
Abstract
Background
Hereditary spastic paraplegia (HSP) represents a group of monogenic neurodegenerative disorders characterized by high clinical and genetic heterogeneity. HSP is characterized by slowly progressing hypertonia of both lower extremities, spastic gait, and myasthenia. The most prevalent autosomal dominant form of HSP, known as spastic paraplegia 4 (SPG4), is attributed to variants in the spastin (SPAST) gene.
Methods and results
Here, a Chinese family presenting with spasticity in both legs and a shuffling gait participated in our investigation. Whole exome sequencing of the proband was utilized to identify the genetic lesion in the family. Through data filtering, Sanger sequencing validation, and co-separation analysis, a novel variant (NM_014946.3: c.1669G > C:p.A557P) of SPAST was identified as the genetic lesion of this family. Furthermore, bioinformatic analysis revealed that this variant was deleterious and located in a highly evolutionarily conserved site.
Conclusion
Our study confirmed the diagnosis of SPG4 in this family, contributing to genetic counseling for families affected by SPG4. Additionally, our study broadened the spectrum of SPAST variants and highlighted the importance of ATPases associated with various cellular activity domains of SPAST.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hereditary spastic paraplegia (HSP) represents a group of hereditary, degenerative, and neurological disorders primarily confined to lower limb weakness, involuntary spasms, and muscle stiffness [1, 2]. Approximately 10% of patients have complicated HSP, which presents with peripheral neuropathy, epilepsy, ataxia, optic neuropathy, hearing loss, and learning and developmental problems [1, 3]. The estimated prevalence of HSP is likely to be 0.1–9.6 per 100,000 individuals reported globally [4]. Current opinions suggest that genetic factors play crucial roles in both pure HSP and complicated HSP. To date, more than 70 related pathogenic genes of HSP have been identified with inheritance patterns divided into three types: autosomal dominant, autosomal recessive and X-linked recessive [1, 5]. In addition, HSP presented with mitochondrial inheritance caused by MT-ATP6 mutation has also been reported in some rare cases [6]. In autosomal dominant HSP, spastic paraplegia 4 (SPG4) is the most common subtype accounting for more than 40% of all autosomal dominant HSP cases resulting from variants in the spastin (SPAST) gene [7, 8]. According to the Human Gene Mutation Database, approximately 1,000 variants including missense mutations, nonsense mutations, frameshift mutations, splice site mutations and complex rearrangements of SPAST have been detected in HSP patients.
The SPAST gene is located on 2p22.3 and encodes a member of the ATPases associated with diverse cellular activity protein families. This family encompasses an ATPase domain and assumes a crucial role in multiple cellular processes, such as organelle biogenesis, membrane trafficking, and intracellular motility, along with protein folding and proteolysis [9, 10]. One isoform of the SPAST gene has been demonstrated to be a microtubule-severing enzyme that regulates the abundance, mobility, and plus-end distribution of microtubules in neurons, which is indispensable for axonal growth and formation [11, 12]. Additionally, SPAST can interact with another HSP pathogenic gene encoding Reticulon-2 [13].
Here, we investigated a Chinese family with spasticity in both legs with a shuffling gait. The objective of this study was to investigate genetic abnormalities in the family via whole exome sequencing and Sanger sequencing. Additionally, bioinformatic analysis was employed to assess the pathogenicity of potential identified variants.
Materials and methods
Subjects
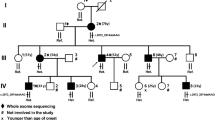
A total of 11 members of the family, with four affected individuals, were investigated in this study (Fig. 1A). Peripheral blood samples were collected from four family members including two affected individuals (II-5 and III-3). The study was approved by the Ethics Committee of the Affiliated Hospital of Yangzhou University. Written informed consent was obtained from all participants.

The clinical data of the family. (A) The family pedigree figure. Squares indicate male members; circles, female members; closed symbols, the affected members; open symbols, unaffected members; arrow, proband. (B) The data filtering strategy of whole exome in this study. (C) The medical history of the proband
Variant detection
Genomic DNA was isolated from the peripheral blood lymphocytes of the participants via a blood DNA kit (D3392-01, OMEGA) according to the manufacturer’s instructions. Whole exome sequencing was employed to analyze the genetic lesion of the proband (II-5). The key experimental procedures, including targeted capture, massive parallel sequencing, mapping, and variant detection, were performed by BerryGenomics Biotech Company (Beijing, China) [14,15,16]. The data filtering strategies are illustrated in Fig. 1B. Variant validation and cosegregation analysis were conducted via polymerase chain reaction with designed primers (forward: 5’-ACTACTTTGGGAGGCTGTGG-3’, reverse: 5’-TGGTTTTAAGGCTGGGCA-3’) and Sanger sequencing on an ABI 3100 Genetic Analyzer (ABI, USA).
