
Abstract
Background
The Brazilian Northeast region is notable for its high prevalence of consanguineous marriages and isolated populations, which has led to a significant prevalence of rare genetic disorders. This study describes the clinical presentation of four affected individuals from the same family, comprising two siblings and their cousins, with ages ranging from 11 to 20 years.
Methods
In a small and isolated community in Northeastern Brazil, affected individuals initially underwent a clinical assessment. Subsequently, written consent was obtained from their legal guardians, and an extensive clinical evaluation was conducted at a medical genetics center. Family data provided the basis for constructing the pedigree, and biological samples (blood or oral swabs) were collected from both affected and unaffected family members. Following informed consent from one patient, Whole Exome Sequencing (WES) was carried out, encompassing exome sequencing, assembly, genotyping, and annotation. A potentially deleterious variant was then singled out for further segregation analysis through Sanger Sequencing, involving both the proband and select family members.
Results and conclusion
These individuals exhibit severe neurodevelopmental delays, encompassing symptoms such as spastic paraplegia, neuropathy, intellectual impairments, and language challenges. Through next-generation sequencing (NGS) techniques, a previously unreported homozygous variant within the ERLIN2 gene linked to spastic paraplegia 18 (SPG18) was identified across all four patients. Also, all patients displayed childhood cataract, expanding the known clinical spectrum of SPG18.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In Brazil, studies conducted since the 1950s have revealed a high occurrence of consanguineous marriages. Particularly in the Northeast region, the frequency has been reported to be approximately 15 times higher than in the Southern region of the country [1]. In 2012, a subsequent study within isolated communities in Northeastern Brazil identified rates of endogamy ranging from 6 to 41.1% [2]. A more recent study conducted in 2021 also linked the low diversity of surnames to a high consanguinity rate in isolated municipalities in the Brazilian northeast [3].
In 2005, a group of researchers identified and described for the first time 26 individuals from consanguineous families in the backlands of Northeastern Brazil who had a complicated autosomal recessive (AR) form of Hereditary Spastic Paraplegia (HSP) characterized by spastic paraplegia, optic atrophy and neuropathy (SPOAN syndrome, OMIM #609541) [4]. After a decade, the etiological diagnosis of SPOAN in more than 70 individuals from this group reinforced the association between the syndrome and levels of inbreeding in these isolated populations [5]. In this work, we identified four patients from the same family (2 siblings and their cousins) with previously unreported homozygous variant in ERLIN2 gene associated with SPG18 (SPG18, OMIM #611225). Furthermore, all four patients present cataract since childhood which represents an expansion of the clinical phenotype for this disorder.
Methods
Clinical analysis and family material
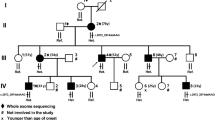
As part of a larger project investigating neurodevelopmental disorders in highly inbred regions of Northeastern Brazil, affected individuals from a small and isolated community underwent clinical evaluation in their hometown. After obtaining written consent from their legal guardians, an extensive clinical evaluation was carried out in a medical genetics center and in an ophthalmology service. The provided family data was used to build the pedigree (Fig. 1). Blood samples or oral swabs were obtained from affected and unaffected family members for further DNA extraction. The data sampling protocol, as well as the consent procedure, were analyzed and approved by the National Research Ethics Committee - CONEP (Process 39674220.1.1001.5013).

Family pedigree showing individuals with spastic paraplegia 18 (SPG18) represented in filled symbols and half-filled symbols indicate heterozygous individuals. Genotyped individuals are underlined
Exome and Sanger Sequencing
Whole Exome Sequencing (WES) was performed after informed consent in one patient (III-7). Genomic DNA was extracted from peripheral blood leukocytes or oral swabs using standard protocols. Exome sequencing, assembly, genotyping, and annotation were performed at Mendelics Genomic Analysis (São Paulo, SP, Brazil). Exome capture was performed using the Illumina Nextera Rapid Capture Exome kit (Illumina, Inc., San Diego, CA, USA). Sequencing was performed in an Illumina HiSeq 2500. Sequence reads were aligned with the reference human genome (UCSC Genome Browser GrCh38/hg38) with the BWA (Burrows-Weeler Aligner) software (2009). Genotyping was performed using the Genome Analysis Toolkit (GATK). Annotation, filtering, and variant prioritization were performed using Mendelics proprietary software, allowing an in silico reduction in candidates for further analysis. Classification of variants follows the guidelines of the American College of Medical Genetics (ACMG). One potentially deleterious variant was selected for further segregation analysis by Sanger Sequencing in the proband and some family members. PCR products were amplified using the following primers: forward: 5′- CACGTTGGACCACACACATT – 3′; reverse: 5′- CTCCCTGTCTCTGCCTTACC – 3′. The reaction products were analyzed in the ABI 3500 DNA Analyzer equipment (Applied Biosystems, Carlsbad, CA, USA). The results were analyzed using the Software Chromas and CLC Sequence Viewer.
