
Abstract
Background
Darbepoetin alfa (DA) is an attractive alternative to recombinant human erythropoietin (rHuEPO) in managing renal anemia. Since DA has not been approved by the appropriate Japanese drug regulatory agencies for the indication of renal anemia in children in Japan, we have conducted a multicenter prospective study to determine the efficacy and safety of DA in Japanese children undergoing peritoneal dialysis (PD).
Methods
Pediatric patients subcutaneously receiving rHuEPO were switched to DA treatment for a period of 28 weeks. The conversion to the initial dose of DA was calculated as 1 μg DA for 200 IU rHuEPO, and DA was administered intravenously once every 2 weeks. The target hemoglobin (Hb) concentration was defined as 11.0 to ≤13.0 g/dL. In some patients, the dose of DA was adjusted appropriately to achieve this target level, and/or the dosing frequency changed to once every 4 weeks.
Results
In the 25 patients switched from rHuEPO to DA the mean Hb concentration increased from 9.9 ± 1.0 to 11.1 ± 1.0 g/dL at 8 weeks following commencement of the DA treatment. The target Hb concentration was achieved in 88 % of these patients, and 60 % maintained this target value on completion of the study. The dosing frequency was extended to once every 4 weeks in 60 % of patients. Twenty-four adverse events were noted in 11 of 25 patients (44 %); however, there was no causality between DA and adverse events.
Conclusions
The results of this study suggest that intravenous administration of DA once every 2 or 4 weeks is an effective and safe treatment for renal anemia in Japanese children undergoing PD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anemia is a prevalent and serious problem among children with chronic kidney disease (CKD). At all stages of CKD, low hemoglobin (Hb) levels have been associated with an increased risk of hospitalization and death, lower cognitive function, increased left ventricular hypertrophy, and decreased left ventricular compliance [1]. Recombinant human erythropoietin (rHuEPO) has become the standard treatment for renal anemia in children [1]; however, to maintain the target Hb concentration (≥11 g/dL) conventional rHuEPO requires two to three injections per week [1]. In contrast, darbepoetin alpha (DA), which has an increased sialic acid carbohydrate content, promotes decreased clearance and has a longer serum half-life than rHuEPO, allowing extended dosing intervals [2]. A number of clinical studies have proven that DA is an effective and safe alternative to rHUEPO for the treatment of renal anemia in adults [3–6]. DA has been also shown to be an attractive alternative to rHuEPO in managing anemia in pediatric patients with CKD because of its comparable efficacy and safety profile, as well as its ability to be administered at extended dosing intervals [7–10]. However, since DA has yet to be approved for the indication of renal anemia in children in Japan, there are no data describing its efficacy and safety profile in Japanese pediatric patients with CKD. A multicenter prospective study was therefore conducted at nine institutions in Japan with the aim to determine the efficacy and safety of intravenous administration of DA in pediatric patients undergoing peritoneal dialysis (PD).
Patients and methods
Study design
This was a multicenter, prospective, single-arm study that enrolled pediatric patients undergoing PD at nine institutions in Japan from January 2009 to December 2011. After an initial 8-week baseline period to determine eligibility, patients receiving rHuEPO subcutaneously once every 2 weeks were switched to DA treatment for a period of 28 weeks. DA was administered intravenously once every 2 weeks. For routine blood sampling, scalp veins were punctured using an infusion scalp vein set; patients also received DA intravenously using the same vein set immediately after the completion of the blood sampling. The initial dose was calculated from the prior dose of rHuEPO according to the following conversion index: 1 μg DA for 200 IU rHuEPO. The target Hb was defined as 11.0 to ≤13.0 g/dL based on the Japanese anemia therapy guideline [11]. To achieve this target Hb level, the DA dosage was appropriately increased when the target Hb was not reached by 8 weeks following commencement of DA treatment. The dosing frequency was changed from once every 2 weeks to once every 4 weeks for some patients whose Hb was assayed at between 11.0 and 13.0 g/dL and, accordingly, the DA dosage was doubled in these patients. The study protocol was approved by the institutional review board of each participating institution. Written informed consent was obtained from patients or their parents before the study-related procedures were performed.
