
Abstract
Clostridium difficile infection control strategies require an understanding of its epidemiology. In this study, we analysed the toxin genotypes of 130 non-duplicate clinical isolates of C. difficile from a university hospital in Tokyo, Japan. Multilocus sequence typing (MLST) and eBURST analysis were performed for these isolates and nine strains previously analysed by polymerase chain reaction (PCR) ribotyping. Minimum inhibitory concentrations (MICs) were determined for six antibiotics, and the bacterial resistance mechanisms were investigated. Ninety-five toxigenic strains (73 %), including seven tcdA-negative, tcdB-positive and cdtA/cdtB-negative strains (A−B+CDT−) and three A+B+CDT+ strains, and 35 (27 %) non-toxigenic strains, were classified into 23 and 12 sequence types, respectively. Of these, sequence type (ST)17 (21.8 %) was the most predominant. MLST and eBURST analysis showed that 139 strains belonged to seven groups and singletons, and most A+B+CDT− strains (98 %, 89/91) were classified into group 1. All isolates were susceptible to metronidazole, vancomycin and meropenem; the ceftriaxone, clindamycin and ciprofloxacin resistance rates were 49, 59 and 99 %, respectively. Resistance rates to ceftriaxone and clindamycin were higher in toxigenic strains than in non-toxigenic strains (P < 0.001). All ST17 and ST81 strains were resistant to these antibiotics. The clindamycin- and fluoroquinolone-resistant strains carried erm(B) and mutations in GyrA and/or GyrB, respectively. To our knowledge, this is the first MLST-based study of the molecular epidemiology of toxigenic and non-toxigenic strains in Japan, providing evidence that non-toxigenic and toxigenic strains exhibit high genetic diversity and that toxigenic strains are more likely than non-toxigenic strains to exhibit multidrug resistance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Clostridium difficile, a Gram-positive anaerobic sporulating bacillus, is the aetiologic pathogen of pseudomembranous colitis; it is also a well-known cause of antibiotic-associated diarrhoea. C. difficile infection (CDI) is of growing concern for its increasing incidence in hospitalised and non-hospitalised patients [1].
C. difficile enterotoxin A (TcdA) and cytotoxin B (TcdB) are largely responsible for the pathogenesis of this organism [2]. Although most pathogenic strains produce TcdA and TcdB, the toxin variant strains that produce only TcdB but not TcdA were also permitted [3]. Epidemic hypervirulent strains with binary toxin (CDT), pulsed-field gel electrophoresis (PFGE) type NAP1/polymerase chain reaction (PCR) ribotype 027 and PCR ribotype 078 have caused nosocomial outbreaks worldwide [4, 5]. The CDT-producing strain was also recently reported in our hospital [6].
Several methods are used to genotype prevalent strains. PCR ribotyping is the main method for identifying C. difficile, but the results are sometimes difficult to interpret and the method does not reveal phylogenetic relationships between the isolates. Although PFGE has been used to study various bacteria, it is a time-consuming process and shows poor data transferability. This has led to the emergence of new genotyping methods such as the sequence typing of surface layer protein genes and multilocus sequence typing (MLST) in recent years [7, 8]. With the support of online resources, MLST has been used for population genetics studies and global epidemiological analysis of various species.
However, in Japan, there have been few epidemiological studies of C. difficile [6, 9, 10], whereas MLST-based studies have not been performed. Previous studies have largely focused on the association between genetic diversity (PCR ribotyping or MLST) and antimicrobial susceptibility in toxigenic strains, including epidemic hypervirulent strains [4, 5, 11–13]. Little is known about the molecular epidemiology and antimicrobial susceptibility of non-toxigenic strains. Effective CDI control strategies must be built on data from non-toxigenic and toxigenic strains. Therefore, we investigated the relationship between genetic diversity and pathogenesis or antimicrobial susceptibility in clinical isolates of C. difficile, including non-toxigenic strains, and investigated the mechanisms of macrolide and fluoroquinolone resistance.
