
Abstract
We determined the complete nucleotide sequence of the plastid genome of the unicellular marine red alga Porphyridium purpureum strain NIES 2140, belonging to the unsequenced class Porphyridiophyceae. The genome is a circular DNA composed of 217,694 bp with the GC content of 30.3 %. Twenty-nine of the 224 protein-coding genes contain one or multiple intron(s). A group I intron was found in the rpl28 gene, whereas the other introns were group II introns. The P. purpureum plastid genome has one non-coding RNA (ncRNA) gene, 29 tRNA genes and two nonidentical ribosomal RNA operons. One rRNA operon has a tRNAAla(UGC) gene between the rrs and the rrl genes, whereas another has a tRNAIle(GAU) gene. Phylogenetic analyses suggest that the plastids of Heterokontophyta, Cryptophyta and Haptophyta originated from the subphylum Rhodophytina. The order of the genes in the ribosomal protein cluster of the P. purpureum plastid genome differs from that of other Rhodophyta and Chromalveolata. These results suggest that a large-scale rearrangement occurred in the plastid genome of P. purpureum after its separation from other Rhodophyta.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plastids, unique organelles in photosynthetic eukaryotes, are capable of oxygenic photosynthesis and have their own genomes and genetic systems. Endosymbiotic cyanobacteria are widely accepted to be the origin of plastids. There are two types of plastids; one is a direct descendent of the primary cyanobacterium endosymbiont (i.e., “primary” plastids) and the other was acquired secondarily from primary plastids, that is, endosymbiosis of plastid-bearing eukaryotes by another eukaryotic host (i.e., “secondary” or “tertiary” plastids) (Kim and Archibald 2009). Primary plastids are found in Archaeplastida, including three phyla, Glaucophyta, Rhodophyta and Viridiplantae.
Rhodophyta (red alga) is divided into two subphyla with seven classes (Yoon et al. 2006): the subphylum Cyanidiophytina contains one class (Cyanidiophyceae) and the subphylum Rhodophytina contains six classes (Florideophyceae, Bangiophyceae, Compsopogonophyceae, Stylonematophyceae, Rhodellophyceae and Porphyridiophyceae). The complete plastid genomes of Rhodophyta were reported in only three classes: two species of Cyanidiophyceae (Glöckner et al. 2000; Ohta et al. 2003), four species of Florideophyceae (DePriest et al. 2013; Hagopian et al. 2004; Janouškovec et al. 2013) and three species of Bangiophyceae (Reith and Munholland 1995; Wang et al. 2013). The detailed phylogenetic relationships of Rhodophyta remain unidentified except in these three classes.
The plastid of Chromalveolata (stramenopiles, Alveolata, Cryptophyta and Haptophyta) originates from Rhodophyta (Cavalier-Smith 1998), but a more accurate phylogenetic relationship remains to be determined, that is, whether the plastid of Chromalveolata originated from the ancestor of Rhodophytina or the ancestor of (Florideophyceae + Bangiophyceae), because the only three classes for the complete plastid genome of Rhodophyta were used in the phylogenetic analyses (Wang et al. 2013). Recently, a draft genome of Porphyridium purpureum CCMP 1328 was reported (Bhattacharya et al. 2013), but the complete plastid genome of this species has not been published.
Here, we report the sequence of the plastid genome of Porphyridium purpureum strain NIES 2140. This strain is a unicellular marine rhodophyte belonging to the unsequenced class Porphyridiophyceae. We analyzed its genome structure and gene content. In addition, using concatenated amino acid alignments of plastid-encoded proteins, we determined the phylogenetic relationship of P. purpureum and the origin of the Chromalveolata plastid.
Materials and methods
DNA sources
P. purpureum strain NIES 2140 was obtained from the collection of the National Institute for Environmental Studies (Tsukuba, Japan). Cells of P. purpureum were cultured in a modified medium similar to enriched sea water medium (ESM) (http://mcc.nies.go.jp/02medium.html#esm) (Okaichi et al. 1982) without soil extract, but including 35 g l−1 of an artificial seawater called Viesalt (MARINETECH, Tokyo, Japan) instead of seawater. The preculture was statically grown at 22 °C under white light (12 h light: 12 h dark; 20 μmol m−2 s−1) for about a month. For DNA isolation, the cells were grown at 30 °C with aeration by 1 % CO2 under white light at 30 μmol m−2 s−1 for 6 days. The cells were harvested by centrifugation, and disrupted with glass beads. DNA was released by treatment with cetyltrimethylammonium bromide (CTAB) and proteinase K, extracted with phenol/chloroform, and purified by CsCl ultracentrifugation.