Bioinformatic analysis
The structure of the SPAST protein was modeled using Swiss-Model software, and local hydrophobicity was analyzed by ProtScale based on this structure. Conservation analysis was carried out through comparison of amino acid sequences across different species.
Results
Clinical description
The proband (II-5), which came from Jangsu Province, China, was admitted to the Department of Neurology, Affiliated Hospital of Yangzhou University Hospital due to spasticity in both legs with a shuffling gait without obvious cause for six years. The neurological examination revealed that the patient presented with scissor gait and hypertonia in both lower limbs, that knee reflexes and ankle reflexes were hyperactive, and that Babinski’s sign was positive in both lower limbs. The patient had no dysarthria, normal muscle strength in both upper limbs, normal deep and superficial sensation, no abnormalities in coordination tests, and normal bladder and bowel function. The patient had a Spastic Paraplegia Rating Scale (SPRS) score of 22. The nerve electrophysiological examination revealed that the amplitudes of the motor potential of both tibial nerve and the peroneal nerve were decreased in the patient. The results of routine laboratory tests and cranial and spinal cord nuclear magnetic resonance imaging did not reveal any abnormal findings. A medical history investigation suggested that the patient had an abnormal gait, but that gait did not affect walking or running at age 8. Running falls occurred around the age of 30. Three years later, the patient experienced stiffness in both lower limbs, and the symptoms were aggravated when the weather was cold or emotional. Approximately five or six years ago, the patient developed spasticity in both lower limbs with a shuffling gait (Fig. 1C). A family history survey indicated that the proband’s mother (I-1) was unable to walk around the age of 60 years and relied on a wheelchair. An aunt (II-1) of the patient developed an abnormal gait at approximately 45 years of age and began to walk with support tools in the past 3 years. The proband’s 22-year-old son (II-3) has been running and falling.
Genetic analysis
After aligning and calling single nucleotide variants, a total of 84,003 variants were detected in the proband. Following the data filtering strategies (Fig. 1B), Sanger sequencing, and coseparation analysis, only 7 variants remained (Table 1). Among these 7 variants, the novel variant (NM_014946.3: c.1669G > C:p.A557P) of SPAST is the most likely genetic lesion for the family with pure HSP (Fig. 2A). Conservation analysis indicated that this novel variant, resulting in the substitution of alanine with proline, was in an evolutionarily conserved site (Fig. 2B). Structural analysis revealed that the p.A524P variant disrupted chemical bonds such as hydrogen bonds between amino acids at site 577 and neighboring amino acids, thereby affecting the structure of the SPAST protein (Fig. 2C). According to the American College of Medical Genetics and Genomics (ACMG) guidelines [15], this novel variant is likely pathogenic because of the following criteria: PM1 + PM2 + PM5 + PP1 + PP3.

The genetic data of the family. (A) Sanger sequencing of SPAST confirmed the c.1669G > C:p.A557P novel variant. (B) Alignment of multiple SPAST protein sequences across species. The affected A577 amino acid is located in a highly conserved region in different mammals (from Ensembl). The red words represent the p.A577 site. (C) Structure prediction of the mutant protein. The wild-type (p.A577) protein structure and the mutant SPAST (p.P577) protein structure are predicted using SWISS-MODEL online software
Discussion
The spastin protein is composed of four domains: (1) a hydrophobic domain; (2) a microtubule-interacting and trafficking domain, which is essential for cytokinesis and endosomal tubule recycling; (3) a microtubule-binding domain, which is responsible for binding spastin to microtubules; and (4) ATPases associated with diverse cellular activities (AAAs), which are responsible for spastin hexamerization and microtubule-severing activity [12, 17, 18]. In our case, the p.A557P variant of SPAST is located in the AAA domain, potentially disrupting its role in microtubule severing activity. This disruption may lead to abnormal structure and function of the microtubules in neurons, affecting neuron development and ultimately causing HSP disease by impacting axonal transport.
