
Abstract
Background
Darbepoetin alfa (KRN321) is a recombinant protein that stimulates erythropoiesis by the same mechanism as endogenous erythropoietin. Due to its longer half-life and greater biological activity than recombinant human erythropoietin (rHuEPO), KRN321 maintains an effective hemoglobin (Hb) level at extended dose intervals compared with rHuEPO. The efficacy and safety of KRN321 administered subcutaneously to patients on peritoneal dialysis (PD) were tested.
Methods
In a multicenter, open-label, single-arm study, KRN321 was administered subcutaneously to patients on PD for 26–28 weeks. Ninety-six patients initially were given a 60 μg subcutaneous dose once every 2 weeks until a target of Hb (11.0–13.0 g/dL) was achieved. Thereafter, their dose was every 2 or 4 weeks.
Results
After the target of Hb was reached in most subjects (96.9%), it was maintained with KRN321 administered every 2 or 4 weeks. On completion of (or withdrawal from) study, 65 subjects (67.7%) maintained the target Hb. Although a number of adverse event related to hypertension occurred, their incidence did not appear to be related to Hb or its rate of increase. These events could be controlled adequately by interrupting or reducing the dose, and/or treatment with antihypertensives.
Conclusions
The efficacy and safety of KRN321 when administered subcutaneously for 28 weeks to PD patients were confirmed. It was suggested that the quality of life of patients can be improved by treatment with KRN321 due to the reduced frequency of administration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
According to a statistical survey by the Japanese Society for Dialysis Therapy (late 2008), there are more than 280,000 patients on dialysis in Japan, among whom about 9,000 are on peritoneal dialysis (PD) [1]. Although progress in dialysis therapy has allowed patients with renal failure to live longer and achieve social rehabilitation, renal anemia is an important complication that affects quality of life (QOL) and life expectancy for dialysis patients.
Recombinant human erythropoietin (rHuEPO) showed exceptional improvements in anemia and dramatically reduced the number of patients who required blood transfusions, and also improved other symptoms associated with anemia, leading to better QOL for patients. However, to improve their anemia, PD and nondialysis (ND) patients must be treated once a week or every other week with rHuEPO. rHuEPO treatments at the level covered by insurance in Japan reportedly fail to improve anemia adequately [2, 3].
Darbepoetin alfa (recombinant, KRN321) is a long-acting erythropoiesis-stimulating agent (ESA) developed by Amgen (US) [4, 5]. In Japan, Kyowa Hakko Kirin worked to develop KRN321, and intravenous administration was approved for the indication of “renal anemia during dialysis.” Development of subcutaneous KRN321 to treat PD and ND patients began in 2001.
This is a report on a study of subcutaneous administration of KRN321 that lasted 28 weeks in PD patients to investigate the efficacy and safety of the drug in Japan.
Subjects and methods
Study design
This multicenter, open-label, single-arm study was conducted at 22 facilities in Japan from November 2005 to 2006. It complied with the Declaration of Helsinki. The protocol was approved by each local institutional review board.
Patients
Patients on PD who received care at the study sites and were at least 20 years of age at consent with baseline Hb <12.0 g/dL were enrolled. However, patients with Hb between 10.0 and 12.0 g/dL were enrolled only if they had been treated with an rHuEPO (weekly dose ≤6,000 IU each week or every 2 weeks) for the previous 8 weeks.
Method of administration
The target Hb was 11.0 to ≤13.0 g/dL.
The initial dose of KRN321 was 60 μg every 2 weeks by subcutaneous injection. The dose/administration generally was not changed, until the target Hb was reached. The dose was increased in steps according to Table 1 (dose adjustment table) when the Hb increased less than 1.0 g/dL over 4 weeks or when the target Hb was not reached after 8 weeks following start of treatment.
After the target Hb was reached, the dose was adjusted to maintain the subject’s Hb at the target.
The dosing frequency was changed from once every 2 weeks to once every 4 weeks according to Table 2 (initial dose when changing dosing frequency) for subjects: (1) whose Hb was controlled between 11.5 and 13.0 g/dL, (2) when the prior dose was less than ≤120 μg, and (3) when the prior dose was the same as that at the last but one.
