
Abstract
This narrative review provides a comprehensive examination of the complex interplay between inflammatory arthritis (IA) and cardiovascular pathology. It particularly illuminates the roles of atherosclerosis initiation, endothelial dysfunction, and glycocalyx shedding. IA not only provokes tissue-specific inflammatory responses, but also engenders a considerable degree of non-specific systemic inflammation. This review underscores the accelerating influence of the chronic inflammatory milieu of IA on cardiovascular disease (CVD) progression. A focal point of our exploration is the critical function of the endothelial glycocalyx (EG) in this acceleration process, which possibly characterizes the earliest phases of atherosclerosis. We delve into the influence of inflammatory mediators on microtubule dynamics, EG modulation, immune cell migration and activation, and lipid dysregulation. We also illuminate the impact of microparticles and microRNA on endothelial function. Further, we elucidate the role of systemic inflammation and sheddases in EG degradation, the repercussions of complement activation, and the essential role of syndecans in preserving EG integrity. Our review provides insight into the complex and dynamic interface between systemic circulation and the endothelium.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chronic inflammation is a critical factor in the pathogenesis of cardiovascular disease (CVD), influencing each aspect from atherosclerosis to arrhythmias and heart failure [1,2,3]. It is well established that inflammatory arthritis (IA) such as rheumatoid arthritis (RA), ankylosing spondylitis (AS), and psoriatic arthritis (PsA), among other conditions on the spondyloarthritis (SpA) spectrum, are systemic diseases associated with an augmented cardiovascular risk [4,5,6,7,8,9]. This suggests a shared pathway for the instigation and progression of endothelial dysfunction, culminating in CVD.
IA not only causes significant joint damage and disability but also leads to systemic inflammation, which has been linked to an increased risk of cardiovascular disease CVD. CVD, a leading cause of mortality globally, presents a significant health burden, with its impact further amplified in individuals with IA. Despite the well-established association between IA and CVD, the underlying mechanisms driving this relationship remain poorly understood. A key area of uncertainty lies in the complex interplay between IA and the initiation of atherosclerosis, endothelial dysfunction, and glycocalyx shedding. This gap in understanding presents a significant problem. Without a clear grasp of these mechanisms, clinicians are limited in their ability to accurately predict which IA patients are at the highest risk of developing CVD. Furthermore, opportunities for targeted therapeutic interventions to mitigate this risk may be missed. Our review addresses this problem by providing a comprehensive examination of the molecular processes underpinning the relationship between IA and CVD. Risk algorithms adjusted for conventional CVD risk factors indicate that inflammation, rather than the specific nosological entity, could be the primary culprit for the heightened CVD burden [5, 7]. IA incites tissue-specific inflammatory responses but also considerable non-specific systemic inflammation. The presence of distinct underlying mechanisms and fluctuating inflammatory load in specific IA can manifest in diverse pathognomic presentations and variable CVD occurrence. This diversity makes it challenging to identify a single causative factor.
The milieu of chronic inflammation lays the foundation for CVD development and progression. This systemic inflammatory environment contributes to a variety of pathophysiological alterations that drive atherosclerosis, including endothelial dysfunction, oxidative stress, and the formation of pro-thrombotic states [10, 11]. The interplay of these factors is complex and dynamic, reflecting the multifactorial nature of CVD and the significant role of inflammation as a common denominator. The significant overlap of pathophysiological processes and the high incidence of CVD morbidity in patients with IA reinforces the emerging paradigm that views atherosclerosis as an autoimmune disease [12, 13].
The induction of atherogenesis is instigated by a chronic inflammatory state, perpetuated by immunocompetent cells, humoral factors, and a myriad of molecular mediators [13, 14]. Such inflammation can have a systemic impact as well as site-specific ramifications, causing alterations in vessel wall functionality, thereby leading to endothelial dysfunction — a critical precursor in the progression towards atherosclerosis. The compromised endothelium, as a result of the aforementioned inflammatory assault, transforms into a site conducive for lipid accumulation and retention, marking the onset of the atherosclerotic process [10, 15]. Such vascular wall disruption can be precipitated by an array of potential triggers, encompassing chemical, physical, and infectious agents; immune responses; ischemia; genetic aberrations; or nutritional insufficiencies. Each of these variables presents a different piece of the complex puzzle of atherosclerosis pathogenesis, highlighting the need for comprehensive and multifaceted strategies in its prevention and management [16,17,18].