Results
Clinical presentation
We evaluated four individuals from the same family (two siblings and their cousins, aged between 11 and 20 years). In all of them, the complicated form of HSP was detected, characterized by earlier onset and severe conditions. They all depend on external help for basic activities, due to neuropsychomotor development delay, verified before 1 year old. We also observed progressive spasticity, which predominates in the lower limbs, associated with lower limb muscle weakness. They also have never walked and make use of a wheelchair. In addition, they have intellectual and language impairments, having never learned to read or write, although they maintain a low level of interaction with the examiner. Furthermore, in proband III-7, an association with epilepsy was observed, and III-6, III-7 and III-8 presents a compromised ability to verbalize. Moreover, probands III-6, III-7 and III-8 were diagnosed with childhood cataract, and more recently III-7 and III-8 underwent surgical correction. The ophthalmological alteration in individuals III-7 and III-8 led us to investigate possible alterations in the remaining individuals. Consequently, subject III-4 (11 years old) underwent his first ophthalmological evaluation, which also revealed the presence of a cataract, described as a pulverulent cataract, a rare subtype. No other family member besides the patients exhibits cataracts. Furthermore, as evident in Table 1, cataracts were also not reported in any other patients described in the literature. This suggests childhood cataract as a phenotypic expansion of SPG18. It also should be emphasized that heterozygous individuals in the family are healthy, characterizing an autosomal recessive inheritance in this family.
The presented table offers a comprehensive overview of families associated with SPG 18 up to the point of writing this article. The clinical presentation of most patients with the complicated form and autosomal recessive inheritance shares common characteristics, always involving intellectual deficiency and motor impairment. This can manifest itself through limb deformities, joint contractures, or an altered gait.
Genetic findings
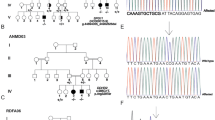
The WES showed a mutation with autosomal recessive inheritance in exon 8 of the ERLIN2 gene: c.47_48delinsAA (Fig. 2A), which results in a premature stop codon in the 16th amino acid cysteine (p.Cys16*), leading to a truncated production of the ERLIN2 protein. This variant cosegregated with the disease (Fig. 2B) and was not present in Brazilian population controls (Mendelics Database) as well as in gnomAD [17], neither in autosomal recessive inheritance, nor in dominant inheritance. Moreover, p.Cys16* is conserved across ERLIN2 orthologues and predicted as deleterious by MutationTaster.

Visual presentation of exome sequencing data made by the Integrative Genomics Viewer (a). Sanger sequencing electropherograms (b) showing homozygous control, heterozygous at position c.47_48 of ERLIN2 gene in a carrier (II-4) and homozygosity in an affected individual (III-4)
Discussion
The approach of combining field investigation in highly inbred regions of Brazil, where clusters of genetic disorders are sought, with advanced molecular techniques has proven to be successful once again in this study. Using this strategy, new causative mechanisms have already been related to two autosomal recessive forms of intellectual disability associated to MED25 and IMPA1 genes [18, 19], as well as a new genetic syndrome associated with spastic paraplegia, known as SPOAN syndrome [5].
Hereditary spastic paraplegias (SPGs) are a group of neurodegenerative diseases with heterogeneous clinical manifestations and genetic etiologies. It is an extremely rare condition with a prevalence ranging from 0.1 to 9.6 cases per 100,000 individuals worldwide [20]. In the case of SPG18, its clinical symptoms include contractures, progressive global muscle weakness, muscle atrophy, increased muscle tone, delayed walking, abnormal gait, spasticity in the lower and upper limbs, extensor plantar responses, hyperreflexia, speech impairment, intellectual disability, seizures, scoliosis, kyphosis, pes cavus, squint, and high-arched palate.
The gene associated with SPG18 is ERLIN2, which was initially identified in 2011, through a mapping of a Turkish family with autosomal recessive intellectual disability, motor impairment, and multiple joint contractures [6]. Around the same time, a family from Saudi Arabia, with the complex form of hereditary spastic paraplegia, was found to have a deletion mutation of ERLIN2. This form of the disease was later classified as SPG18 [7].
To date, different presentations of the disease have been described, both in its pure and complicated form, from a total of seventeen families/cases investigated. Among them, nine have autosomal recessive SPG18, five have autosomal dominant SPG18, and three are sporadic cases [6,7,8,9,10,11,12]. All of these patients with different types of SPG were from Asia (Middle East, Iran, Turkey, China, and Korea) and Europe (France and Norway). None of them were from the American continent. Seven of the eight families with autosomal recessive SPG18 had reported consanguineous marriages (7 out of 8). It is crucial to underscore the significant association between the autosomal recessive inheritance pattern and the complex phenotype of the disease, considering that all complicated cases exhibit this inheritance pattern. At this stage, it is possible to notice that all cases with autosomal recessive inheritance and complicated forms consistently manifest intellectual deficiency. Notably, only two cases demonstrate both an autosomal recessive pattern and a pure phenotype: a family from Iran [13] and another from China [10]. It is pertinent to mention that all reported autosomal dominant and sporadic cases are pure.