Materials and methods
Bacterial strains and culture conditions
In all, 130 non-duplicate C. difficile clinical isolates were recovered from 130 patients with suspected CDI at the Medical Hospital of Tokyo Medical and Dental University (a 763-bed teaching hospital with 33 clinical departments and 33 central clinical centres) between April 2012 and March 2013. These were the first clinical isolates from each patient. Nine strains from a previous study [6] were also examined by MLST. The PCR ribotypes a, e, f, h, j, n, q, x and 138 corresponded to PFGE types N, H, V, AF, K, AF, AL, S and Z described previously [6]. Strains were identified with the C. DIFF QUIK CHEK COMPLETE test (Alere Medical, Tokyo, Japan). Isolated strains were also identified by 16S rRNA gene sequencing using primers 10F (5′-GTTTGATCCTGGCTCA-3′) and 800R (5′-TACCAGGGTATCTAATCC-3′). Bacteria were anaerobically cultured at 37 °C on Gifu anaerobic medium (GAM) agar plates (Nissui Pharmaceutical, Tokyo, Japan). The isolates were suspended in 10 % skimmed milk for storage at −80 °C.
DNA isolation
A single colony isolated from GAM agar was emulsified in lysis solution (1 M Tris–HCl [pH 8.0], 4.5 % Nonidet P-40, 4.5 % Tween 20, 10 mg/mL proteinase K) and heated for 10 min at 60 °C and for 10 min at 100 °C. DNA was stored at −20 °C.
Determination of toxigenic type and tcdC sequencing
Multiplex PCR was used to detect tcdA encoding toxin A, tcdB encoding toxin B, cdtA and cdtB encoding binary toxin (CDT), and the 16S rRNA gene [14]. The length of tcdA was confirmed as previously described [3]. We also sequenced tcdC, a negative regulator of tcdA and tcdB [14].
Cytotoxicity assay
Vero cells (9.0 × 103 cells/mL) were grown in D-MEM (Wako Pure Chemical Industries, Osaka, Japan) with 10 % foetal bovine serum in 96-well plates. The isolates were cultured in brain–heart infusion broth (Oxoid, Basingstoke, UK) at 37 °C for 24 h. The filtered supernatants (10 μL) of C. difficile were added to 96-well plates. After incubation for 48 h, cytotoxicity was determined in replicates of three or four by using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Optical density was measured in a microplate reader (Wako, Osaka, Japan) at 450 nm and normalised to the untreated and blank groups. The results are presented as the cell viability percentage [mean ± standard deviation (SD)].
MLST
MLST was performed using seven loci (adk, atpA, dxr, glyA, recA, sodA and tpi) as previously described, with some modification [7]. Briefly, adk was amplified with primers adk1F2 (5′-CGTTGTTGGAGTTGCTTTGG-3′) and adk1R2 (5′-TGTCAGCAACTATTTTACCTGCT-3′), which were designed to match the sequence of C. difficile strain 630 (GenBank accession number NC_009089). PCR was performed in 2× EmeraldAmp MAX PCR Master Mix (Takara Bio, Shiga, Japan), and the products were then purified and sequenced. DNA sequences were submitted to the PubMLST sequence query page (http://pubmlst.org/cdifficile/) to obtain the sequence type (ST); a newly identified allele was deposited in the PubMLST database. The genetic diversity represented by the MLST data was analysed in eBURST version 3 (http://eburst.mlst.net/).
Antimicrobial susceptibility testing
The minimum inhibitory concentrations (MICs) of metronidazole, vancomycin, meropenem, ceftriaxone, clindamycin and ciprofloxacin were determined by the agar dilution method according to the Clinical and Laboratory Standards Institute (CLSI) guidelines, M100-S22 [15]. In 18 isolates, the MICs of moxifloxacin were also determined by the Etest (AB BIODISK, Solna, Sweden). The interpretation of breakpoints was based on CLSI criteria. The breakpoints of vancomycin and ciprofloxacin were ≥32 and ≥8 mg/L, respectively. Bacteroides fragilis ATCC 25285 was used for quality control. Multidrug resistance was defined as resistance to at least three classes of antibiotics.