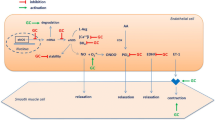
The vascular endothelial layer, particularly its pericellular matrix — the glycocalyx, seems to be the primary site for the initial molecular reactions that contribute to cardiovascular damage [19]. The endothelial glycocalyx (EG) is composed of complex proteoglycan polymers, which can be rapidly shed under the influence of factors present in chronic inflammatory conditions (Fig. 1). This shedding may be the initiating event, with upregulated repair processes potentially playing a key role in lipoprotein deposition and immune cell recruitment. By-products of EG turnover are detectable in the serum of patients with IA and may serve as markers for disease activity [20, 21]. Accelerated EG degradation and subsequent endothelial dysfunction create a pathological feedback loop through impaired nitric oxide (NO) synthesis, mechanotransduction, and activation of sheddases, further facilitating vascular damage [22]. Aberrant EG degradation in critical illness like sepsis, polytrauma, and chronic kidney disease is associated with altered metabolic profile, increased inflammation, coagulation, and mortality [23].

Depiction of endothelial cells under normal homeostatic conditions with intact glycocalyx. The glycocalyx, represented by the dense, brush-like layer on the surface of the endothelial cells, plays a crucial role in vascular health by providing a protective barrier against harmful substances, regulating vascular permeability, and mediating cell–cell interactions. In this state, the cellular interactions with the endothelium are restricted, preventing unnecessary activation of inflammatory pathways and maintaining vascular integrity. VL, vessel lumen; EG, endothelial glycocalyx; EC, endothelial cells
This narrative review aims to summarize the current knowledge on chronic systemic inflammation’s role in vascular damage, particularly in relation to the EG, a dynamic interface between systemic blood flow and the endothelium. We focus on its degradation and subsequent dysfunction, leading to early-stage atherosclerosis and promoting CVD progression. While our primary focus is on chronic inflammatory conditions such as IA, the pathways and processes discussed are relevant to similar inflammatory disorders. By examining the inflammatory milieu in IA, we aim to elucidate the damage to the EG, explore atherosclerosis initiation and progression, and understand how these processes accelerate CVD development. Our central interest lies in investigating the potentially deleterious effects of chronic inflammatory immune responses on vascular structures, with emphasis on the endothelial glycocalyx.
Search methodology
To understand the full scope of cardiovascular damage by the chronic inflammatory milieu, we performed a comprehensive literature review as of June 2023. We utilized the strategy from the previously published recommendations for writing a narrative review [24]. MEDLINE database via Pubmed and Scopus were queried for studies reporting potential mechanisms and biomarkers for subclinical atherosclerosis, endothelial dysfunction, and glycocalyx shedding in inflammatory arthritis. The search encompassed articles in English language from inception up to June 30, 2023, for MEDLINE, and July 1, 2023, for Scopus.
The initial query was set to include terms such as “endothelial dysfunction,” “glycocalyx,” “microparticles,” “cholesterol,” “atherosclerosis,” “microtubules,” “cell,” “adhesion molecule,” “sheddase,” and “chemotaxis.” To incorporate the context of inflammatory arthritis, the terms “inflammatory joint disease,” “inflammatory arthritis,” “rheumatoid arthritis,” “spondyloarthritis,” “ankylosing spondylitis,” “psoriatic arthritis,” and “reactive arthritis” were connected using the “OR” operator. These terms were then combined with the cardiovascular terms using the “AND” operator to narrow down the search results and identify articles that discussed both cardiovascular health and inflammatory arthritis. References of retrieved studies and relevant reviews were hand-searched for a further supplement. The final selection of studies for the present narrative review was based on authors’ professional expertise, experience, existing theories, and models, creating an integrated data interpretation [24].