Concomitant medication and treatment
Red blood cell transfusions, concomitant rHuEPO, and other anemia-correcting medications were prohibited during the trial. Iron was supplemented preferably intravenously, and at the discretion of the investigators, to maintain transferrin saturation (TSAT) ≥20% or ferritin ≥100 ng/mL.
Observation/examination parameters
Observations and examinations were conducted according to the schedule given in Table 3.
Statistical analysis
The sample size of 80 patients was estimated to investigate the safety of the long-term use of KRN321 and to reduce the standard error of mean Hb.
The primary endpoints were Hb profiles, which comprised rate of increase in Hb, percentage and time to reach target Hb, and percentage of subjects who maintained the target Hb. The rate of increase in Hb and the percentage and time to reach target Hb were calculated for subjects with Hb at enrollment less than 10.0 g/dL.
As a secondary endpoint of efficacy, QOL was assessed at baseline and week 12 using Short Form-36 (SF-36) ver 2 acute form, which was validated in advance [6], and Functional Assessment of Chronic Illness Therapy (FACIT) fatigue scale. For intragroup comparisons the change in QOL score (2nd − 1st QOL score) was calculated and compared within groups by a single-sample t test.
Hb profiles and change in weekly KRN321 dose were also analyzed for subjects who changed to a dosing frequency of once every 4 weeks.
For safety analysis, the number and percentage of subjects who experienced adverse events or adverse drug reactions, and the number of incidents, were tallied by event coded by MedDRA/J 8.1.
Results
Subject disposition
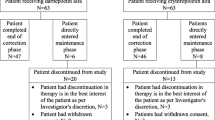
The subject flowchart is shown in Fig. 1. Of the 99 subjects who gave informed consent, 96 were eligible and were enrolled in the study. After KRN321 treatment was initiated, six subjects withdrew, and 90 completed the study. Of the six subjects who withdrew, one withdrew consent, three withdrew because of adverse events (unstable angina pectoris, peritonitis), and two changed from PD to hemodialysis (HD) (because dialysis was insufficient).

Allocation of subjects
Of 96 subjects, 67 subjects whose Hb at enrollment was less than 10.0 g/dL were measured for rate of increase in Hb, and 94 subjects completed the first and second QOL questionnaires.
Table 4 presents the characteristics of the subjects used for the efficacy and safety analyses.
Primary endpoints
Hb profiles are shown in Fig. 2. Hb at start of KRN321 treatment was 9.6 ± 1.1 g/dL (mean ± SD). At week 10, Hb was 11.1 ± 1.3 g/dL, which was above the target Hb of a lower limit of 11.0 g/dL. The Hb then increased, remained at around 12 g/dL at week 16 and thereafter, and was 11.9 ± 1.2 g/dL at end of treatment (or withdrawal).

Hb profiles. Hb was examined at time of completion or discontinuation of study medication
Figure 3 shows the rate of increase in Hb for each subject (with Hb at enrollment <10.0 g/dL) plotted by individual, as well as mean ± SD. The mean rate of increase in Hb was 0.18 g/dL/week [95% confidence interval (CI) 0.14–0.22 g/dL/week]; the rate of increase in Hb was more than 0.5 g/dL/week in 2 subjects. Of 67 subjects with Hb at time of enrollment <10.0 g/dL, 63 reached the target Hb lower limit (11.0 g/dL); the cumulative proportion reaching the target Hb, as determined by the Kaplan–Meier method, was 96.9% (95% CI 92.7–100%). The point at which 25, 50, or 75% reached the target Hb of a lower limit of 11.0 g/dL was 8, 12, or 16 weeks, respectively. The shortest period was 2 weeks.

Rate of increase in Hb
Figure 4 shows the profiles for the percentage of subjects who maintained the target Hb. At the time treatment began, 9 of 96 subjects (9.4%) had already been within the target Hb range. This proportion increased after treatment started. Fifty percent or more maintained the target Hb after 10 weeks, and more than 70% after 20 weeks. At the end of treatment (or withdrawal) Hb was within the target range in 65 subjects (67.7%). At the end of treatment (or withdrawal), 19 of the 96 subjects (19.8%) were below the target Hb range, and 12 (12.5%) were above.