DNA sequencing
Genomic DNA (5.5 μg) was sheared by ultrasonic treatment and sequenced with a genome sequencer FLX instrument (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer’s protocol (this is usually referred to as ‘454 sequencing’). Total genome data of P. purpureum strain NIES 2140 were assembled with Newbler Matrics version 2.5.3 into 3,355 contigs (average length = 5,775 bp). The contigs had a peak depth of 27.0 and totaled 19.4 Mbp. To find its genomic origin, namely, nuclear genome or organelle genomes, each contig was analyzed by blastn software version 2.2.18 against the sequences of the organelles in algae and plants as well as the nuclear genome. The sequenced reads corresponding to plastid sequences were assembled into 13 contigs. PCR experiments were performed to fill the gaps. This plastid genome has a pair of nearly identical inverted repeat sequences. To validate the sequence differences, each of the repeat sequences was separately amplified by long PCR. PCR products were sequenced by conventional Sanger sequencing, using the sequencing services of FASMAC Co. Ltd. (Atsugi, Japan). The complete plastid genome is available under the GenBank Accession Number AP012987.
Genome annotations and data analyses
To detect the open reading frames (ORFs), we used the MetaGeneAnnotator program (Noguchi et al. 2008). Then, we used the tblastn program, which was run against the cluster data of CyanoClust (Sasaki and Sato 2010) prepared by the Gclust software (Sato 2009). Homology search using tblastn was also run against the plastid genomes of seven species of Heterokontophyta: Ectocarpus siliculosus, Fistulifera sp. JPCC DA0580, Fucus vesiculosus, Heterosigma akashiwo, Synedra acus, Thalassiosira oceanica and Vaucheria litorea (Supplementary material 1). Finally, we searched for ORFs that were longer than 30 codons and started with ATG or GTG. Homology search of ORFs with orthologs in other plastid genomes of algae and plants, and cluster analysis of proteins were performed using the described database (Supplementary material 1). To detect the RNA genes, we used the blastn program against the plastid genomes of Rhodophyta and Glaucophyta (Supplementary material 1). To search for tRNA genes, we used the tRNAscan-SE program (Lowe and Eddy 1997). To detect introns, we used the RNAweasel program (Lang et al. 2007), EST-data of related species (Chan et al. 2011) and the result of alignment analysis. We compared the rRNA operon structure of P. purpureum with that of sequenced algae and plants in the GOBASE database (O’Brien et al. 2009). In addition, the plastid genomes of the following species were used as references: Paulinella chromatophora, seven species of Heterokontophyta (as described above), and four species of Rhodophyta (Pyropia haitanensis, Calliarthron tuberculosum, Chondrus crispus, and Grateloupia taiwanensis) (Supplementary material 1). Processing of DNA and protein sequences was performed with the SISEQ software version 1.59 (Sato 2000). Sequence alignment of tRNA(CAU) genes was constructed with ClustalX software version 2.0.9 (Larkin et al. 2007). Other sequence alignments were constructed with ClustalW/ClustalX software version 1.8.3 (Thompson et al. 1994) or Muscle software version 3.6 (Edgar 2004). The genomic sequence was manipulated using the Artemis software version 13.0 (Rutherford et al. 2000).
Phylogenetic analyses of Rhodophyta and Chromalveolata
For phylogenetic analyses of Rhodophyta and Chromalveolata, four types of sequence alignments of proteins were constructed: three types used sequences that are conserved in six species of Rhodophyta, 13 species of Chromalveolata and 17 species of cyanobacteria, and one type used sequences that are conserved in six species of Rhodophyta, 13 species of Chromalveolata, 17 species of cyanobacteria, six species of Viridiplantae and one species of Glaucophyta. The analyzed proteins are listed in Supplementary material 2. The four alignments of concatenated protein data are described in Supplementary material 3a–d, respectively. We used maximum likelihood (ML), neighbor-joining (NJ) and Bayesian inference (BI) methods for phylogenetic inference. ML analysis was performed using TREEFINDER software version March 2011 (Jobb et al. 2004). The evolutionary model was selected with the ProtTest program version 2.4 (Abascal et al. 2005). The LG model was selected, but because it was not implemented in MEGA and MrBayes, the best available model was used for each respective analysis. NJ analysis was carried out using MEGA software version 5.05 (Tamura et al. 2011) with the JTT model. BI analysis was performed using MrBayes version 3.2.1 (Ronquist and Huelsenbeck 2003) with the WAG model. Two chains were run; trees were sampled at every 200 for 1,000,000 generations and the first 2,000 trees were discarded as burn-in.