Cardiovascular disease burden
Patients with IA have increased vascular calcium deposition and atherosclerotic burden [25, 26]. RA with its high levels of chronic inflammation has been extensively studied to elucidate the potential mechanisms of accelerated CVD [27]. Impaired endothelial function and subclinical atherosclerosis correlated with increased epicardial adipose tissue thickness, intima-media thickness (IMT), and impaired flow-mediated dilation in SpA patients [28]. Prevalence of atherosclerotic burden and IMT is observed even in RA patients with inactive disease [29], although disease severity, rheumatoid factor isotypes, and anti-citrullinated protein antibodies are associated with increased incidence of CVD [30].
To elucidate the preceding events of the classical stages of atherosclerosis, it is essential to understand the initiator of lipid deposition and subsequent retention, which remains elusive. Retained lipoprotein modification incites the recruitment of immune cells, the release of chemoattractants, and the upregulation of adhesion molecules [31] Utilization of advanced imaging like positron emission tomography–computed tomography (PET-CT), coronary CT angiography (CCTA), and serum-based assays have been used identify subclinical coronary disease in PsA. Several potential contributors have been identified such as elevated IL-6 and increased uptake of fluorodeoxyglucose (FDG) in the liver, spleen, bone marrow, and fat. The findings reveal that PsA is characterized by metabolic dysregulation, systemic inflammation, and subclinical coronary artery disease compared to age-sex-matched volunteers. The severity of these conditions is found to be worse in subjects with moderate-severe skin disease, suggesting that the combination of severe skin inflammation and joint disease may be particularly atherogenic [32].
Individuals with arthritis are found to have a heightened risk of CVD, independently of obesity status, whereas anti-rheumatic drugs have a positive impact on reducing the risk [33]. This suggests that the onset of arthritis symptoms could serve as a critical point for healthcare providers to screen for latent CVD risk factors. By concurrently managing both arthritis and CVD risk factors, there is a potential to enhance the prognosis for both conditions.
Glycocalyx shedding: causes and consequences
Under pathological conditions such as the systemic chronic milieu in IA, the EG can undergo degradation, a process known as shedding. This leads to a loss of homeostatic protective functions and contributes to vascular dysfunction (Fig. 2).

Detailed depiction of the endothelial cell membrane with an attached glycocalyx under different conditions. Panel A: Homeostatic condition. The endothelial cell membrane is shown with an intact glycocalyx. The expression of adhesion molecules such as PECAM-1, ICAM-1, and VCAM-1 is minimal, reflecting the low level of cellular interactions in this state. Panel B: milieu present in inflammatory arthritis. The endothelial glycocalyx is shown under the influence of inflammatory mediators such as C-reactive protein and tumor necrosis factor, matrix metalloproteinases, and sheddases. These factors contribute to the degradation of the glycocalyx, leading to an increase in the expression of adhesion molecules. The damaged glycocalyx is represented as a sparse layer, indicating the loss of its protective function and the increased vulnerability of the endothelium. VL, vessel lumen; EG, endothelial glycocalyx; EC, endothelial cells; MT, microtubules; TJ, tight junction; PM, phospholipid endothelial cell membrane; MP, microparticles; MI, mediators of inflammation
Sheddases: key enzymes in EG shedding
EG shedding in IA is a complex process involving various enzymes known as “sheddases” [34]. Sheddases, a group of enzymes that facilitate the shedding of cell surface proteins, play a crucial role in the regulation of the endothelial EG. These enzymes, which include matrix metalloproteinases (MMPs), A disintegrin and metalloproteinase (ADAM) family, heparanase, and hyaluronidase, are responsible for the cleavage of core proteins and the degradation of the EG [35, 36]. The activity of sheddases is often upregulated in inflammatory conditions, leading to increased EG shedding. For instance, heparanase and hyaluronidase, key sheddases, are known to be upregulated in IA and contribute to EG degradation [37].