Patients
Patients receiving PD and aged between 1 and 18 years were eligible for enrolment in this study. Patients were excluded if they had undergone major surgery within 4 weeks before providing signed informed consent, were scheduled for a living donor kidney transplant within 16 weeks following consent, or received a red blood cell transfusion within 4 weeks prior to providing signed informed consent. Patients with uncontrolled hypertension, cardiac failure, malignancy and/or hematological disease, and known resistance to rHuEPO were also excluded.
Patients were required to have been receiving stable rHuEPO therapy administered subcutaneously for at least 8 weeks prior to the switch to DA therapy. They were also required to have a baseline Hb concentration of <11.0 g/dL before starting DA treatment, a baseline transferrin saturation (TSAT) ≥20 %, and/or a serum ferritin level of ≥100 ng/mL.
Endpoints
The primary endpoints were the Hb profiles, which comprised changes in Hb concentration, the rate of increase in Hb concentration, the percentage and time taken to reach the target Hb concentration, and the percentage of patients who maintained the target Hb concentration. Additionally, changes in Hb and changes in DA dose per week were also analyzed in some patients who underwent a change in the initial dosing frequency to once every 4 weeks. Safety was assessed by monitoring adverse events (both treatment-related and unrelated), laboratory parameters, and vital signs.
Statistical analysis
For continuous variables, descriptive statistics were computed, namely, the mean, median, range, and standard deviation (SD). Percentages and sample sizes were used to summarize categorical variables. The Kaplan–Meier method was employed to assess the percentage and time taken to reach the target Hb. For safety analyses, counts and percentages were calculated for adverse events.
Results
Patient disposition
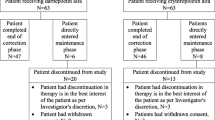
A patient flowchart is shown in Fig. 1. Of the 28 patients who were enrolled in this study, 25 were eligible for the switch to DA therapy. The remaining three were judged to be ineligible because of unstable rHuEPO therapy during the 8 weeks prior to conversion. Of those who successfully underwent conversion to DA treatment, 23 completed the study; the remaining two withdrew due to kidney transplantation.

Patient allocation
Patient demographics and baseline characteristics
Patient demographics and baseline characteristics are summarized in Table 1. Males represented 68 % of the patient population, and the mean age at the start of DA therapy was 11.2 ± 5.7 years. The most common etiology among the study patients was hypoplastic/dysplastic kidneys (12 patients, 48 %). The mean duration of PD before the switch to DA was 18.9 ± 16.6 months. At baseline, the mean weekly rHuEPO dose (IU/kg per week) was 140.6 ± 116.0, the mean Hb concentration (g/dL) was 9.9 ± 1.0, and the mean values of ferritin and TSAT were 192.4 ± 123.6 ng/mL and 42.2 ± 16.4 %, respectively.
Efficacy
Hemoglobin profiles are shown in Fig. 2. At the start of DA treatment, Hb was 9.9 ± 1.0 g/dL (mean ± SD), while at week 8 it was 11.1 ± 1.0 g/dL, which was above the lower limit of the target Hb (11.0 g/dL). Thereafter, the mean Hb remained above this lower limit, reaching 11.3 ± 0.9 g/dL at the end of treatment.

Hemoglobin (Hb) profiles. The shaded area shows the target Hb concentration
The mean DA dose at the time of the switch to DA therapy and at the end of treatment (withdrawal) was 0.93 ± 0.99 (median 0.54, range 0.15–4.20) and 0.99 ± 0.87 (median 0.69, range 0.18–3.22) μg/kg per week, respectively. Figure 3 shows the dose of DA/kg per injection at the end of treatment (withdrawal) for each patient plotted by individual. The dose per injection varied largely, with nine patients (36 %) needing >3 μg/kg of DA per injection.

Darbepoetin doses per injection. The plot shows the individual values
Figure 4 shows the rate of increase in Hb concentration during the 8 weeks following commencement of DA therapy for each patient plotted by individual. Although one patient showed a rate of increase of >0.5 g/dL per week, the mean rate of increase was 0.12 ± 0.13 g/dL per week.