Phylogenetic analyses of introns
For phylogenetic analysis of P. purpureum maturases, homologous proteins were obtained from the dataset “Cyanoclust4” (Cluster No. 5 and Cluster No. 4454) of the Gclust database (Sato 2009). We used sequences that were more than 350 amino acids in length. The phylogenetic relationship was analyzed by the ML method using the LG model. The alignment of maturases is provided in Supplementary material 3e.
Phylogenetic analysis of group II introns was performed by the ML method with the GTR + G + I model. The alignment of group II introns is provided in Supplementary material 3f.
Results
Structure of the plastid genome of P. purpureum
We sequenced the plastid genome of P. purpureum strain NIES 2140 using Roche 454 GS FLX. The sequenced reads were assembled into 13 contigs, and gaps were filled completely by Sanger sequencing. The plastid genome of P. purpureum is a circular molecule composed of 217,694 bp, with a GC content of 30.3 % (Table 1). It is larger than the plastid genomes of other species of Rhodophyta due to the presence of many introns (total length: 30,433 bp) in P. purpureum. The plastid genome of P. purpureum encodes one non-coding RNA (ncRNA) (rnpB), six rRNAs (two rrs, two rrl, and two rrf), 29 tRNAs and 224 proteins, including unidentified ORFs (Table S1, Fig. 1a).

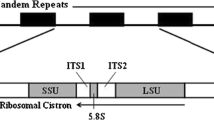
Structure of the plastid genome of P. purpureum. a A gene map of the plastid genome of P. purpureum. The two internal sectors represent RNA genes, whereas the two external sectors represent protein-coding genes. The outer rings and inner rings of each set represent genes on the clockwise and counterclockwise strands, respectively. Protein-coding genes are color-coded by their function, as shown at the bottom left corner. The belts in protein-coding regions indicate intron regions. b rRNA operon structure of the plastid genome. The region between the two IRs is called the small single copy (SSC) region. IR1 is shown to the right. Each single capital letter indicates the tRNA gene corresponding to the amino acid abbreviation
Generally, plastid genomes have an inverted repeat (IR) structure consisting of a pair of identical sequences containing rRNA operons. The rRNA operons of plastid genomes of most algae contain three rRNA genes (rrs, rrl, and rrf) and two tRNA genes, namely, tRNAIle(GAU) and tRNAAla(UGC). The tRNAIle(GAU) and the tRNAAla(UGC) genes are located between the rrs (encoding 16S rRNA) and the rrl (encoding 23S rRNA) genes (Fig. 1b, Fig. S1). rRNA operons of plastid genomes in Bangiophyceae are a pair of direct repeat structures. In Cyanidiophyceae and Florideophyceae, the plastid genome has only one rRNA operon.
In the plastid genome of P. purpureum, the rRNA genes exist as a pair of nearly identical IRs. One of them (IR1, right) has a tRNAAla(UGC) gene between the rrs and the rrl genes, while another (IR2, left) has a tRNAIle(GAU) gene (Fig. 1a). This was confirmed by sequencing of the two IRs, which were amplified separately. According to the EST-data (Chan et al. 2011), the plastid genome of a related species (Porphyridium purpureum CCMP 1328) holds a similar structure of nonidentical IRs. This type of IR architecture involving nonidentical rRNA operons is unique to the class of Porphyridiophyceae among all sequenced plastid genomes (Fig. 1b, Fig. S1).
Comparison of protein-coding genes
We conducted cluster analysis of all proteins in the plastid genome of P. purpureum (Fig. S2). We performed all-against-all blastp by using a data set including all of the plastid-encoded proteins of P. purpureum, five species of Rhodophyta, 13 species of photosynthetic Chromalveolata, a Glaucophyta (Cyanophora paradoxa), 40 species of Viridiplantae, and all the proteins of 41 species of cyanobacteria (Supplementary material 1). 37 protein genes of P. purpureum do not have a homolog in any other analyzed genome. Five genes (mat1a, mat1b, mat1c, mat1d and mat1e) are homologs of known maturase genes that are conserved in Rhodomonas salina and some cyanobacteria. Except for these and ycf12 and ycf80, all protein genes encoded by the plastid genome of P. purpureum are conserved in cyanobacteria. The 57 genes, which are conserved in all phototrophs, include genes encoding RNA polymerases, ribosomal proteins, cytochrome complexes, ATP synthases and photosystems I and II. The genes for subunits of allophycocyanin (apcA, apcB and apcD), phycocyanin (cpcA and cpcB) and phycobilisome (apcE and apcF) are conserved in Rhodophyta and C. paradoxa, but not in Chromalveolata. The genes for ribonucleases (rne and ycf56), phycoerythrin-related proteins (cpeA and cpeB) and tRNA synthetases (syfB and syh) are conserved in Rhodophytina. The homologs of manganese ABC transporter genes (mntA and mntB) are conserved in C. paradoxa and P. purpureum.