ADAMs and MMPs are proteolytic enzymes capable of cleaving and degrading the macromolecules comprising the glycocalyx. ADAMs, particularly ADAM17, have been implicated in EG shedding through their ability to cleave EG components such as syndecans and hyaluronan synthase 2 (HAS2), leading to the disruption of the EG structure. ADAM17, also known as tumor necrosis factor-alpha converting enzyme (TACE), is a key member of the ADAM family involved in the shedding of cell surface proteins and glycoproteins [38]. Its activation and subsequent proteolytic activity contribute to the shedding of EG components, compromising the integrity and function of the EG. MMPs, such as MMP-2 and MMP-9, have been shown to degrade EG components and contribute to glycocalyx shedding in various pathological conditions. The increased activity of MMPs in response to inflammation and oxidative stress can lead to the breakdown of the EG and impairment of endothelial function. The dysregulation of ADAMs and MMPs in glycocalyx shedding has been associated with endothelial dysfunction, increased vascular permeability, and inflammation [39]. This process results in the exposure of adhesion molecules and the release of chemokines, promoting leukocyte adhesion and extravasation, key steps in the initiation of atherosclerosis [40].
Heparanase and hyaluronidase, key sheddases, are known to be upregulated in IA and contribute to EG degradation [37, 41]. This process results in the exposure of adhesion molecules and the release of chemokines, promoting leukocyte adhesion and extravasation, key steps in the initiation of atherosclerosis [39]. Adhesion molecules, such as selectins and integrins, are also implicated in the process of EG shedding. These molecules mediate the adhesion and transmigration of leukocytes across the endothelium, a process that can lead to EG degradation. The exposure of adhesion molecules following EG shedding can promote further leukocyte adhesion, creating a vicious cycle of inflammation and EG degradation [36, 41].
Interplay between complement system and EG shedding
C3a and C5a are important components of the complement system, a part of the immune system involved in inflammation and immune responses. In the context of IA, C3a and C5a play significant roles in the pathophysiology and progression of the disease, contributing to the inflammatory processes and immune dysregulation. They can stimulate the production of matrix MMPs and other proteolytic enzymes, which contribute to the degradation of cartilage [42]. Activation of this particular component of complement system has been shown to induce EG shedding and bind to their respective receptors on endothelial cells, triggering intracellular signaling pathways that lead to the activation of enzymes responsible for EG degradation, such as heparanase and hyaluronidase.
Furthermore, the membrane attack complex (MAC), the end product of complement activation, can cause direct damage to the endothelium and contribute to EG shedding. Therefore, complement activation and EG shedding represent interconnected processes in the pathogenesis of IA and other inflammatory conditions [43].
Implications of EG shedding in inflammatory processes
Syndecans, a family of transmembrane proteoglycans, are a major component of the endothelial EG. They play a crucial role in maintaining the structural integrity of the EG and mediating its various functions. Syndecans consist of a core protein with covalently attached heparan sulfate chains, which contribute to the negative charge and hydration of the EG, thereby influencing its barrier function and interaction with circulating cells and molecules. The shedding of syndecans from the EG is a key event that contributes to glycocalyx degradation and the subsequent onset of inflammation and vascular diseases. This shedding process is primarily mediated by sheddases, such as heparanase and MMPs, which cleave the syndecan core protein and release the ectodomains into the circulation [20, 44].
Interestingly, these shed syndecan ectodomains are not merely by-products of EG degradation, but can act as effector molecules themselves. They have been found to modulate various biological processes, including inflammation, coagulation, and vascular permeability. For instance, shed syndecan-1 ectodomains can bind to inflammatory cytokines and chemokines, thereby modulating their bioavailability and activity. They can also interact with growth factors and coagulation factors, influencing cell proliferation, tissue repair, and blood clotting [45].
Cathepsin, a lysosomal cysteine protease, has been implicated in the pathogenesis of IA. It is known to be involved in the degradation of extracellular matrix proteins and is overexpressed in synovial fibroblasts, the cells that line the joints. This overexpression is thought to contribute to the joint damage seen in these conditions by promoting the degradation of cartilage and bone [46]. In addition to its role in joint tissue degradation, studies have suggested that cathepsin may also be involved in the shedding of the EG, a protective layer on the surface of endothelial cells. While the exact mechanisms are still being elucidated, it is thought that cathepsin may contribute to glycocalyx shedding by degrading its protein components, leading to a loss of integrity and function [47].