Rate of Hb increase. The safe level of increase in Hb is reportedly <0.5 g/dL per week
As shown in Fig. 5, the cumulative proportion who reached the target Hb concentration, as determined by the Kaplan–Meier method, was 88 %. At week 8, 68 % of patients had reached the lower limit of the target concentration (11.0 g/dL). The point at which 25, 50, and 75 % reached the lower limit was 2, 6, and 12 weeks, respectively.

Cumulative proportion reaching target Hb concentration
Figure 6 shows the profiles of the percentage of patients who maintained the target Hb concentration. The proportion of these patients increased after commencement of DA treatment. Eight weeks after the start of DA therapy, ≥50 % maintained the target Hb. At the end of treatment, 60 % of patients were within the target Hb range, 40 % were below it, and none were above it.

Change in the percentage of patients who maintained the target Hb concentration
Fifteen patients (60 %) underwent a change in dosing frequency from once every 2 weeks to once every 4 weeks. Figure 7 shows the change in Hb and change in DA dose in these 15 patients. The mean Hb value when treatment was switched to once every 4 weeks was 12.2 ± 0.60 g/dL. After the switch, Hb remained within the target range, being 11.3 ± 0.5 g/dL at the end of the treatment. The mean weekly doses at the time of the switch and at the end of treatment (withdrawal) were 0.89 ± 0.87 (median 0.63, range 0.12–3.05) and 0.63 ± 0.33 (median 0.75, range 0.25–0.88) μg/kg per week, respectively.

Change in Hb and darbepoetin alfa dose after the switch to medication once every 4 weeks. The shaded area shows the target Hb concentration
Safety
Twenty-four adverse events were noted in 11 (44 %) of 25 patients. All those that occurred in patients receiving DA are listed in Table 2. Among the adverse events, no adverse drug reactions indicating causality with DA treatment were observed. In addition, no clear correlation was found between the incidence of adverse events and Hb values at the time they occurred. There were no adverse events that led to death in this study. No changes in laboratory findings were noted other than in parameters related to erythropoiesis.
Discussion
The objective of this study was to determine the efficacy and safety of intravenous administration of DA in Japanese pediatric patients undergoing PD. Since DA has yet to be approved by Japanese regulatory agencies for the indication of renal anemia in children in Japan, the study protocol was approved by the institutional review board of each participating institution. This multicenter prospective study was conducted at nine medical institutions, and a total of 28 pediatric PD patients were enrolled. Although the number of patients enrolled was too small to allow definite conclusions, given the annual report from the Japanese Society for Dialysis Therapy indicating that the number of dialysis patients aged <15 years was 103 at the end of 2008 [12], our analysis of the efficacy and safety of DA treatment in Japanese children undergoing PD was deemed possible.
It has been suggested that the psychological stress imposed by repeated painful injections in chronically ill children may result in reduced medical adherence [13]. It has been reported that subcutaneous injections of DA are more painful than those of rHuEPO in the majority of pediatric patients [8, 14] and, therefore, an intravenous route of administration is commonly chosen to reduce stress among both the children and medical staff. Additionally, the plasma disappearance half-life of DA has been found to increase by two- to threefold relative to rHuEPO after both subcutaneous and intravenous administration in both adults [15] and children [2]. It has also been reported that there are no differences in patient responses to DA administered intravenously or subcutaneously [8]. Taking these results into account, we chose the intravenous route of DA administration in our study.
The findings of our study indicate that in many cases rHuEPO treatment at the level covered by healthcare insurance in Japan (subcutaneous administration of 100–200 IU/kg once every 2 weeks) is inadequate for improvements in anemia to a target Hb level of >11.0 g/dL. The same worrying findings were also found in adult PD patients in Japan [16]. Furthermore, the North American Pediatric Renal Transplant Cooperative Study (NAPRTCS) reported that two to three injections of rHuEPO per week were required to maintain target Hb levels (≥11 g/dL) in pediatric PD patients [17]. Compared to patients undergoing hemodialysis, PD patients have the advantage that fewer hospital visits are needed for their therapy. DA is therefore an attractive alternative to rHuEPO in managing renal anemia of patients undergoing PD.