We analyzed the loss of plastid protein-coding genes during the evolution of Rhodophyta (Fig. S3). Nineteen genes are conserved only in Cyanidiophytina. The genes for protochlorophyllide reductase proteins (chlB, chlL and chlN) are conserved only in Bangiophyceae. The genes for the subunits of 3-isopropylmalate dehydratase (leuC and leuD) were considered to be acquired by horizontal gene transfer into the plastid genome only in Gracilaria tenuistipitata (Janouškovec et al. 2013). The genes for acetyl-CoA carboxylase (ACCase) (accA, accB and accD), acetylglutamate kinase (argB) and ycf29 are conserved in the plastid genomes of Rhodophyta, except P. purpureum. P. purpureum has only the homolog of the nuclear-encoded single-subunit eukaryotic form of ACCase in the nuclear genome (data not shown), and lacks the prokaryotic form that is commonly encoded in the plastid genomes, as reported in Gramineae (Konishi et al. 1996) and Chromalveolata (Livne and Sukenik 1990; Roessler and Ohlrogge 1993).
Introns and maturase genes
We detected introns by various means, namely, using the RNAweasel program (Lang et al. 2007), comparison with EST-data of related species (Chan et al. 2011) and alignment analysis of putative protein sequences. Among the 224 protein-coding genes, 29 genes contain one or multiple intron(s) (Table S2). A group I intron was found in rpl28. This intron has a secondary structure typical for group I introns (pseudoknot structure) (Cech 1988; Lang et al. 2007) (Fig. S4). Others were group II introns. Introns in dnaK, infC, gltB and rpoC2 encode a maturase. In total, the P. purpureum plastid genome has five maturase genes (Fig. S5). mat1a is located in the intergenic region between atpE and orf44. All maturases contain RT domain and X domain, and only Mat1d contains a Zn domain (nuclease domain). Mat1a and Mat1b lost the YxDD motif (polymerase active site, in domain 5) (Zimmerly et al. 2001). The intron within the dnaK gene, which is written as int.b (dnaK) in the present article, contains mat1b, but this intron was not detected using the RNAweasel program. Almost all land plants have intronic maturase genes (matK) within the plastid trnK genes (Neuhaus and Link 1987), but the maturases of P. purpureum were not homologous to MatK. According to phylogenetic analysis, two types of maturases are encoded by the plastid genome of P. purpureum (Fig. S6). One type contains Mat1a, Mat1c, Mat1d and Mat1e. Mat1a is closely related to the plastid-encoded maturase of Rhodomonas salina. Mat1b differs from these. Mat1d seems to retain mobility, and the introns encoding this type of maturase may have been present as several copies inserted at difference sites. According to the phylogenic analysis of introns (Fig. S7), int (mntA), int.b (rpoC2) and int.b (atpI) are closely related to the intron encoding the mobile maturase [int (gltB)], to which other introns are distantly related. The total number of base pairs of the introns is 30,433 bp, which corresponds to about 14 % of the entire P. purpureum plastid genome.
Identification of the three tRNA species containing the CAU anticodon
Usually, at most one tRNA species exists per anticodon, except for tRNA genes in the repeat region. The CAU anticodon, however, has three tRNA species: tRNAMet(CAU), tRNAfMet(CAU) and tRNAIle(CAU), which is also the case in the plastid genome of P. purpureum (Table S3).
tRNAfMet(CAU) functions as the initiator tRNA. In tRNAfMet(CAU), AU base pairs at positions 11 and 25, GC base pairs at positions 12 and 24 in the D-loop stem, and two or three GC base pairs in the anticodon stem are conserved in plastids and cyanobacteria (Alkatib et al. 2012). tRNA(CAU) in region 126,118–126,045 was identified as tRNAfMet(CAU).