Detection of methylase genes and sequencing of gyrA and gyrB
The presence of methylase genes, erm(A), erm(B), erm(C) and erm(F), was determined as previously described [16, 17] in 18 isolates, including two clindamycin- and ciprofloxacin-susceptible strains. Among them, mutations in gyrA and gyrB that mediate fluoroquinolone resistance were also assessed as previously described [18].
Statistical analysis
Categorical variables and differences for comparisons were evaluated by the Chi-square or Fisher’s exact test and the Mann–Whitney U-test, respectively (JMP software, version 11, SAS Institute Japan, Tokyo, Japan). In all analyses, P < 0.05 was considered significant.
Results
Information about the clinical isolates
Among the 130 strains, 82 strains (63 %) were isolated from patients over 60 years old and 74 strains (57 %) were from male patients. Twelve strains (9 %) were isolated from outpatients. These 130 strains were frequently isolated from the gastroenterology and hepatology department (20 strains, 15 %), followed by the acute care medical centre department (13 strains, 10 %) and the paediatric department (10 strains, 8 %).
Toxigenic types and sequences of tcdC
Of the 130 strains, 85 (66 %) were tcdA-positive, tcdB-positive and cdtA/cdtB-negative (A+B+CDT−); 35 (27 %) were tcdA-negative, tcdB-negative and cdtA/cdtB-negative (A−B−CDT−); and seven (5 %) isolates were tcdA-negative, tcdB-positive and cdtA/cdtB-negative (A−B+CDT−). The remaining three (2 %) isolates were tcdA-positive, tcdB-positive and cdtA/cdtB-positive (A+B+CDT+). Of the nine previously described isolates [6], six were A+B+CDT−, two were A−B+CDT− and one was A+B+CDT+. In three A+B+CDT+ strains, tcdC carried an 18-bp deletion at nucleotide positions 330–347.
Cytotoxicity assay
Cytotoxicity assays were performed on three A+B+CDT+ and four A+B+CDT− strains. At 48 h, there was no significant difference in cytotoxicity between the A+B+CDT− and A+B+CDT+ strains (74.7 ± 11.4 vs. 74.3 ± 7.6; P = 0.8) (Fig. 1).

The cytotoxicity of A+B+CDT− (n = 4) and A+B+CDT+ (n = 3) strains
ST and toxin genotypes
The 130 strains were classified into 33 STs (Table 1). Fourteen strains were assigned novel STs, including ST182, ST185 and ST205, with new combinations of allelic profiles; ST183, ST184, ST201, ST202 and ST203 with one or two new alleles; and ST204 with seven new alleles. ST17 (28 isolates, 21.5 %) was the most frequent of these 33 STs, followed by ST2 (13 isolates, 10 %) and ST8 (12 isolates, 9.2 %; Table 1). ST2 and ST8 were three- and two-allelic variants of ST17, respectively. Ninety-five toxigenic strains and 35 non-toxigenic strains were classified into 23 and 12 STs, respectively (Table 1). ST3 and ST48 strains included both toxigenic and non-toxigenic types. All ST17, ST2 and ST8 strains were A+B+CDT− strains; all seven A−B+CDT− strains were classified into ST81. Three A+B+CDT+ strains were both ST5 and ST201, respectively. The nine previously identified strains included PCR ribotypes a, e, f, h, j, n, q, x and 138 [6] and were classified into ST2, ST8, ST17, ST37, ST3, ST81, ST35, ST63 and ST97, respectively. ST37 (A−B+CDT−), ST63 (A+B+CDT−) and ST97 (A+B+CDT+) were not identified among the strains collected from April 2012 to March 2013. After combining these data with the data from the clinical isolates, we found that, although three strains classified as ST39 (A−B−CDT−), ST42 (A+B+CDT−) and ST182 (A+B+CDT−) were isolated from outpatients, there was no evidence of CDI outbreaks caused by toxigenic types with specific STs in any clinical department during the study period.
eBURST analysis
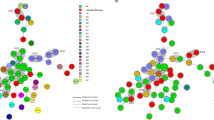
Genetic diversity was characterised using eBURST version 3 and the MLST data for 139 strains and all previously registered STs (275 as of 6 June 2014; Fig. 2). We used the default eBURST setting, which defined a group as one in which all members shared identical alleles at more than six of seven loci with at least one other member of the group; the primary founder of a group was defined as the ST that differed from the largest number of other STs at only a single locus.