Change in percentage of subjects who maintained target Hb
Secondary endpoints
Figure 5 shows the change in QOL score [mean (95% CI)]. At the second survey the mean (±SD) Hb was 11.6 ± 1.2 g/dL, an increase over the Hb of 9.6 ± 1.1 g/dL at the first survey. The QOL scores were higher for all parameters except social functioning (SF), role-emotional (RE), and mental health (MH) at the second survey, but not statistically significantly so.

Change in quality of life (QOL) score (2nd − 1st QOL score)
Figure 6 shows the change in Hb and change in KRN321 dose in subjects who switched to treatment once every 4 weeks. Hb when treatment was switched to once every 4 weeks was 12.3 ± 0.45 g/dL (mean ± SD). There was no clinical change in Hb after the switch; Hb remained at around 12.0 g/dL. At end of treatment (or withdrawal), Hb was 11.7 ± 0.92 g/dL. The weekly dose when treatment was switched to once every 4 weeks was 34.3 ± 9.0 μg.

Change in Hb and KRN321 dose after switching to medication once every 4 weeks. Hb was examined at time of completion or discontinuation of study medication
Safety
Three hundred seventy-four adverse events were noted in 91 of 96 subjects (94.8%, 95% CI 88.3–98.3%). The most frequent adverse events was nasopharyngitis, which occurred as 40 events in 32 subjects (33.3%). Adverse events that occurred in 10% or more of subjects comprised 23 events of “hypertension” in 22 subjects (22.9%), 19 “catheter site infections” in 16 subjects (16.7%), and 10 events of “diarrhea” in 10 subjects (10.4%). No clear correlation was found between incidence of adverse events and Hb at the time they occurred. There were no adverse events that led to death in this study. No clinical changes in laboratory findings were noted other than in parameters related to erythropoiesis (the pharmacological action of this drug).
Among adverse events, adverse drug reactions which indicates the causality between adverse events and KRN321 was not excluded were 33 events in 23 of 96 subjects (24.0%, 95% CI 15.8–33.7%). The major adverse drug reactions was 11 events of “hypertension” in 11 subjects (11.5%). “Increased blood pressure” and “increased eosinophil count” each occurred twice in two subjects (2.1%). Other events that occurred frequently were 8 “investigations” including “blood pressure increased” in 8 subjects (8.3%). No clear correlation was found between incidence of adverse drug reactions and Hb recorded at the time they occurred.
Discussion
PD patients with renal anemia were treated for 26–28 weeks with an initial 60 μg subcutaneous dose of KRN321 once every 2 weeks, regardless of Hb, until the target Hb (11.0–13.0 g/dL) was achieved. Thereafter, the dose and frequency were adjusted to maintain their Hb within the target range. As a result, the target Hb was maintained in nearly all subjects. In subjects with Hb at enrollment <11.0 g/dL, the weekly rate of increase in Hb after treatment at the initial dose was 0.18 g/dL/week. It was reported that all four patients who showed low responses to the initial dose had colds, but the Hb increased when the dose was increased. This rate of increase of Hb meets US, European, and Japanese anemia therapy guidelines [7–9], which recommend ESA therapy with rate of Hb increase of 0.5 g/dL/week or less.
After treatment at the initial dose, Hb reached the target range in most subjects. After that, Hb was maintained near a mean of 12.0 g/dL in most subjects. Approximately 60% of the subjects were switched to treatment once every 4 weeks. There were no clinical changes in Hb or dose after the switch, and mean Hb remained at around 12.0 g/dL.
PD patients are provided with the advantage that the number of hospital visits is reduced compared with HD patients. However, once PD patients develop renal anemia, they are forced to visit every week or once every 2 weeks to be treated with rHuEPO due to its dose and dosage, so PD patients cannot make full use of this advantage of PD. In this study, subcutaneous administration of KRN321 once every 2–4 weeks sufficiently improved anemia and allowed patients to make full use of this advantage of PD as compared with rHuEPO by reducing the number of hospital visits and injections.