A number of clinical studies have proven DA to be effective and safe in treatment of renal anemia in adults [3–6]. Additionally, several publications on the administration of DA in children with CKD have found it to be an effective agent in controlling renal anemia [7–10]. In our study, pediatric PD patients receiving rHuEPO subcutaneously once every 2 weeks were switched to DA therapy, with the initial dose of DA being based on the prior dose of rHuEPO according to the conversion index: 1 μg DA for 200 IU rHuEPO. DA was administered intravenously once every 2 weeks, followed by an extension of the dosing interval with concomitant dose adjustment.
In our study, the mean Hb concentration increased from 9.9 ± 1.0 g/dL at the start of DA treatment to 11.1 ± 1.0 g/dL at 8 weeks following commencement. The weekly rate of increase in Hb after treatment at the initial dose for 8 weeks was 0.12 ± 0.13 g/dL per week. This rate of increase meets the Japanese anemia therapy guideline [11], which recommends erythropoiesis-stimulating agent (ESA) therapy that achieves an increase rate of ≤0.5 g Hb/dL per week. Thus, the conversion index of 1 μg DA for 200 IU rHuEPO appears to have been satisfactory in our pediatric patients undergoing PD.
During the maintenance period, the mean Hb concentration remained above the lower limit of the target Hb (11.0 g/dL) and was 11.3 ± 0.9 g/dL at the end of treatment. The target Hb concentration was achieved in 88 % of patients, with 60 % maintaining this level upon completion of this study. The dosing frequency was extended to once every 4 weeks in 60 % of patients, in line with a report on adult PD patients [6]. The median DA dose at the end of treatment (withdrawal) was 0.69 μg/kg per week and was similar to the dose reported in another study on children undergoing PD [18]. Notably, the dose of DA largely varied among the patients in our study, and nine patients (36 %) needed >3 μg/kg of DA per injection to maintain the target Hb concentration. Thus, it is essential that the dose of DA is adjusted on an individual basis, as reported previously [19].
The tolerance to DA was excellent among the patients in our study, and none discontinued treatment due to adverse events. One patient complained of headache and another developed hypertension; however, no clear correlation was found between the incidence of adverse events and Hb concentration at the time they occurred. Since hypertension and headache are common in patients undergoing treatment with ESA and can develop after a rapid rise in the Hb concentration [11], it is essential that DA be administered carefully to prevent such a rapid increase.
Pain perception is known to be higher in children than in adults [14]. In fact, it has been reported that the most prevalent treatment-related adverse event in children administered DA subcutaneously is the injection-site pain [8, 9]. In contrast, there were no adverse events related to injection-site pain in our study. Therefore, the intravenous administration of DA, which helps prevent stress in both children and the medical staff compared with pain related to subcutaneous administration, seems to be the suitable administration route for pediatric patients.
In conclusion, the results of this study suggest that the intravenous administration of DA once every 2 weeks or once every 4 weeks is an effective and safe treatment option for renal anemia in Japanese children undergoing PD.
References
Koshy SM, Geary DF. Anemia in children with chronic kidney disease. Pediatr Nephrol. 2008;23:209–19.
Gary Lerner, Kale AS, Warady BA, Jabs K, Bunchman TE, Heatherington A. Pharmacokinetics of darbepoetin alfa in pediatric patients with chronic kidney disease. Pediatr Nephrol. 2002;17:933–7.
Brunkhorst R, Bommer J, Braum J, Hagg-Weber M, Gill C, Wagner J, et al. Darbepoetin alfa effectively maintains hemoglobin concentrations at extended dose intervals relative to intravenous or subcutaneous recombinant human erythropoietin in dialysis patients. Nephrol Dial Transplant. 2004;19:1224–30.
Jadoul M, Vanrenterghem Y, Foret M, Walker R, Gray SJ. Darbepoetin alfa administered once monthly maintains hemoglobin levels in stable dialysis patients. Nephrol Dial Transplant. 2004;19:898–903.