The rough sketch produced by eBURST representing the C. difficile population and the positions of 139 strains, including nine previously identified strains [6]. These strains were classified into seven groups and singletons. Singletons were omitted. □, A+B+CDT−; △, A−B+CDT−; ■, A+B+CDT+; ○, A−B−CDT−
All 275 STs were classified into 16 groups and singletons. Our 139 strains belonged to groups 1, 2, 3, 6, 7, 9, 14 and singletons (Fig. 2): (i) group 1, the largest group (74 %, 103/139), comprising 156 STs such as ST17, ST2 and ST8, ST28 being the founder; (ii) group 2, comprising eight STs including ST5 and ST201; (iii) group 3, seven STs, including ST37 and ST81; (iv) groups 6, 7, 9 and 14 included ST39, ST109, ST55 and ST202, respectively.
Most A+B+CDT− strains (98 %, 89/91) were classified into group 1 (Fig. 2), while most A−B−CDT− strains (60 %, 21/35) belonged to groups 6, 7, 14 and singletons. Among the three A+B+CDT+ strains, ST5 and ST201 were in group 2; however, a strain of ST97, assigned to PCR ribotype 138 in a previous study, was a singleton and had no genetic relationship to ST1, which was assigned to the NAP1/027 strain [7].
Antimicrobial resistance profiles
The MIC90 values of metronidazole, vancomycin, meropenem, ceftriaxone, clindamycin and ciprofloxacin were 0.5, 2, 4, >256, >256 and >32 mg/L, respectively (Table 2). All 130 strains were susceptible to metronidazole and vancomycin; one strain was classified as intermediate susceptibility to meropenem (MIC = 8 mg/L). In contrast, 49, 59 and 99 % of the 130 strains were resistant to ceftriaxone, clindamycin and ciprofloxacin, respectively. All ST17 and ST81 strains showed high-level resistance to ceftriaxone (MIC ≥128 mg/L), clindamycin (MIC ≥32 mg/L) and ciprofloxacin (MIC ≥32 mg/L) (Table 1, Fig. 3). Toxigenic strains showed higher resistance rates to ceftriaxone and clindamycin than in non-toxigenic strains (P < 0.001, Table 2). However, although multidrug-resistant strains such as ST17 and ST81 were isolated from many clinical departments, there was no relationship between ST and antibiotic resistance in any clinical department or outpatient during the study period.

Antimicrobial susceptibility to ceftriaxone (CRO), clindamycin (CLI) and ciprofloxacin (CIP) of the predominant STs and toxigenic types found
Mechanism of resistance to clindamycin and ciprofloxacin
The presence of methylase genes and mutations in GyrA and GyrB were investigated in 18 isolates with varying MICs for clindamycin and ciprofloxacin. erm(B) was detected in 13 clindamycin-resistant isolates, in addition to one clindamycin-susceptible and one clindamycin-intermediate isolate (MIC = 2 and 4 mg/L, respectively; Table 3). We observed no correlation between the presence of erm(B) and ST. However, nonsense mutations Glu201X or Gln153X were identified in susceptible isolates. The tested isolates did not carry erm(A), erm(C) or erm(F).