It has been reported that the AAA domain shares homology across diverse species and that alterations in the AAA domain of SPAST are imperative causes of SPG4 [11, 19]. The AAA domain contains 10 exons from exon 7 to exon 17, and most pathogenic variants of SPAST occur in the AAA domain, indicating its functional importance [20, 21]. Here, the p.A557P variant was also located in the AAA domain, which may further result in defects in microtubule severing and ATPase activity, and ultimately lead to SPG4 [17]. Simultaneously, we also found that the p.A557V and p.A557G variants of SPAST, which presented the same mutation site as our study, have been recorded in the ClinVar database. The p.A557G variant was identified in a patient with spastic paraparesis, but the condition of the p.A557V carrier was not described. Both the p.A557V and p.A557G variants are likely pathogenic. Our study findings align with records in the ClinVar database, which collectively indicate that the AAA domain serves as a mutational hotspot for SPAST.
Usually, variants in the SPAST gene may result in pure HSP, however, an increasing number of patients with SPG4 have been found to exhibit nonmotor symptoms such as pain, fatigue, and depression as the disease progresses [20, 22]. SPG4 demonstrates significant heterogeneity and can present at any age, with the most common onset typically occurring at approximately 10 years of age or between 30 and 50 years of age [1]. Concurrently, a growing number of reports indicate that the age of onset of SPG4 is progressively advancing in successive generations in families, which is called genetic anticipation [23]. In addition, some studies have reported that patients with pure HSP may experience symptoms such as depression, pain, and restless legs [22, 24]. The mental disorders including depression and emotional issues may result from the fact that patients with severe motor impairments find it difficult to engage in normal social activities. These mental disorders may manifest as severe somatic symptoms and could exacerbate weakness in the limbs, and some relief of symptoms can be achieved in these patients using antidepressants [24]. In our study, the proband (II-5) also presented emotional problems, which can sometimes aggravate symptoms, and the patient’s son (III-3) presented running falls much earlier than the proband did. Our study suggested that mental disorder existed in HSP patients and genetic anticipation also existed in our family.
HSP typically presents with a protracted disease course and slow progression. The patient in this study exhibited this pattern, as the onset of his disease occurred approximately 30 years of age and gradually progressed [1, 23]. Owing to its slow progression, individuals often do not seek medical attention at the outset of the disease, as was the case with our patient who was not diagnosed until the age of 46. The clinical manifestations of HSP are diverse, posing challenges for diagnosis. For example, some reports suggested that the pure HSP patients can also exhibit abnormal electromyography findings [25, 26], which was consistent with our patient. Consequently, genetic testing is essential. When symptoms such as difficulty walking, unsteady gait, and bilateral lower-extremity spasticity manifest, suspicion for HSP should arise and gene sequencing should be pursued. Therapeutic options for HSP are limited, increasing the importance of gene therapy in its treatment [27]. The novel variant we discovered expands the spectrum of pathogenic variants associated with HSP, providing valuable information for genetic diagnosis and offering a new strategy for gene therapy.
Conclusion
In conclusion, we identified a novel variant (NM_014946.3: c.1669G > C/p.A557P) of SPAST in a Chinese family with pure hereditary spastic paraplegia (HSP) through whole exome sequencing. According to the ACMG guidelines, this variant is classified as likely pathogenic (PM1 + PM2 + PM5 + PP1 + PP3). Our study not only broadens the spectrum of SPAST variants and highlights the significance of the AAA domain of SPAST but also confirms the diagnosis within the family, contributing to genetic counseling for families affected by SPG4.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- HSP:
