate was completely complemented by expression of the pdc and aldB genes (Fig. 4; Table 2). This finding strongly suggests that the metabolic defects in the als-negative mutant are caused by accumulation of the metabolic intermediate, pyruvate, and may be recovered by metabolic engineering aimed at removing this accumulation (e.g., via the expression of pdc and aldB).

Pyruvate analysis following fed-batch fermentation of the K. pneumoniae mutant strains. Symbols: closed circles, K. pneumoniae ΔldhA harboring pBR322; open circles, K. pneumoniae Δ(ldhA als) harboring pBR322; closed triangles, K. pneumoniae Δ(ldhA als) harboring pBR-aldB-pdc
Effect of aeration rate on fed-batch fermentation of the Δ(ldhA als)-transfected pBR-aldB-pdc mutant
Finally, we evaluated the effect of different aeration rates on glycerol metabolism by the Δ(ldhA als)/pBR-aldB-pdc strain (Fig. 6). When the aeration rate was increased to 3.0 vvm from 2.0 vvm, the production level of 1,3-PD was slightly increased (Figs. 4c, 6b). The productivity and conversion rate were also enhanced by the fermentation condition, from 1.32 to 1.42 (g L−1 h−1) and from 0.51 to 0.63 (mol mol−1), respectively (Table 2). However, production of by-product 2,3-BD was also slightly increased. Contrastively, when aeration rate was reduced, 1,3-PD production significantly decreased. The decrease of 1,3-PD production might be due to the reduced cell growth. Low aeration rate also increased the ethanol level, further accompanying with the decrease of acetate level. Collectively, our results indicate that optimization of fermentation parameters together with the further metabolic engineering, such as the disruption of other als homologs, could further elevate 1,3-PD production in the als-negative mutant.

Effect of aeration rate during fed-batch fermentation on the glycerol metabolism of K. pneumoniae Δ(ldhA als) harboring pBR-aldB-pdc. a 0.5 vvm, and b 3.0 vvm
Discussion
The by-products of the fermentation, such as ethanol and acetate can be easily separated from 1,3-PD. However, 2,3-BD is a major by-product during the synthesis of 1,3-PD and it may serve as an obstacle for obtaining a high purity of 1,3-PD in downstream processes because of its similar boiling point [15].
In this study, we successfully used genetic engineering to enhance the production of 1,3-PD in the K. pneumoniae from glycerol while minimizing 2,3-BD production. We hypothesized that glycerol metabolism in the als-deficient mutant was affected by accumulation of the intermediate metabolite, pyruvate, and showed that heterologous expression of the Z. mobilis pdc and aldB genes efficiently recovered the metabolic defect and improved 1,3-PD production with cell growth in the als-deficient mutant by converting pyruvate to ethanol. However, the 1,3-PD production levels on glycerol is still slightly lower than ΔldhA mutant and production of by-product ethanol was strikingly increased from 0.55 to 8.21 (g L−1) (Table 1). This indicated that most of the NADH was consumed for ethanol production than 2,3-BD pathway and thus reduced the 1,3-PD production levels [19, 20]. These results may prove to be a promising alternative for enhancing the industrial production of 1,3-PD from crude glycerol while minimizing 2,3-BD production.
We have previously identified and characterized an aldehyde/alcohol dehydrogenase (AdhE) involved in production of ethanol from glycerol in K. pneumoniae [21]. We also expressed adhE in the als-deficient mutant. However, 1,3-PD production was not recovered in the recombinant strain (data not shown). K. pneumoniae AdhE catalyzes synthesis of ethanol from acetyl-CoA not from pyruvate (Fig. 1). The results indicated that removal of accumulated pyruvate is crucial for the recovery of glycerol metabolism in Δ(ldhA als)/pBR-aldB-pdc strain.