A high level of cross-resistance to moxifloxacin and ciprofloxacin was observed (Table 4). Twelve fluoroquinolone-resistant isolates with ciprofloxacin and moxifloxacin MICs of >4 mg/L carried a single GyrA mutation (three isolates), double GyrA mutations (one isolate), a single GyrB mutation (one isolate) or both GyrA and GyrB mutations (seven isolates) (Table 4). Isolates with Thr82Ile mutations in GyrA showed high-level resistance to moxifloxacin (MICs of >32 mg/L) and ciprofloxacin (MICs of 32–128 mg/L), while isolates with Thr82Val or Thr82Ala mutations showed moxifloxacin MICs of 16 mg/L and ciprofloxacin MICs of 16–64 mg/L. Asp426Asn, Asp426Val, Gln434Lys and Glu466Lys mutations in GyrB were detected only in fluoroquinolone-resistant isolates; an isolate with a single Glu466Lys mutation and no GyrA mutations had a moxifloxacin MIC of 8 mg/L (Table 4). The fluoroquinolone-susceptible isolate, B-13-23, had three or four mutations in GyrA and GyrB (ciprofloxacin MIC = 0.25 mg/L and moxifloxacin MIC = 0.38 mg/L, respectively). No significant relationship was determined between mutations in GyrA and/or GyrB and ST or toxigenic types.
Discussion
In a previous study at our hospital, 148 toxigenic strains were recovered during the five-year period from November 1999 to October 2004 [6]. We obtained 95 toxigenic isolates from April 2012 to March 2013, suggesting that, although CDI was estimated by isolated numbers and toxin assays, the frequency of CDI cases may have increased in our hospital, similar to that in Japan and overseas [2, 19].
Compared with MLST studies in China [20] and Spain [13], the prevalence of A−B+CDT− (5 %) and A+B+CDT+ (2 %) strains in Japan were very low. Although the reason for this difference is not clear, our findings were similar to a previous report in Japan [10]. CDT-producing strains such as NAP1/027 produce excess toxins A and B due to deletions (18-bp, 39-bp or 54-bp) in tcdC, a negative regulator of tcdA and tcdB [14]. In this study, however, we detected only an 18-bp deletion in three A+B+CDT+ strains that exhibited similar cytotoxicity in comparison to randomly selected A+B+CDT− strains. Therefore, our results also suggest that the truncated tcdC may not be associated with toxin production, as has been previously proposed [21].
Sawabe et al. found that predominant toxigenic clones shifted from PCR ribotype a to f between 2000 and 2004 [6]. In this study, ST17 (21.5 %) corresponded to PCR ribotype f and was the predominant clone in our hospital, followed by ST2 (10 %), corresponding to PCR ribotype a. A previous domestic report showed that ST17, which is classified as smz by PCR ribotyping, is predominant in some Japanese hospitals [9]. Although we could not determine whether ST17 was responsible for the outbreak in this study, our findings suggest that ST17 may have spread through the country over the past decade. Other researchers have reported that PCR ribotype 018, corresponding to the smz type in Austria, Spain and Slovenia [22, 23], has been the most prevalent clone in Italy since 2007 [24]. Therefore, we suggest that ST17 strains, classified as smz type (corresponding to PCR ribotype 018), are prevalent worldwide. Although ST2 strains related to PCR ribotype 014 [25] have also been recovered from various European countries, this type may be globally widespread [13, 26].
In this study, MLST and eBURST analysis revealed that most A+B+CDT− strains, including ST17, ST2 and ST8, were classified into group 1, and may be derived from presumptive ST28 (Table 1 and Fig. 2). Furthermore, because ST3 and ST48 strains in group 1 include toxigenic and non-toxigenic types, we suggest these STs may be easy to lose or obtain toxin synthesis-related genes through genetic shift. Moreover, our results provide the first evidence that toxigenic strains (classified into 23 STs) and non-toxigenic strains (classified into 12 STs) exhibit high genetic diversity. Therefore, we suggest that selective pressures such as antibiotics, the human immune system and environmental conditions may drive the genetic rearrangements in non-toxigenic and toxigenic strains. On the other hand, all A−B+CDT− strains were assigned to ST81 and carried a single allelic variant (atpA) in comparison to ST37, which was the predominant type (corresponding to PCR ribotype h) in a previous study at our hospital [6]. Although ST81 may have come from elsewhere, our findings suggest that the genetic shift from ST37 to ST81 occurred in our hospital. Among A+B+CDT+ strains, there was no genetic relationship between the present A+B+CDT+ strains (ST5 and ST201), the previous A+B+CDT+ strain (ST97) and NAP1/027 (ST1) [7].