Advancements in non-invasive imaging tools, such as GlycoCheck, have made it possible to directly measure EG shedding. By assessing the perfused boundary region, an indicator of EG health, these tools can provide valuable insights into the extent of EG shedding in IA patients [48, 49]. Further research into the mechanisms of endothelial EG shedding and its implications in IA and CVD could lead to novel therapeutic strategies and improved patient outcomes. Understanding the regulation and involvement of sheddases in EG degradation may provide valuable therapeutic targets for the treatment of IA and associated CVD, enhancing disease monitoring and evaluating the effectiveness of interventions.
Mediators of inflammation and microtubule dynamics
Microtubules, as integral components of the cellular cytoskeleton, are implicated in a myriad of cellular functions, encompassing cell division, intracellular transport, and the maintenance of cell structure and motility. Their significance becomes particularly pronounced in the context of IA, where they play a pivotal role in the migration and activation of immune cells.
Microtubules: role in cellular functions and disease processes
The chemotaxis of leukocytes, the primary mediators of the immune response, is heavily reliant on microtubules. The polarization of these cells and their subsequent movement towards inflammation sites is contingent upon the reorientation of the microtubule-organizing center. This reorientation facilitates the directed secretion of inflammatory mediators, thereby modulating the immune response. Moreover, microtubule dynamics are instrumental in regulating the stiffness of endothelial cells. These cells undergo extensive morphological changes during angiogenesis, a process integral to both the progression of atherosclerosis and healing. Atherosclerotic regions are often characterized by aberrant angiogenesis, a process governed by the intricate balance between microtubule polymerization and depolymerization. Disruption of microtubule dynamics in endothelial cells has been linked to the induction of a pro-inflammatory state and increased leukocyte adhesiveness, a critical step in the initiation of atherosclerosis. Thus, the role of microtubules extends beyond structural support, influencing key cellular processes and disease progression [50, 51].
EG-microtubule crosstalk: signaling and cellular processes
The EG serves as a crucial interface for maintaining vascular homeostasis. It is increasingly recognized that the EG can signal to the cell interior and influence the organization and dynamics of microtubules, which are key components of the cytoskeleton. The EG can modulate microtubule behavior through various signaling pathways. For instance, shear stress forces detected by the EG can activate integrins, which in turn can stimulate Rho GTPases such as RhoA and Rac1. These molecules are known to regulate microtubule stability and dynamics, thus influencing cell shape, polarity, and motility. Additionally, the EG can influence the activity of microtubule-associated proteins (MAPs), which can stabilize or destabilize microtubules. Moreover, the EG can also influence the activity of endothelial nitric oxide synthase (eNOS), which produces nitric oxide (NO), a molecule known to influence microtubule dynamics. NO can nitrosylate tubulin, the building block of microtubules, leading to changes in microtubule stability. Therefore, through these and potentially other mechanisms, the EG can signal to microtubules and influence their organization and dynamics, thereby affecting various cellular processes such as cell shape changes, migration, and response to mechanical forces. However, the precise mechanisms of EG-microtubule crosstalk are still not fully understood and are an active area of research [52, 53].
Mechanotransduction, by the EG, is the process of converting mechanical stimuli into biochemical signals. This mechanotransduction is particularly important in the context of shear stress, a force exerted by blood flow on the endothelial cells lining the blood vessels. The EG, particularly its component heparan sulfate (HS), acts as a mechanotransducer, transmitting the shear stress signals to the endothelial cells. This mechanotransduction process regulates various cellular functions, including the expression of angiopoietin-2 (Ang-2), a key mediator of vascular disease. Cleavage of HS from the EG impairs shear stress-related AMPK/FoxO1 signaling, leading to increased expression of Ang-2. This finding suggests that the disruption of the EG, specifically the cleavage of HS, can alter the mechanotransduction process, leading to dysregulated cellular responses such as increased Ang-2 expression. This process could potentially contribute to the pathogenesis of sepsis and other vascular diseases. Furthermore, the study found that the plasma levels of HS, a marker of EG degradation, peaked before the plasma levels of Ang-2 in both children and mice with sepsis. This observation suggests a temporal association between EG damage and the subsequent upregulation of Ang-2, further highlighting the potential mechanistic link between EG injury and vascular disease progression [54, 55].