When the study began there were no guidelines for anemia therapy for PD patients in Japan, and anemia therapy was based on a target Hb of 10.0 g/dL, as set forth in the package inserts for rHuEPO. In contrast, Kidney Disease Outcomes Quality Initiative and European Best Practice Guidelines anemia therapy guidelines [8, 9] have been published in the USA and Europe, and they recommend target Hb ≥11.0 g/dL, based on clinical studies using survival rate, QOL, cardiac function, and other indicators. The target Hb in this study was set at 11.0–13.0 g/dL based on these guidelines, and we evaluated these recommendations on the basis of QOL. There were no significant changes in QOL score for all parameters, while the scores for 6 of the 9 parameters were higher at the second QOL survey. In late phase II [10] and phase III studies [11] of KRN321 on ND patients in Japan, treatment with this drug was found to increase Hb to 11.0 g/dL or higher, which resulted in significantly increased vitality (VT) scores. VT scores also increased during the second QOL survey in this study, although not significantly. These data showed that the drug improved anemia by maintaining Hb between 11.0 and 13.0 g/dL, while no significant differences were found for SF-36 and FACIT fatigue, but since six of the nine items showed increases in the score and the frequency of KRN321 administration could be reduced to less than that for existing ESAs, which may lead to reduction of patients’ hospital visits, it was suggested that treatment with KRN321 was also useful in improving the QOL of patients.
On the other hand, there were many adverse events related to hypertension, although none were severe enough to cause patients to withdraw from the study. These events often are found during treatment with other ESAs and can be controlled well by interrupting or reducing dose, and/or by treatment with antihypertensives. However, since the possibility that improvements of anemia may lead to increase in blood pressure still cannot be ruled out, caution might be required on administration of darbepoetin alfa in the same way as with other ESAs [7]. In this study, some subjects had increased Hb above 13.0 g/dL, which was the upper limit of the target Hb. However, there were no new or clinically significant adverse events that had not previously been reported with this drug or other rHuEPO.
Recently, the result of the large-scale clinical Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) was released. TREAT was originally planned as a multinational, randomized, placebo-controlled, double-blind trial to determine whether treatment of anemia with darbepoetin alfa would reduce the risk of death and major cardiovascular events in patients with type 2 diabetes mellitus and chronic kidney disease. As a result, there was no significant difference regarding the primary endpoint (composite outcomes of death or a cardiovascular event or death or a renal event), however the occurrence of fatal and nonfatal stroke was higher in the darbepoetin alfa group [12]. In our study of PD subjects, there were no subjects who suffered stroke or other lethal adverse events.
Conclusions
We confirmed that Hb could be maintained safely between 11.0 and 13.0 g/dL, when KRN321 was administered in a subcutaneous dose of 60 μg once every 2 weeks to PD patients with renal anemia in Japan and the dose was then adjusted as needed. The Hb could also be maintained by subcutaneous administration once every 4 weeks, which suggested that this drug could reduce the frequency of hospital visits for PD patients to treat anemia compared with rHuEPO, and suggested the possibility of QOL improvement as well.
References
An overview of dialysis treatment in Japan (as of Dec. 31, 2008). In: The Statistical Survey Committee of the Japanese Society for Dialysis Therapy, editor. The Japanese Society for Dialysis Therapy; 2009.
Hiramatsu M, Kubota M, Yamamoto H. Limitation of rHuEPO treatment for target Hb in peritoneal dialysis patients. Kidney Dial. 2007;63:915–22. (Japanese).
Kuriyama S, Otsuka Y, Uetake D, Shirai I, Hosotani T. Current management of renal anemia in patients with chronic kidney disease at the predialysis stage. J Jpn Soc Nephrol. 2007;49:505–10. Japanese.
Egrie JC, Dwyer E, Browne JK, Hitz A, Lykos MA. Darbepoetin alfa has a longer circulating half-life and greater in vivo potency than recombinant human erythropoietin. Exp Hematol. 2003;31:290–9.