Previous studies have shown reduced susceptibility to antibiotics such as fluoroquinolone and macrolides in clinical isolates of C. difficile, including PCR ribotypes 001, 078 and NAP1/027 [4, 5, 12, 13]; our results were consistent with these findings (Fig. 3). Moreover, although there is no difference in the MICs of metronidazole, vancomycin, meropenem and ciprofloxacin, we demonstrated that toxigenic strains are more likely than non-toxigenic strains to acquire resistance to ceftriaxone and clindamycin. We also suggest that multidrug resistance is common in toxigenic strains such as ST17 and ST81.
The mechanisms of clindamycin resistance involve active efflux of the antibiotic and modification of the ribosomal target by enzymes such as rRNA methylase or via mutations in the 23S rRNA gene [27, 28]. Previous reports of C. difficile have demonstrated the association between Erm(B) and clindamycin resistance [12]; however, these reports have been limited. Although clindamycin-susceptible and -intermediate strains (MIC ≤4 mg/L) expressed Erm(B) nonsense mutants (Glu201X and Gln153X), specific genetic lineages with high-level resistance to clindamycin are likely to carry Erm(B); our results also indicated that Erm(B) may be associated with clindamycin resistance (MIC) in C. difficile, independent of the ST or toxigenic type.
On the other hand, mutations of GyrA and GyrB (DNA gyrase subunit A and B) are important for acquiring resistance to fluoroquinolone in various bacteria, including C. difficile [29]. In this study, a mutation at Thr82 in GyrA and/or mutations in GyrB were detected in fluoroquinolone-resistant strains (MIC ≥8 mg/L), similar to previous reports [11, 18, 30, 31]. Specific genetic lineages with high-level resistance to fluoroquinolones are likely to carry mutations in GyrA and GyrB, and mutations at Thr82 in GyrA or Glu466 in GyrB play an important role in fluoroquinolone resistance in C. difficile, independent of the ST or toxigenic type. Furthermore, our observations indicated that mutations in GyrA and GyrB synergistically contribute to the acquisition of high-level resistance to ciprofloxacin, and that Thr82Ile mutations in GyrA may be associated only with high-level resistance to moxifloxacin. Although one fluoroquinolone-susceptible isolate carried mutations in GyrA and GyrB, we suggest that there was no relationship between fluoroquinolone resistance and these mutations. A previous report demonstrated that a Ser416Ala mutation is responsible for fluoroquinolone resistance [11]. In our study, this mutation was associated with a fluoroquinolone-susceptible strain; however, a site-directed mutagenesis study is needed in order to further investigate the relationship between fluoroquinolone resistance and target mutations.
To the best of our knowledge, this is the first molecular epidemiological study to evaluate toxigenic and non-toxigenic strains by MLST. We showed that ST17, which has been isolated from many countries, has been the prevalent strain in our hospital since 2004. MLST and eBURST analysis showed the first evidence that most A+B+CDT− strains, including ST17, were classified into group 1, and that both toxigenic and non-toxigenic strains exhibit high genetic diversity. Moreover, toxigenic strains, particularly those belonging to ST17 and ST81, are more likely to exhibit multidrug resistance in comparison to non-toxigenic strains. Our findings indicate that Erm(B) and mutations in GyrA and/or GyrB play an important role in resistance to clindamycin and fluoroquinolone, respectively; however, further work is needed to understand the relationship between these mechanisms and the ST or toxigenic type. Although infection control strategies of CDI must be built on investigations of the changing genetic diversity of C. difficile, we also suggest that MLST is useful for monitoring nosocomial strains and specific genetic lineages worldwide.