tRNAIle(CAU) contains a lysidine, a modified cytidine, at the first anticodon by the action of TilS (tRNAIle-lysidine synthetase) (Soma et al. 2003), which is encoded by the ycf62 gene universally conserved in the plastid genomes of Rhodophyta. This modification in tRNAIle(CAU) switches the codon-recognition from AUG to AUA, which corresponds to the change in amino acid specificity from methionine to isoleucine. TilS recognizes C33 and A39 of tRNAIle(CAU) (Nakanishi et al. 2009). Curiously, the other two tRNA(CAU) genes also have identical nucleotides at these positions. The RNA sequence of a spinach chloroplast tRNAIle(CAU) was confirmed experimentally (Francis and Dudock 1982). The alignment analysis using spinach sequences as the reference indicated that tRNA(CAU) in region 114,922–114,837 is tRNAIle(CAU). The remaining tRNA(CAU) in region 19,649–19,577 is tRNAMet(CAU). We also determined and corrected the identity of some tRNA(CAU)s of other Rhodophyta and C. paradoxa plastid genomes that were misannotated (Fig. S8).
Phylogenetic analyses of Rhodophyta and Chromalveolata
For phylogenetic analyses of Rhodophyta and Chromalveolata, we constructed an amino acid sequence alignment of 76 concatenated proteins (20,088 sites) that are conserved in Rhodophyta, Chromalveolata and cyanobacteria (Fig. S9a). Qiu et al. (2012) suggested that serially duplicated genes might provide a misleading phylogenetic signal due to the concerted evolution and differential gene loss that accompanies genome reduction. In addition, they suggested that short sequence alignments lack a phylogenetic signal. Therefore, sequences of <75 amino acids in alignment length were removed. Accordingly, an alignment of 65 concatenated proteins was constructed (18,905 sites) (Fig. 2). Additionally, we constructed an amino acid sequence alignment of 34 concatenated proteins (10,570 sites) that are conserved in Rhodophyta, Chromalveolata and cyanobacteria (Fig. S9b). We also constructed an amino acid sequence alignment of the same concatenated proteins (10,524 sites), including Viridiplantae/Glaucophyta OTUs (Fig. S9c). We used maximum likelihood (ML), neighbor-joining (NJ) and Bayesian inference (BI) methods. The phylogenetic relationships of Rhodophyta, as shown in Fig. 2 and Fig. S9, were supported in all data sets by the three analyses. These relationships are consistent with the results of Yoon et al. (2006) (see also Verbruggen et al. 2010).

Phylogenetic tree of plastids and cyanobacteria based on concatenated sequences of 65 conserved proteins. The bar above the tree shows the scale for the branch length (substitutions per site). The numbers at each branch indicate the confidence level, bootstrap value and posterior probability, provided by ML (first value), NJ (second value) and BI (third value) analyses, respectively. Each “-” indicates that this branch is not supported by NJ analysis. The tree topology as shown here was identical in both ML and BI analyses. The thick lines represent the branches that were supported by the three analyses
The results of ML and BI analyses in the all datasets suggest that the plastids of Chromalveolata are sister groups of Rhodophytina, but the result of NJ analysis suggests that Cryptophyta is separated from the rest of Chromalveolata, and is a sister group of Rhodophyta as a whole. The differences in the results of the NJ analysis could result from the known problem that NJ anlyses tend to be affected by long-branch attraction, especially in multigene phylogenetic analyses. The ML tree of 76 concatenated proteins (Fig. S9a) did not support the monophyletic relationship of Chromalveolata, which was supported by other trees (Fig. 2, Fig. S9b, c). The sequences of the proteins of serially duplicated genes (psbA and psbD) were suggested to contribute to this difference. The tree of 34 concatenated proteins, including Viridiplantae/Glaucophyta OTUs (Fig. S9c), supported the monophyletic relationship of Cryptophyta and Haptophyta, which was not supported by the trees for the 65 concatenated proteins (Fig. 2) and 34 concatenated proteins without Viridiplantae/Glaucophyta OTUs (Fig. S9b).