Macdougall IC, Gray SJ, Elston O, Breen C, Jenkins B, Egrie J. Pharmacokinetics of novel erythropoiesis stimulating protein compared with epoetin alfa in dialysis patients. J Am Soc Nephrol. 1999;10:2392–5.
Hasegawa T, Suzukamo Y, Akizawa T. Validation of the Japanese SF-36 v2 acute form in patients with chronic kidney disease. Nippon Jinzo Gakkai Shi. 2008;50:42–50.
2008 Japanese Society for Dialysis Therapy Guideline for Renal Anemia in Chronic Kidney Disease. J Jpn Soc Dial Ther 2008;41:661–716.
KDOQI clinical practice guidelines and clinical practice recommendation for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis 2007;50:471–530.
Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transpl 2004;19(Suppl 2):1–47.
Hirakata H, Tsubakihara Y, Gejyo F, Nishi S, Iino Y, Watanabe Y, et al. Maintaining high hemoglobin levels improved left ventricular mass index (LVMI) and quality of life (QOL) scores in pre-dialysis Japanese chronic kidney disease (CKD) patients. Clin Exp Nephrol. 2010;14:28–35.
Akizawa T, Gejyo F, Nishi S, Iino Y, Watanabe Y, Suzuki M, et al. Target level for hemoglobin correction by Darbepoetin Alfa (KRN321) for patients with chronic kidney disease (CKD) not on dialysis in a randomized controlled study; from the Viewpoint of the efficacy. J Am Soc Nephrol. 2007;18:762A. (abstract SUPO804).
Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, Zeeuw D, Eckardt KU, et al. A trial of Darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019–32.
Acknowledgments
The authors thank Prof. E Uchida (Showa University) and Dr. Y Tsukamoto (Shuwa General Hospital) for their advice on this study. This study was supported by Kyowa Hakko Kirin Co. Ltd.
Conflict of interest
None.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
KRN321 SCA10 Study Group members and their affiliations are listed in Appendix.
Appendix
Appendix
The KRN321 SCA10 Study Group comprises the following members and institutions: Katsuo Suzuki (Hakodate Koseiin Hakodate Goryokaku Hospital), Motoyuki Yamashita and Yoshiko Nishizawa (Oji Hospital), Yasuhiro Komatsu (St. Luke’s International Hospital), Yoshitaka Maeda (Toride Kyodo General Hospital), Mizuya Fukasawa (University of Yamanashi Hospital), Masaki Nagasawa (Nagano Prefectural Federation of Agricultural Cooperatives for Health and Welfare, Shinonoi General Hospital), Takahiko Aoki (Ogaki Municipal Hospital), Daijo Inaguma and Kei Kurata (Tosei General Hospital), Enyu Imai (Osaka University Graduate School of Medicine), Hirofumi Hasegawa (Kinki University Hospital), Akihiro Yasui (Kadoma Clinic), Tsutomu Tabata (Inoue Hospital), Masato Kasahara and Akihiro Yoshimoto (Kobe City Medical Center General Hospital), Masahiro Takeda (Komatsu Municipal Hospital), Makoto Hiramatsu (Okayama Saiseikai General Hospital), Masaki Fukushima (Kurashiki Central Hospital), Shigeaki Hayashida (Tokuyama Central Hospital), Akira Numata (Takamatsu Red Cross Hospital), Kei Hori (Munakata Medical Association Hospital), Masanobu Miyazaki, Katsushige Abe, and Akira Furusu (Nagasaki University Hospital), Jiro Machida (Saiseikai Kumamoto Hospital), and Masahito Yamakawa (Kyoritsu General Hospital).
About this article
Cite this article
Kubota, M., Hiramatsu, M., Yamakawa, M. et al. Darbepoetin alfa (KRN321) is safe and effective when administered subcutaneously once every 2 or 4 weeks to patients on peritoneal dialysis in Japan. Clin Exp Nephrol 15, 884–892 (2011). https://doi.org/10.1007/s10157-011-0527-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-011-0527-9