References
Depestel DD, Aronoff DM (2013) Epidemiology of Clostridium difficile infection. J Pharm Pract 26:464–475
George RH, Symonds JM, Dimock F, Brown JD, Arabi Y, Shinagawa N, Keighley MR, Alexander-Williams J, Burdon DW (1978) Identification of Clostridium difficile as a cause of pseudomembranous colitis. Br Med J 1:695
Kato H, Kato N, Watanabe K, Iwai N, Nakamura H, Yamamoto T, Suzuki K, Kim SM, Chong Y, Wasito EB (1998) Identification of toxin A-negative, toxin B-positive Clostridium difficile by PCR. J Clin Microbiol 36:2178–2182
Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC (2005) Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 366:1079–1084
Goorhuis A, Bakker D, Corver J, Debast SB, Harmanus C, Notermans DW, Bergwerff AA, Dekker FW, Kuijper EJ (2008) Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin Infect Dis 47:1162–1170
Sawabe E, Kato H, Osawa K, Chida T, Tojo N, Arakawa Y, Okamura N (2007) Molecular analysis of Clostridium difficile at a university teaching hospital in Japan: a shift in the predominant type over a five-year period. Eur J Clin Microbiol Infect Dis 26:695–703
Griffiths D, Fawley W, Kachrimanidou M, Bowden R, Crook DW, Fung R, Golubchik T, Harding RM, Jeffery KJM, Jolley KA, Kirton R, Peto TE, Rees G, Stoesser N, Vaughan A, Walker AS, Young BC, Wilcox M, Dingle KE (2010) Multilocus sequence typing of Clostridium difficile. J Clin Microbiol 48:770–778
Kato H, Yokoyama T, Arakawa Y (2005) Typing by sequencing the slpA gene of Clostridium difficile strains causing multiple outbreaks in Japan. J Med Microbiol 54:167–171
Kato H, Kato N, Watanabe K, Yamamoto T, Suzuki K, Ishigo S, Kunihiro S, Nakamura I, Killgore GE, Nakamura S (2001) Analysis of Clostridium difficile isolates from nosocomial outbreaks at three hospitals in diverse areas of Japan. J Clin Microbiol 39:1391–1395
Oka K, Osaki T, Hanawa T, Kurata S, Okazaki M, Manzoku T, Takahashi M, Tanaka M, Taguchi H, Watanabe T, Inamatsu T, Kamiya S (2012) Molecular and microbiological characterization of Clostridium difficile isolates from single, relapse, and reinfection cases. J Clin Microbiol 50:915–921
Liao CH, Ko WC, Lu JJ, Hsueh PR (2012) Characterizations of clinical isolates of Clostridium difficile by toxin genotypes and by susceptibility to 12 antimicrobial agents, including fidaxomicin (OPT-80) and rifaximin: a multicenter study in Taiwan. Antimicrob Agents Chemother 56:3943–3949
Kim J, Kang JO, Pai H, Choi TY (2012) Association between PCR ribotypes and antimicrobial susceptibility among Clostridium difficile isolates from healthcare-associated infections in South Korea. Int J Antimicrob Agents 40:24–29
Weber I, Riera E, Déniz C, Pérez JL, Oliver A, Mena A (2013) Molecular epidemiology and resistance profiles of Clostridium difficile in a tertiary care hospital in Spain. Int J Med Microbiol 303:128–133
Persson S, Torpdahl M, Olsen KEP (2008) New multiplex PCR method for the detection of Clostridium difficile toxin A (tcdA) and toxin B (tcdB) and the binary toxin (cdtA/cdtB) genes applied to a Danish strain collection. Clin Microbiol Infect 14:1057–1064
Clinical and Laboratory Standards Institute (CLSI) (2012) Performance standards for antimicrobial susceptibility testing; Twenty-second informational supplement. CLSI document M100-S22. CLSI, Wayne, PA
Nonaka S, Matsuzaki K, Kazama T, Nishiyama H, Ida Y, Koyano S, Sonobe K, Okamura N, Saito R (2014) Antimicrobial susceptibility and mechanisms of high-level macrolide resistance in clinical isolates of Moraxella nonliquefaciens. J Med Microbiol 63:242–247
Chung WO, Werckenthin C, Schwarz S, Roberts MC (1999) Host range of the ermF rRNA methylase gene in bacteria of human and animal origin. J Antimicrob Chemother 43:5–14