-
Hereditary spastic paraplegia
- AD:
-
Autosomal dominant
- SPG4:
-
Spastic paraplegia 4
- SPAST:
-
Spastin
- ACMG:
-
American College of Medical Genetics and Genomics
- AAA:
-
ATPases associated with a variety of cellular activities
References
Mereaux JL, Banneau G, Papin M, Coarelli G, Valter R, Raymond L, Kol B, Ariste O et al (2022) Clinical and genetic spectra of 1550 index patients with hereditary spastic paraplegia. Brain 145(3):1029–1037. https://doi.org/10.1093/brain/awab386
Awuah WA, Tan JK, Shkodina AD, Ferreira T, Adebusoye FT, Mazzoleni A, Wellington J, David L et al (2024) Hereditary spastic paraplegia: novel insights into the pathogenesis and management. SAGE Open Med 12:20503121231221941. https://doi.org/10.1177/20503121231221941
Freua F, Almeida MEC, Nobrega PR, Paiva ARB, Della-Ripa B, Cunha P, Macedo-Souza LI, Bueno C et al (2022) Arginase 1 deficiency presenting as complicated hereditary spastic paraplegia. Cold Spring Harb Mol Case Stud 8(6). p. a006232
Ortega Suero G, Abenza Abildua MJ, Serrano Munuera C, Rouco Axpe I, Arpa Gutierrez FJ, Adarmes Gomez AD, Rodriguez FJ, de Rivera B, Quintans, Castro et al (2023) Epidemiology of ataxia and hereditary spastic paraplegia in Spain: a cross-sectional study. Neurologia (Engl Ed) 38(6):379–386. https://doi.org/10.1016/j.nrleng.2023.04.003
Shribman S, Reid E, Crosby AH, Houlden H, Warner TT (2019) Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol 18(12):1136–1146. https://doi.org/10.1016/S1474-4422(19)30235-2
Verny C, Guegen N, Desquiret V, Chevrollier A, Prundean A, Dubas F, Cassereau J, Ferre M et al (2011) Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion 11(1):70–75. https://doi.org/10.1016/j.mito.2010.07.006
Varghaei P, Estiar MA, Ashtiani S, Veyron S, Mufti K, Leveille E, Yu E, Spiegelman D et al (2022) Genetic, structural and clinical analysis of spastic paraplegia 4. Parkinsonism Relat Disord 98:62–69. https://doi.org/10.1016/j.parkreldis.2022.03.019
Jacinto-Scudeiro LA, Rothe-Neves R, Dos Santos VB, Machado GD, Burguez D, Padovani MMP, Ayres A, Rech RS et al (2023) Dysarthria in hereditary spastic paraplegia type 4. Clin (Sao Paulo) 78:100128. https://doi.org/10.1016/j.clinsp.2022.100128
Solowska JM, Baas PW (2015) Hereditary spastic paraplegia SPG4: what is known and not known about the disease. Brain 138(9):2471–2484. https://doi.org/10.1093/brain/awv178
Elert-Dobkowska E, Stepniak I, Radziwonik-Fraczyk W, Jahic A, Beetz C, Sulek A (2024) SPAST Intragenic CNVs lead to Hereditary Spastic Paraplegia via a haploinsufficiency mechanism. Int J Mol Sci 25(9):5008. https://doi.org/10.3390/ijms25095008
Salinas S, Carazo-Salas RE, Proukakis C, Schiavo G, Warner TT (2007) Spastin and microtubules: functions in health and disease. J Neurosci Res 85(12):2778–2782. https://doi.org/10.1002/jnr.21238
Sakoe K, Shioda N, Matsuura T (2021) A newly identified NES sequence present in spastin regulates its subcellular localization and microtubule severing activity. Biochim Biophys Acta Mol Cell Res 1868(1):118862. https://doi.org/10.1016/j.bbamcr.2020.118862
Montenegro G, Rebelo AP, Connell J, Allison R, Babalini C, D’Aloia M, Montieri P, Schule R et al (2012) Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J Clin Invest 122(2):538–544. https://doi.org/10.1172/JCI60560
Liu L, Luo H, Sheng Y, Kang X, Peng H, Luo H, Fan LL (2023) A novel mutation of CTC1 leads to telomere shortening in a Chinese family with interstitial lung disease. Hereditas 160(1):37. https://doi.org/10.1186/s41065-023-00299-4
Liu YX, Hu YQ, Zhang SY, Guo YD, Chen YQ, Fan LL, Jin JY, Xiang R (2024) Identification of Arrhythmia-Associated Gene mutations in Chinese patients with primary Electrical disorders or Sudden Cardiac Death. Cardiovasc Innovations Appl 9(1):pe966 Artn 3410.15212/Cvia.2024.0018
Wang C, Xiao J, Su Y, Fan LL, Zhu L, Xiang R (2024) Identification of a Novel DSP variant in a patient with Sudden Cardiac Death through Post-mortem Genetic Investigation. Cardiovasc Innovations Appl 9(1):e937. https://doi.org/10.15212/CVIA.2024.0043