Hiramatsu M, Kubota M, Iwasaki M, Akizawa T, Kosikawa, and the KRN321 A09 Study Group. Darbepoetin alfa (KRN321) administered intravenously once monthly maintain hemoglobin levels in peritoneal dialysis patients. Ther Apher Dial. 2008;12:19–27.
Kubota M, Hiramatsu M, Yamakawa M, Fukuhara S, Morita S, Iwasaki M, et al. Darbepoetin alfa (KRN321) is safe and effective when administered subcutaneously once every 2 or 4 weeks to patients on peritoneal dialysis in Japan. Clin Exp Nephrol. 2011;15:884–92.
De Palo T, Giordano M, Palumbo F, Bellantuono R, Messina G, Colella V, et al. Clinical experience with darbepoetin alfa (NESP) in children undergoing hemodialysis. Pediatr Nephrol. 2004;19:337–40.
Geary DF, Keating LE, Vigneux A, Stephens D, Hebert D, Harvey EA. Darbepoetin alfa (AranespTM) in children with chronic renal failure. Kidney Int. 2005;68:1759–65.
Warady BA, Arar MY, Lerner G, Nakanishi AM, Stehman-Breen C. Darbepoetin alfa for the treatment of anemia in pediatric patients with chronic kidney disease. Pediatr Nephrol. 2006;21:1144–52.
Andre JL, Deschenes G, Boudailliez B, Broux F, Fischbach M, Gagnadoux M-F, et al. Darbepoetin, effective treatment of anaemia in paediatric patients with chronic renal failure. Pediatr Nephrol. 2007;22:708–14.
Japanese Society for Dialysis Therapy. 2008 Japanese Society for Dialysis Therapy guideline for renal anemia in chronic kidney disease. J Jpn Soc Dial Ther 2008; 41:661–716.
Japanese Society for Dialysis Therapy. An overview of dialysis treatment in Japan (as of Dec. 31, 2008). J Jpn Soc Dial Ther 2010; 43:1–35.
Cummings EA, Reid GJ, Finley GA, McGrath PJ, Ritchie JA. Prevalence and source of pain in pediatric inpatients. Pain. 1996;68:25–31.
Schmitt CP, Nau B, Brummer C, Rosenkranz J, Schaefer F. Increase injection pain with darbepoetin-α compared to epoetin-β in paediatric dialysis patients. Nephrol Dial Transplant. 2006;21:3520–4.
Macdougal IC, Gray SJ, Elston O, et al. Pharmacokinetics of novel erythropoiesis stimulating protein compared with epoetin alfa in dialysis patients. J Am Soc Nephrol. 1999;10:2392–5.
Hiramatsu M, Kubota M, Yamamoto H. Limitation of rHuEPO treatment for target Hb in peritoneal dialysis patients (in Japanese). Kidney Dial. 2007;63:915–22.
North American Pediatric Renal Transplant Cooperative Study (NAPRTCS). 2004 Annual Report. NAPRTCS Administrative Office, Boston, 2004.
Rijk Y, Raaijmakers R, Kar N, Schroder C. Intraperitoneal treatment with darbepoetin for children on peritoneal dialysis. Pediatr Nephrol. 2007;22:436–40.
Durkan AM, Keating LE, Vigneux A, Geary DF. The use of darbepoetin in infants with chronic renal impairment. Pediatr Nephrol. 2006;21:694–7.
Acknowledgments
The authors gratefully acknowledge Drs. Tomoyuki Sakai and Kenji Ishikura (Department of Nephrology, Tokyo Metropolitan Children’s Medical Center, Tokyo, Japan) and Yasushi Tsutumi (Department of Pediatrics, Kyushu University) for advice during this study. This study was supported by a grant from the Japan Dialysis Outcome Group (Grant #07009).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Hattori, M., Matsunaga, A., Akioka, Y. et al. Darbepoetin alfa for the treatment of anemia in children undergoing peritoneal dialysis: a multicenter prospective study in Japan. Clin Exp Nephrol 17, 582–588 (2013). https://doi.org/10.1007/s10157-012-0714-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-012-0714-3