Drudy D, Quinn T, O’Mahony R, Kyne L, O’Gaora P, Fanning S (2006) High-level resistance to moxifloxacin and gatifloxacin associated with a novel mutation in gyrB in toxin-A-negative, toxin-B-positive Clostridium difficile. J Antimicrob Chemother 58:1264–1267
Zilberberg MD, Shorr AF, Kollef MH (2008) Increase in adult Clostridium difficile-related hospitalizations and case–fatality rate, United States, 2000–2005. Emerg Infect Dis 14:929–931
Yan Q, Zhang J, Chen C, Zhou H, Du P, Cui Z, Cen R, Liu L, Li W, Cao B, Lu J, Cheng Y (2013) Multilocus sequence typing (MLST) analysis of 104 Clostridium difficile strains isolated from China. Epidemiol Infect 141:195–199
Murray R, Boyd D, Levett PN, Mulvey MR, Alfa MJ (2009) Truncation in the tcdC region of the Clostridium difficile PathLoc of clinical isolates does not predict increased biological activity of Toxin B or Toxin A. BMC Infect Dis 9:103
Collins DA, Hawkey PM, Riley TV (2013) Epidemiology of Clostridium difficile infection in Asia. Antimicrob Resist Infect Control 2:21
Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M, Monnet DL, van Dissel JT, Kuijper EJ; ECDIS Study Group (2011) Clostridium difficile infection in Europe: a hospital-based survey. Lancet 377:63–73
Spigaglia P, Barbanti F, Dionisi AM, Mastrantonio P (2010) Clostridium difficile isolates resistant to fluoroquinolones in Italy: emergence of PCR ribotype 018. J Clin Microbiol 48:2892–2896
Knetsch CW, Terveer EM, Lauber C, Gorbalenya AE, Harmanus C, Kuijper EJ, Corver J, van Leeuwen HC (2012) Comparative analysis of an expanded Clostridium difficile reference strain collection reveals genetic diversity and evolution through six lineages. Infect Genet Evol 12:1577–1585
Barbut F, Mastrantonio P, Delmée M, Brazier J, Kuijper E, Poxton I; European Study Group on Clostridium difficile (ESGCD) (2007) Prospective study of Clostridium difficile infections in Europe with phenotypic and genotypic characterisation of the isolates. Clin Microbiol Infect 13:1048–1057
Trieu-Cuot P, Poyart-Salmeron C, Carlier C, Courvalin P (1990) Nucleotide sequence of the erythromycin resistance gene of the conjugative transposon Tn1545. Nucleic Acids Res 18:3660
Gregory ST, Dahlberg AE (1999) Erythromycin resistance mutations in ribosomal proteins L22 and L4 perturb the higher order structure of 23 S ribosomal RNA. J Mol Biol 289:827–834
Hooper DC, Wolfson JS, Ng EY, Swartz MN (1987) Mechanisms of action of and resistance to ciprofloxacin. Am J Med 82:12–20
Spigaglia P, Barbanti F, Mastrantonio P, Brazier JS, Barbut F, Delmée M, Kuijper E, Poxton IR; European Study Group on Clostridium difficile (ESGCD) (2008) Fluoroquinolone resistance in Clostridium difficile isolates from a prospective study of C. difficile infections in Europe. J Med Microbiol 57:784–789
Walkty A, Boyd DA, Gravel D, Hutchinson J, McGeer A, Moore D, Simor A, Suh K, Taylor G, Miller M, Mulvey MR; Canadian Nosocomial Infection Surveillance Program (2010) Molecular characterization of moxifloxacin resistance from Canadian Clostridium difficile clinical isolates. Diagn Microbiol Infect Dis 66:419–424
Conflict of interest
The study did not receive financial support from any third party. All authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kuwata, Y., Tanimoto, S., Sawabe, E. et al. Molecular epidemiology and antimicrobial susceptibility of Clostridium difficile isolated from a university teaching hospital in Japan. Eur J Clin Microbiol Infect Dis 34, 763–772 (2015). https://doi.org/10.1007/s10096-014-2290-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-014-2290-9