Analysis of the gene cluster for ribosomal proteins in the plastid genome
We compared the gene cluster for ribosomal proteins in the plastid genome of P. purpureum with those in the genomes of Synechocystis sp. PCC 6803 and Anabaena sp. PCC 7120, as well as the plastid genomes of Glaucophyta, other Rhodophyta and Chromalveolata (Fig. S10). dnaK is located upstream of rpl3 and oriented in the reverse direction with respect to other genes in Rhodophyta and Chromalveolata, except Phaeophyceae. The Haptophyta (Emiliania huxleyi) plastid genome lacked some genes in the gene cluster for ribosomal proteins, namely rpl4, rpl29, rpl24, rpl18 and rpl13. The gene called ycf88, which was only detected in diatoms, is located between rps19 and rpl22.
Despite these differences in gene content of the gene cluster for ribosomal proteins, the order of genes is highly conserved in Rhodophyta and Chromalveolata, in which the rps12–rps7 segment is located downstream of the rpl36–rpl31 segment, a result of rhodophyte-specific translocation (Ohta et al. 1997). This is not the case in C. paradoxa and cyanobacteria.
In P. purpureum, the gene cluster for ribosomal proteins is divided into four parts, possibly by further translocation: the sites of disjunction are shown by arrowheads in Fig. S10 and are found between rps5 and secY, between secY and rpl36, and between rps7 and tufA. secY is isolated from the cluster and is located between orf57c and rpoC2. secY, rps10 and tufA are oriented in the reverse direction with respect to the other genes. The disjunction between secY and rpl36 was also found in the plastid genome of C. paradoxa but not in the genomes of cyanobacteria (Stirewalt et al. 1995). These data suggest that the plastid genome of P. purpureum underwent large-scale rearrangement after separation of this species from other red algae.
Discussion
We sequenced the complete plastid genome of P. purpureum and found some unique features in this plastid genome. First, the P. purpureum plastid genome has many introns. Second, it has a unique nonidentical structure of rRNA operons. Third, it has a highly rearranged gene order in the ribosomal protein gene cluster.
Introns have been reported in several plastid genes of Rhodophyta (Bernard et al. 1992; Janouškovec et al. 2013; Yoon et al. 2006), but in these cases, only one or two introns existed in the entire plastid genome. Some species of Rhodellophyceae, Porphyridiophyceae and Stylonematophyceae have an intron in the psaA gene (Yoon et al. 2006). In P. purpureum, the psaA gene has an intron, but the insertion position of the intron is different. In Florideophyceae, an intron encodes a reverse transcriptase in tRNA(CAU) (Janouškovec et al. 2013).
In Chromalveolata, which has secondary plastids originating from Rhodophyta, the rRNA operons exist as a pair of identical IRs, but this symmetric structure has not been found in any Rhodophyta analyzed so far. The plastid rRNA operons of P. purpureum are a pair of nonidentical IRs, each containing a different tRNA gene between the rrs and the rrl genes, respectively.
A previous report described the phylogenetic relationship of plastids in Rhodophyta and Chromalveolata (Wang et al. 2013). Unfortunately, only three classes of Rhodophyta were used in the analysis. Whether the plastids of Chromalveolata originated from the ancestor of Rhodophytina or the ancestor of (Florideophyceae + Bangiophyceae) remains to be determined.
We added a new species to the analysis. Our phylogenetic analyses included Porphyridiophyceae, which is a different clade from Florideophyceae and Bangiophyceae (Verbruggen et al. 2010). The results suggest that the plastids of Chromalveolata originated from Rhodophytina. Previous reports showed that the monophyly of the red algae-derived secondary/tertiary plastids (Harper and Keeling 2003; Yoon et al. 2002) and our phylogenetic analyses using concatenated proteins without the proteins of serially duplicated genes support this hypothesis. Our phylogenetic analyses suggest that the phylogenetic position of Haptophyta was affected by taxon sampling (presence or absence of Viridiplantae/Glaucophyta OTUs). Adding more Haptophyta taxa or new lineages with potentially intermediate positions will be helpful in resolving the phylogenetic relationships of Chromalveolata (Burki et al. 2012). Data from a larger number of rhodophytan taxa will be helpful in resolving a more detailed and accurate phylogenetic relationship between Rhodophyta and secondary/tertiary plastids.
The results obtained in the present study imply the following evolutionary scenario regarding the structure of the plastid genome. First, the ancestor of Rhodophyta had two inverted rRNA operon copies. One of the copies was lost in Cyanidiophyceae after its separation from Rhodophytina. Then secondary endosymbiosis events occurred one or several times in Rhodophytina; the secondary plastids conserved two copies of inverted rRNA operons. In Bangiophyceae, the rRNA operons were rearranged to become direct repeats and one of the copies was lost in Florideophyceae. In Porphyridiophyceae, large-scale genome rearrangement occurred and the two rRNA operons became nonidentical, perhaps due to asymmetric selective loss of tRNA genes after separation from other classes. The rRNA operon structure of the other three classes (Compsopogonophyceae, Stylonematophyceae and Rhodellophyceae) remains to be determined. Elucidation of the complete plastid genome in all classes of Rhodophyta will be helpful in understanding the evolution of Rhodophyta and Chromalveolata.