Lim JH, Kang HM, Jung HR, Kim DS, Noh KH, Chang TK, Kim BJ, Sung DH et al (2018) Missense mutation of SPAST protein (I344K) results in loss of ATPase activity and prolonged the half-life, implicated in autosomal dominant hereditary spastic paraplegia. Biochim Biophys Acta Mol Basis Dis 1864(10):3221–3233. https://doi.org/10.1016/j.bbadis.2018.07.009
Taylor JL, White SR, Lauring B, Kull FJ (2012) Crystal structure of the human spastin AAA domain. J Struct Biol 179(2):133–137. https://doi.org/10.1016/j.jsb.2012.03.002
Yao L, Cao Y, Zhang C, Huang X, Tian W, Cao L (2024) Clinical and genetic characteristics in a Chinese cohort of complex spastic paraplegia type 4. Clin Genet 106(1):56–65. https://doi.org/10.1111/cge.14510
Verriello L, Lonigro IR, Pessa ME, Betto E, Pauletto G, Fogolari F, Gigli GL, Curcio F (2021) Amplifying the spectrum of SPAST gene mutations. Acta Biomed 92(S1):e2021220. https://doi.org/10.23750/abm.v92iS1.11608
Chen X, Li X, Tan Y, Yang D, Lu L, Deng Y, Xu R (2023) Identification of c.1495C > T mutation in SPAST gene in a family of Han Chinese with hereditary spastic paraplegia. Neurosci Lett 812:137399. https://doi.org/10.1016/j.neulet.2023.137399
Rattay TW, Boldt A, Volker M, Wiethoff S, Hengel H, Schule R, Schols L (2020) Non-motor symptoms are relevant and possibly treatable in hereditary spastic paraplegia type 4 (SPG4). J Neurol 267(2):369–379. https://doi.org/10.1007/s00415-019-09573-w
Hashemi SS, Hajati R, Davarzani A, Rohani M, DanaeeFard F, Rahimi Bidgoli MM, Fatehi F, Kariminejad A et al (2022) Anticipation can be more common in Hereditary Spastic Paraplegia with SPAST mutations than it appears. Can J Neurol Sci 49(5):651–661. https://doi.org/10.1017/cjn.2021.188
Vahter L, Braschinsky M, Haldre S, Gross-Paju K (2009) The prevalence of depression in hereditary spastic paraplegia. Clin Rehabil 23(9):857–861. https://doi.org/10.1177/0269215509337186
Narendiran S, Debnath M, Shivaram S, Kannan R, Sharma S, Christopher R, Seshagiri DV, Jain S et al (2022) Novel insights into the genetic profile of hereditary spastic paraplegia in India. J Neurogenet 36(1):21–31. https://doi.org/10.1080/01677063.2022.2064463
Chen J, Zhao Z, Shen H, Bing Q, Li N, Guo X, Hu J (2022) Genetic origin of patients having spastic paraplegia with or without other neurologic manifestations. BMC Neurol 22(1):180. https://doi.org/10.1186/s12883-022-02708-z
Mohan N, Qiang L, Morfini G, Baas PW (2021) Therapeutic strategies for mutant SPAST-Based Hereditary Spastic Paraplegia. Brain Sci 11(8):1081. https://doi.org/10.3390/brainsci11081081
Acknowledgements
We are very grateful to the family members for their participation in this study.
Funding
This study was supported by the National Natural Science Foundation of China (82201394 and 82470297), Hunan Province Natural Science Foundation (2023JJ20078) and Innovation and entrepreneurship training program for college students (20240031020034).
Author information
Authors and Affiliations
Contributions
Yu-Han Jin and Yang-Ziyu Xiang wrote the draft of the manuscript. Yu-Han Jin and Yang-Ziyu Xiang performed the genetic analysis and Sanger sequencing. Mei-Fang Zhao conducted the bioinformatic analysis. Yi-Hui Liu and Xiao-Cong Li enrolled the samples. Xiao-Cong Li and Liang-Liang Fan designed the project, revised the draft and supported the study. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval
The study was approved by the Ethics Committee of the affiliated Hospital of Yangzhou University (No. 2022-YKL02-G008) and was performed according to the principles of the Declaration of Helsinki. Written informed consent was obtained from all participants.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jin, YH., Xiang, YZ., Zhao, MF. et al. A novel variant (p.A524P) in Spastin is responsible for a Chinese family with hereditary spastic paraplegia. Mol Biol Rep 51, 951 (2024). https://doi.org/10.1007/s11033-024-09880-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09880-0