TNF-α and CRP: impact on microtubule dynamics and EG integrity
Upregulation and overexpression of tumor necrosis factor alpha (TNF-α) is present in IA by various distinct cell types such as macrophages, myeloid cells, and T and B lymphocytes among others that modulate disease development through diverse mechanisms [56]. TNF-α is a crucial participant in the initiation of multiple molecular cascades and maintenance of systemic chronic inflammation. Of particular interest is the exertion of direct effects on cell microtubule and EG dynamics. It exerts direct effects on cell microtubule and extracellular matrix dynamics, including EG degradation, induction of cytoskeleton destabilization, and the formation of intercellular gaps [57, 58]. Elevated levels of osteoprotegerin (OPG), a member of the TNF receptor superfamily, are detected in sera from patients with IA [59, 60]. In the past OPG has been portrayed as an anti-resorptive cytokine but now its function is acknowledged in the pathophysiology of vascular, tumor, and immune diseases. EG and other extracellular and vascular components like syndecan-1, von Willebrand factor/factor VIII complex, heparin, RANKL, glycosaminoglycans, and proteoglycans can act as ligands for OPG and promote cell adhesion and migration. It takes part in bone metabolism regulation and atherosclerosis initiation and progression. Increased circulation of OPG is independently positively associated with higher artery calcium and subclinical atherosclerosis [61,62,63].
C-reactive protein (CRP) is a prototypical marker of inflammation and has been implicated in direct mediation of lipid uptake, complement activation, expression of adhesion molecules, monocyte infiltration, NO inhibition, endothelial dysfunction, and atherogenesis [64]. CRP has widespread clinical use in rheumatology for monitoring disease activity and therapeutic outcomes [65]. It has been observed that CRP in a dose-dependent matter impairs EG structural and functional integrity inducing endothelial dysfunction [66]. Elevated constituents of the EG, suggestive of shedding like hyaluronan, syndecan, and heparan sulfate, have been detected and correlated with IL-6 and CRP in sepsis. Although there is a bacterial etiology, the activation of similar inflammatory pathways can be extrapolated [67]. CRP can also bind to low-density lipoprotein (LDL), and may play a role in the development of atherosclerosis by promoting the uptake of LDL by macrophages in the arterial wall, leading to the formation of foam cells, a hallmark of atherosclerotic plaques [68]. The NLRP3 inflammasome is a multi-protein complex component of the innate immune system and is implicated in the pathogenesis IA. Activation of the NLRP3 inflammasome leads to the release of pro-inflammatory cytokines like IL-1β and IL-18, contributing to chronic inflammation in these conditions. Increased NLRP3 expression and inflammasome activation have been observed in the synovium and skin lesions [69, 70]. In the context of atherosclerosis, activation of the NLRP3 inflammasome in macrophages and endothelial cells can contribute to plaque formation and progression. NLRP3 inflammasome activation leads to increased production of pro-inflammatory cytokines and chemokines, promoting leukocyte adhesion and extravasation, and disrupting contractility and intercellular connections. This can lead to endothelial dysfunction. In addition, NLRP3 inflammasome activation can lead to pyroptosis, a form of inflammatory cell death, further contributing to plaque instability and the risk of plaque rupture [71].
Cellular interactions
IA is characterized by a complex interplay of cellular and molecular events that contribute to the pathophysiology of the disease. Among these, the role of various immune cells, including macrophages, thrombocytes, and neutrophils, is of particular interest. These cells not only contribute to the inflammatory milieu characteristic of IA but also play a crucial role in the process of EG shedding.