In anaerobic and microaerobic conditions, acetate production is closely related with cell growth, which can supply ATP [22]. The cultivations carried out in this study were maintained at microaerobic condition irrespective of aeration rate. Agreed with, production of acetate was observed at growth stage, and then the production was stopped with the cessation of growth. The lower growth of Δ(ldhA als)/pBR-aldB-pdc strain and subsequent decreased production of 1,3-PD at lower aeration rate condition might be due to the stimulated production of ethanol from pyruvate, inhibiting acetate synthesis (Table 2). This means that precise optimization of cultivation condition is necessary for the best performance of engineered strain considering metabolic balance. Maximal production of 1,3-PD was observed at 3.0 vvm aeration rate by the Δ(ldhA als)/pBR-aldB-pdc. It has been also reported that growth of K. pneumoniae under microaerobic conditions (compared to anaerobic conditions) results in higher levels of 1,3-PD production [23].
In the als-deficient mutant of K. pneumoniae, 2,3-BD was still produced although the level was significantly decreased. It could be explained by presence of homologs of Als enzymes. Agreed with, genes annotated as acetolactate synthase I, II, and III were identified from the whole-genome sequences of K. pneumoniae. Differ to the als, the genes were not clustered with genes encoding acetolactate decarboxylase (Adc) and 2,3-BD dehydrogenase/acetoin reductase (Ard) involved in 2,3-BD biosynthesis. The gene products may be majorly involved in synthesis of branched chain amino acids such as valine in K. pneumoniae, in which, the biosynthetic pathway is branched out of acetolactate. The als homologs might be logical targets for further engineering in a future study.
References
Johnson DT, Taconi KA (2007) The glycerin glut: options for the value-added conversion of crude glycerol resulting from biodiesel production. Environ Prog 26:338–348
da Silva GP, Mack M, Contiero J (2009) Glycerol: a promising and abundant carbon source for industrial microbiology. Biotechnol Adv 27:30–39. doi:10.1016/j.biotechadv.2008.07.006
Hong WK, Kim CH, Heo SY, Luo LH, Oh BR, Seo JW (2010) Enhanced production of ethanol from glycerol by engineered Hansenula polymorpha expressing pyruvate decarboxylase and aldehyde dehydrogenase genes from Zymomonas mobilis. Biotechnol Lett 32:1077–1082. doi:10.1007/s10529-010-0259-z
Luo LH, Seo JW, Heo SY, Oh BR, Kim DH, Kim CH (2013) Identification and characterization of Klebsiella pneumoniae aldehyde dehydrogenases increasing production of 3-hydroxypropionic acid from glycerol. Bioprocess Biosys Eng 40:1319–1326. doi:10.1007/s00449-012-0880-4
Oh BR, Seo JW, Choi MH, Kim CH (2008) Optimization of culture conditions for 1,3-propanediol production from crude glycerol by Klebsiella pneumoniae using response surface methodology. Biotechnol Bioprocess Eng 13:666–670
Pagliaro M, Ciriminna R, Kimura H, Rossi M, Della Pina C (2007) From glycerol to value-added products. Angew Chem Int Ed Engl 46:4434–4440. doi:10.1002/anie.200604694
Skraly FA, Lytle BL, Cameron DC (1998) Construction and characterization of a 1,3-propanediol operon. Appl Environ Microbiol 64:98–105
Sun J, Heuvel J, Soucaille P, Qu Y, Zeng AP (2003) Comparative genomic analysis of dha regulon and related genes for anaerobic glycerol metabolism in bacteria. Biotechnol Prog 19:263–272. doi:10.1021/bp025739m
Hao J, Wang W, Tian J, Li J, Liu DH (2008) Decrease of 3-hydroxypropionaldehyde accumulation in 1,3-propanediol production by over-expressing dhaT gene in Klebsiella pneumoniae TUAC01. J Ind Microbiol Biotechnol 35:735–741. doi:10.1007/s10295-008-0340-y
Nakamura CE, Whited GM (2003) Metabolic engineering for the microbial production of 1,3-propanediol. Curr Opin Biotechnol 14:454–459. doi:10.1016/j.copbio.2003.08.005