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105
Alkatib S, Fleischmann TT, Scharff LB, Bock R (2012) Evolutionary constraints on the plastid tRNA set decoding methionine and isoleucine. Nucleic Acids Res 40:6713–6724
Bernard C, Thomas JC, Mazel D, Mousseau A, Castets AM, Tandeau de Marsac N, Dubacq JP (1992) Characterization of the genes encoding phycoerythrin in the red alga Rhodella violacea: evidence for a splitting of the rpeB gene by an intron. Proc Natl Acad Sci USA 89:9564–9568
Bhattacharya D, Price DC, Chan CX, Qiu H, Rose N, Ball S, Weber APM, Arias MC, Henrissat B, Coutinho PM, Krishnan A, Zäuner S, Morath S, Hilliou F, Egizi A, Perrineau MM, Yoon HS (2013) Genome of the red alga Porphyridium purpureum. Nat Commun 4:1941
Burki F, Okamoto N, Pombert JF, Keeling PJ (2012) The evolutionary history of haptophytes and cryptophytes: phylogenomic evidence for separate origins. Proc Biol Sci 279:2246–2254
Cavalier-Smith T (1998) A revised six-kingdom system of life. Biol Rev 73:203–266
Cech TR (1988) Conserved sequences and structures of group I introns: building an active site for RNA catalysis––a review. Gene 73:259–271
Chan CX, Yang EC, Banerjee T, Yoon HS, Martone PT, Estevez JM, Bhattacharya D (2011) Red and green algal monophyly and extensive gene sharing found in a rich repertoire of red algal genes. Curr Biol 21:328–333
DePriest MS, Bhattacharya D, López-Bautista JM (2013) The plastid genome of the red macroalga Grateloupia taiwanensis (Halymeniaceae). PLoS One 8:e68246
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Francis MA, Dudock BS (1982) Nucleotide sequence of a spinach chloroplast isoleucine tRNA. J Biol Chem 257:1195–1198
Glöckner G, Rosenthal A, Valentin K (2000) The structure and gene repertoire of an ancient red algal plastid genome. J Mol Evol 51:382–390
Hagopian JC, Reis M, Kitajima JP, Bhattacharya D, Oliveria MC (2004) Comparative analysis of the complete plastid genome sequence of the red alga Gracilaria tenuistipitata var. liui provides insights into the evolution of Rhodoplasts and their relationship to other plastids. J Mol Evol 59:464–477
Harper JT, Keeling PJ (2003) Nucleus-encoded, plastid-targeted glyceraldehyde-3-phosphate dehydrogenase (GAPDH) indicates a single origin for chromalveolate plastids. Mol Biol Evol 20:1730–1735
Janouškovec J, Liu SL, Martone PT, Carré W, Leblanc C, Collén J, Keeling PJ (2013) Evolution of red algal plastid genomes: ancient architectures, introns, horizontal gene transfer, and taxonomic utility of plastid markers. PLoS One 8:e59001
Jobb G, von Haeseier A, Strimmer K (2004) TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol 4:18
Kim E, Archibald JM (2009) Diversity and evolution of plastids and their genomes. The chloroplast––interactions with the environment. In: Aronsson H, Sandelius AS (eds) Plant cell monographs, vol 13. Springer, Berlin, pp 1–39
Konishi T, Shinohara K, Yamada K, Sasaki Y (1996) Acetyl-CoA carboxylase in higher plants: most plants other than gramineae have both the prokaryotic and the eukaryotic forms of this enzyme. Plant Cell Physiol 37:117–122
Lang BF, Laforest MJ, Burger G (2007) Mitochondrial introns: a critical view. Trends Genet 23:119–125
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Livne A, Sukenik A (1990) Acetyl-coenzyme-A carboxylase from the marine prymnesiophyte Isochrysis galbana. Plant Cell Physiol 31:851–858
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Nakanishi K, Bonnefond L, Kimura S, Suzuki T, Ishitani R, Nureki O (2009) Structural basis for translational fidelity ensured by transfer RNA lysidine synthetase. Nature 461:1144–1148
Neuhaus H, Link G (1987) The chloroplast tRNALys(UUU) gene from mustard (Sinapis alba) contains a class II intron potentially coding for a maturase-related polypeptide. Curr Genet 11:251–257