Furthermore, it is relevant to highlight a discernible pattern of genetic alteration in pure forms resulting from autosomal dominant inheritance and sporadic cases, all originating from amino acid substitutions. In contrast, complicated phenotypes, represented by autosomal recessive inheritance, arise from genetic variants in homozygosity, encompassing large deletions, insertions, duplications, splicing mutations, and point mutations.
Here, we present two affected siblings and their two cousins with a novel variant in the ERLIN2 gene (c.47_48delinsAA). It’s important to notice that the children’s fathers are brothers and their mothers are cousins to each other, all from an isolated region in the Brazilian northeast. Once the same homozygous variant was found in all four patients, it possibly suggests that they share a common ancestor. This is the first case report about a family with SPG18 in the Americas and the first worldwide to report childhood cataract in SPG18, representing an expansion of this disorder’s clinical phenotype.
Conclusion
In conclusion, this study marks the first documented case of SPG18 in the Americas, identified through the assessment of four individuals within the same family. The findings highlight a consistent presentation of the complicated form of Hereditary Spastic Paraplegia (HSP) and a notable association with childhood cataracts, suggesting a phenotypic expansion of SPG18. Importantly, this case introduces a novel variant in the ERLIN2 gene, contributing to our evolving understanding of the genetic factors underlying SPG18.
Data availability
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to personal identifying information.
References
Freire-Maia N (1957) Inbreeding in Brazil. Am J Hum Genet 9(4):284–298
Weller M, Tanieri M, Pereira JC, Almeida EDS, Kok F, Santos S (2012) Consanguineous unions and the burden of disability: a population-based study in communities of Northeastern Brazil. Am J Human Biol 24(6):835–840. https://doi.org/10.1002/ajhb.22328
Cardoso-dos-Santos AC, Ramallo V, Zagonel-Oliveira M, Veronez MR, Navarro P, Monlleó IL, Valiati VH, Dipierri JE, Schuler-Faccini L (2021) An invincible memory: what surname analysis tells us about history, health and population medical genetics in the Brazilian Northeast. J Biosoc Sci 53(2):183–198. https://doi.org/10.1017/S0021932020000127
Macedo-Souza LI, Kok F, Santos S, Amorim SC, Starling A, Nishimura A, Lezirovitz K, Lino AMM, Zatz M (2005) Spastic paraplegia, optic atrophy, and neuropathy is linked to chromosome 11q13. Ann Neurol 57(5):730–737. https://doi.org/10.1002/ana.20478
Melo US, Macedo-Souza LI, Figueiredo T, Muotri AR, Gleeson JG, Coux G, Armas P, Calcaterra NB, Kitajima JP, Amorim S, Olávio TR, Griesi-Oliveira K, Coatti GC, Rocha CRR, Martins-Pinheiro M, Menck CFM, Zaki MS, Kok F, Zatz M, Santos S (2015) Overexpression of KLC2 due to a homozygous deletion in the non-coding region causes SPOAN syndrome. Hum Mol Genet 24(24):6877–6885. https://doi.org/10.1093/hmg/ddv388
Yıldırım Y, Orhan EK, Iseri SAU, Serdaroglu-Oflazer P, Kara B, Solakoğlu S, Tolun A (2011) A frameshift mutation of ERLIN2 in recessive intellectual disability, motor dysfunction and multiple joint contractures. Hum Mol Genet 20(10):1886–1892. https://doi.org/10.1093/hmg/ddr070
Alazami AM, Adly N, Al Dhalaan H, Alkuraya FS (2011) A nullimorphic ERLIN2 mutation defines a complicated hereditary spastic paraplegia locus (SPG18). Neurogenetics 12(4):333–336. https://doi.org/10.1007/s10048-011-0291-8
Al-Saif A, Bohlega S, Al-Mohanna F (2012) Loss of ERLIN2 function leads to juvenile primary lateral sclerosis. Ann Neurol 72(4):510–516. https://doi.org/10.1002/ana.23641
Wakil SM, Bohlega S, Hagos S, Baz B, Al Dossari H, Ramzan K, Al-Hassnan ZN (2013) A novel splice site mutation in ERLIN2 causes hereditary spastic paraplegia in a Saudi family. Eur J Med Genet 56(1):43–45. https://doi.org/10.1016/j.ejmg.2012.10.003