Oh BR, Hong WK, Heo SY, Luo LH, Kondo A, Seo JW, Kim CH (2013) The production of 1,3-propanediol from mixtures of glycerol and glucose by a Klebsiella pneumoniae mutant deficient in carbon catabolite repression. Bioresour Technol 130:719–724. doi:10.1016/j.biortech.2012.12.076
Tong IT, Liao HH, Cameron DC (1991) 1,3-propanediol production by Escherichia coli expressing genes from the Klebsiella pneumoniae dha regulon. Appl Env Microbiol 57:3541–3546
Zheng P, Wereath K, Sun J, van den Heuvel J, Zeng AP (2006) Overexpression of genes of the dha regulon and its effects on cell growth, glycerol fermentation to 1,3-propanediol and plasmid stability in Klebsiella pneumoniae. Process Biochem 41:2160–2169
Oh BR, Seo JW, Heo SY, Hong WK, Luo LH, Kim SH, Park DH, Kim CH (2012) Optimization of culture conditions for 1,3-propanediol production from glycerol using a mutant strain of Klebsiella pneumoniae. Appl Biochem Biotechnol 166:127–137. doi:10.1007/s12010-011-9409-6
Xiu ZL, Zeng AP (2008) Present state and perspective of downstream processing of biologically produced 1,3-propanediol and 2,3-butanediol. Appl Microbiol Biotechnol 78:917–926. doi:10.1007/s00253-008-1387-4
Oh BR, Seo JW, Heo SY, Hong WK, Luo LH, Joe MH, Park DH, Kim CH (2011) Efficient production of ethanol from crude glycerol by a Klebsiella pneumoniae mutant strain. Bioresour Technol 102:3918–3922. doi:10.1016/j.biortech.2010.12.007
Fournet-Fayard S, Joly B, Forestier C (1995) Transformation of wild type Klebsiella pneumoniae with plasmid DNA by electroporation. J Microbiol Methods 24:49–54
Postma E, Verduyn C, Scheffers WA, Van Dijken JP (1989) Enzymic analysis of the crabtree effect in glucose-limited chemostat cultures of Saccharomyces cerevisiae. Appl Environ Microbiol 55:468–477
Ahren K, Menzel K, Zeng AP, Deckwer WD (1998) Kinetic, dynamic, and pathway studies of glycerol metabolism by Klebsiella pneumoniae in anaerobic continuous culture: III enzymes and fluxes of glycerol dissimilation and 1,3-propanediol formation. Biotechnol Bioeng 59:544–552
Biebl H, Zeng AP, Menzel K, Deckwer WD (1998) Fermentation of glycerol to 1,3-propanediol and 2,3-butanediol by Klebsiella pneumoniae. Appl Microbiol Biotechnol 50:248–249
Oh BR, Hong WK, Heo SY, Joe MH, Seo JW, Kim CH (2013) The role of aldehyde/alcohol dehydrogenase (AdhE) in ethanol production from glycerol by Klebsiella pneumoniae. J Ind Microbiol Biotechnol 40:227–233. doi:10.1007/s10295-012-1224-8
Xue X, Li W, Li Z, Xia Y, Ye Q (2010) Enhanced 1,3-propanediol production by supply of organic acids and repeated fed-batch culture. J Ind Microbiol Biotechnol 37:681–687. doi:10.1007/s10295-010-0711-z
Cheng KK, Liu DH, Sun Y, Liu WB (2004) 1,3-Propanediol production by Klebsiella pneumoniae under different aeration strategies. Biotechnol Lett 26:911–915
Cheng K-K, Liu H-J, Liu D-H (2005) Multiple growth inhibition of Klebsiella pneumoniae in 1,3-propanediol fermentation. Biotechnol Lett 27:19–22. doi:10.1007/s10529-004-6308-8
Acknowledgments
This subject was supported by Korea Ministry of Environment as “Converging technology project” and by Korea Research Council of Fundamental Science and Technology.
Author information
Authors and Affiliations
Corresponding authors
Additional information
The authors S.-M. Lee and W.-K. Hong are co-first authors and contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lee, SM., Hong, WK., Heo, SY. et al. Enhancement of 1,3-propanediol production by expression of pyruvate decarboxylase and aldehyde dehydrogenase from Zymomonas mobilis in the acetolactate-synthase-deficient mutant of Klebsiella pneumoniae . J Ind Microbiol Biotechnol 41, 1259–1266 (2014). https://doi.org/10.1007/s10295-014-1456-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1456-x