Noguchi H, Taniguchi T, Itoh T (2008) MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res 15:387–396
O’Brien EA, Zhang Y, Wang E, Marie V, Badejoko W, Lang BF, Burger G (2009) GOBASE: an organelle genome database. Nucleic Acids Res 37:D946–D950
Ohta N, Sato N, Nozaki H, Kuroiwa T (1997) Analysis of the cluster of ribosomal protein genes in the plastid genome of a unicellular red alga Cyanidioschyzon merolae: translocation of the str cluster as an early event in the Rhodophyte–Chromophyte lineage of plastid evolution. J Mol Evol 45:688–695
Ohta N, Matsuzaki M, Misumi O, Miyagishima S, Nozaki H, Tanaka K, Shin-I T, Kohara Y, Kuroiwa T (2003) Complete sequence and analysis of the plastid genome of the unicellular red alga Cyanidioschyzon merolae. DNA Res 10:67–77
Okaichi T, Nishio S, Imatomi Y (1982) Collection and mass culture. In: Jpn Fish Soc (ed) In toxic phytoplankton––occurrence, mode of action, and toxins. Koseisya-Koseikaku, Tokyo, pp 22–34
Qiu H, Yang EC, Bhattacharya D, Yoon HS (2012) Ancient gene paralogy may mislead inference of plastid phylogeny. Mol Biol Evol 29:3333–3343
Reith M, Munholland J (1995) Complete nucleotide sequence of the Porphyra purpurea chloroplast genome. Plant Mol Biol Rep 13:333–335
Roessler PG, Ohlrogge JB (1993) Cloning and characterization of the gene that encodes acetyl-coenzyme A carboxylase in the alga Cyclotella cryptica. J Biol Chem 268:19254–19259
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B (2000) Artemis: sequence visualization and annotation. Bioinformatics 16:944–945
Sasaki NV, Sato N (2010) CyanoClust: comparative genome resources of cyanobacteria and plastids. Database 2010:bap025
Sato N (2000) SISEQ: manipulation of multiple sequence and large database files for common platforms. Bioinformatics 16:180–181
Sato N (2009) Gclust: trans-kingdom classification of proteins using automatic individual threshold setting. Bioinformatics 25:599–605
Soma A, Ikeuchi Y, Kanemasa S, Kobayashi K, Ogasawara N, Ote T, Kato J, Watanabe K, Sekine Y, Suzuki T (2003) An RNA-modifying enzyme that governs both the codon and amino acid specificities of isoleucine tRNA. Mol Cell 12:689–698
Stirewalt VL, Michalowski CB, Löffelhardt W, Bohnert HJ, Bryant DA (1995) Nucleotide sequence of the cyanelle genome from Cyanophora paradoxa. Plant Mol Biol Rep 13:327–332
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Verbruggen H, Maggs CA, Saunders GW, Le Gall L, Yoon HS, De Clerck O (2010) Data mining approach identifies research priorities and data requirements for resolving the red algal tree of life. BMC Evol Biol 10:16
Wang L, Mao Y, Kong F, Li G, Ma F, Zhang B, Sun P, Bi G, Zhang F, Xue H, Cao M (2013) Complete sequence and analysis of plastid genomes of two economically important red algae: Pyropia haitanensis and Pyropia yezoensis. PLoS One 8:e65902
Yoon HS, Hackett JD, Pinto G, Bhattacharya D (2002) The single, ancient origin of chromist plastids. Proc Natl Acad Sci USA 99:15507–15512
Yoon HS, Müller KM, Sheath RG, Ott FD, Bhattacharya D (2006) Defining the major lineages of red algae (Rhodophyta). J Phycol 42:482–492
Zimmerly S, Hausner G, Wu XC (2001) Phylogenetic relationships among group II intron ORFs. Nucleic Acids Res 29:1238–1250
Acknowledgments
The present study was supported in part by a grant for the Global COE Program, “From the Earth to “Earths””, from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by a CREST grant from the Japan Science and Technology Agency.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Tajima, N., Sato, S., Maruyama, F. et al. Analysis of the complete plastid genome of the unicellular red alga Porphyridium purpureum . J Plant Res 127, 389–397 (2014). https://doi.org/10.1007/s10265-014-0627-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-014-0627-1