Tian W-T, Shen J-Y, Liu X-L, Wang T, Luan X-H, Zhou H-Y, Chen S-D, Huang X-J, Cao L (2016) Novel mutations in endoplasmic reticulum lipid raft-associated protein 2 gene cause pure hereditary spastic paraplegia type 18. Chin Med J 129(22):2759–2761. https://doi.org/10.4103/0366-6999.193444
Amador, M.-D.-M., Muratet, F., Teyssou, E., Banneau, G., Danel-Brunaud, V., Allart, E., Antoine, J.-C., Camdessanché, J.-P., Anheim, M., Rudolf, G., Tranchant, C., Fleury, M.-C., Bernard, E., Stevanin, G., & Millecamps, S. (2019). Spastic paraplegia due to recessive or dominant mutations in ERLIN2 can convert to ALS. Neurology: Genetics, 5(6), e374. https://doi.org/10.1212/NXG.0000000000000374
Srivastava S, D’Amore A, Cohen JS, Swanson LC, Ricca I, Pini A, Fatemi A, Ebrahimi-Fakhari D, Santorelli FM (2020) Expansion of the genetic landscape of ERLIN2-related disorders. Ann Clin Transl Neurol 7(4):573–578. https://doi.org/10.1002/acn3.51007
Sadr Z, Rohani M, Jamali P, Alavi A (2023) A case report of concurrent occurrence of two inherited axonopathies within a family: the benefit of whole-exome sequencing. Int J Neurosci 1–6. https://doi.org/10.1080/00207454.2023.2260091
Rydning SL, Dudesek A, Rimmele F, Funke C, Krüger S, Biskup S, Vigeland MD, Hjorthaug HS, Sejersted Y, Tallaksen C, Selmer KK, Kamm C (2018) A novel heterozygous variant in ERLIN2 causes autosomal dominant pure hereditary spastic paraplegia. Eur J Neurol 25(7):943-e71. https://doi.org/10.1111/ene.13625
Park J-M, Lee B, Kim J-H, Park S-Y, Yu J, Kim U-K, Park J-S (2020) An autosomal dominant ERLIN2 mutation leads to a pure HSP phenotype distinct from the autosomal recessive ERLIN2 mutations (SPG18). Sci Rep 10(1):1. https://doi.org/10.1038/s41598-020-60374-y
Chen S, Zou J-L, He S, Li W, Zhang J-W, Li S-J (2021) More autosomal dominant SPG18 cases than recessive? The first AD-SPG18 pedigree in Chinese and literature review. Brain Behav 11(12):e32395. https://doi.org/10.1002/brb3.2395
Gnom AD (n.d.) Retrieved December 23, 2023, from https://gnomad.broadinstitute.org
Figueiredo T, Melo US, Pessoa ALS, Nobrega PR, Kitajima JP, Correa I, Zatz M, Kok F, Santos S (2015) Homozygous missense mutation in MED25 segregates with syndromic intellectual disability in a large consanguineous family. J Med Genet 52(2):123–127. https://doi.org/10.1136/jmedgenet-2014-102793
Figueiredo T, Melo US, Pessoa ALS, Nobrega PR, Kitajima JP, Rusch H, Vaz F, Lucato LT, Zatz M, Kok F, Santos S (2016) A homozygous loss-of-function mutation in inositol monophosphatase 1 (IMPA1) causes severe intellectual disability. Mol Psychiatry 21(8):1125–1129. https://doi.org/10.1038/mp.2015.150
Meyyazhagan A, Orlacchio A (2022) Hereditary spastic paraplegia: an update. Int J Mol Sci 23(3):1697. https://doi.org/10.3390/ijms23031697
Acknowledgements
The authors wish to thank the patients and their family members for participation in this study.
Funding
This research was funded by the Research Program for the Unified Health System (PPSUS): shared management in health (Process Number: 60030.0000000215/2021).
Author information
Authors and Affiliations
Contributions
Conceptualization, T.F.; methodology, T.F. and F.K..; investigation, T.F., F.K., G.S., M.M., M.S., M.F., J.A. and D.R.; writing—original draft preparation, T.F., G.S., M.M. and M.S..; writing—review and editing, all authors; supervision, T.F.; project administration, T.F.; funding acquisition, T.F. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Fernando Kok is a shareholder and Medical Director of Mendelics Genomic Analysis and Associated Editor for Neurogenetics of Arquivos de Neuropsiquiatria.
Ethics approval and consent to participate
The data sampling protocol, as well as the consent procedure, were analyzed and approved by the National Research Ethics Committee—CONEP (Process 39674220.1.1001.5013).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
de Souza, G.C., Malta, M.C., Santos, M.R.S. et al. Novel ERLIN2 variant expands the phenotype of Spastic Paraplegia 18. Neurol Sci 45, 2705–2710 (2024). https://doi.org/10.1007/s10072-023-07271-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-023-07271-0