Thrombocytes, traditionally known for their role in hemostasis, have been implicated in chronic inflammatory processes as well. In a hypercoagulable state, often promoted by chronic inflammation, there is an excessive activation of the coagulation cascade, coupled with a chronic inhibition of the anti-coagulation and fibrinolytic pathways. In this state they can contribute to inflammation through the release of pro-inflammatory mediators, creating a feed-forward loop that further exacerbates the inflammatory response. The interaction of platelets with the endothelium, facilitated by selectins and other adhesion molecules, is also critical for immune cell recruitment and activation. This process, known as margination, plays an essential role in inflammation. These cellular interactions can upregulate immune responses, further promoting the chronic inflammation typically seen in IA and cardiovascular disease [72, 73]. Macrophages, as key immune cells, play a significant role in the pathogenesis of IA and contribute to the process of endothelial EG shedding and endothelial dysfunction. In IA, macrophages infiltrate the synovium and become activated, releasing pro-inflammatory cytokines such TNF-α and IL-1β. These cytokines promote the degradation of the EG. The loss of the EG exposes the underlying endothelial cells and their adhesion molecules, facilitating the adhesion and transmigration of leukocytes, including macrophages, into the inflamed joints. Furthermore, macrophages themselves can directly contribute to EG shedding through the release of MMPs, including MMP-2 and MMP-9, which degrade the EG components. This disruption of the EG integrity and subsequent endothelial dysfunction further exacerbate the inflammatory process and contribute to the pathophysiology of CVD [39, 74,75,76].
miRNA
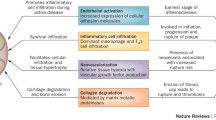
Epigenetic mechanisms, specifically involving microRNAs (miRNAs), are increasingly implicated in the pathophysiological processes underlying both IA and CVD. miRNAs are emerging as not only promising biomarkers for disease onset, activity, progression, and therapeutic response but also as pivotal regulators in the intercellular communication network displaying hormone-like activities [77,78,79]. However, the precise transport mechanisms by which miRNAs traverse the cell membrane remain elusive. Several theories exist, including clathrin or lipid raft endocytosis, phagocytosis, direct membrane fusion, and toll-like or specific miRNA receptors [80]. In normal conditions the sialic acid-containing glycoproteins of the EG and its anionic effects impede the approach of exogenous nucleic acids [81]. It can be speculated that inflammatory driven EG shedding can escalate miRNA transport and expression. Even after adjusting for traditional risk factors, a plasma miRNA panel demonstrates a high prediction rate for coronary artery calcium prevalence in RA, indicating a direct role of miRNAs in the cardiovascular complications of RA [82]. It is now evident that miRNAs engage in intricate molecular circuits that influence disease pathology. Altered expression of miRNAs in a specific tissue due to disease state can inadvertently trigger systemic damaging effects, thereby instigating comorbidities [83]. MiRNAs regulate the production of pro-inflammatory cytokines such as TNF-α and IL-1β. This can contribute to cardiovascular pathology by promoting endothelial dysfunction, vascular inflammation, and atherogenesis (Fig. 3). In experimental models, it has been demonstrated that inhibition of TNF-α prevents specific miRNA upregulation and subsequently improves vasorelaxation, hinting at the vast therapeutic potential of targeting miRNA pathways [84]. Additionally, high-intensity interval training has been shown to induce changes in the EG and associated miRNAs, which may serve as a tool for monitoring early vasculoprotective adaptations to physical activity [85].

Consequences of glycocalyx degradation on endothelial cell function and cellular interactions. The degradation of the glycocalyx leads to microtubule reorganization within endothelial cells, resulting in decreased junctional integrity. This facilitates increased leukocyte adhesion and migration, platelet margination and activation, and enhanced microRNA traffic. These changes in endothelial cell function and cellular interactions are key preluding factors to the development of atherosclerosis, underscoring the critical role of the endothelial glycocalyx in vascular health. VL, vessel lumen; EG, endothelial glycocalyx; EC, endothelial cells; PM, phospholipid endothelial cell membrane
Dysregulated lipid metabolism
Alterations in lipid profiles, including increased levels of total cholesterol, low-density lipoprotein (LDL), and triglycerides, as well as decreased levels of high-density lipoprotein (HDL), have been observed in patients with IA. These lipid abnormalities are believed to result from chronic inflammation, immune dysregulation, and systemic effects of the diseases. The dysregulation of lipid metabolism in IA can have significant implications for cardiovascular health. Elevated levels of LDL cholesterol and triglycerides, along with reduced levels of HDL cholesterol, contribute to the development of atherosclerosis, a major cardiovascular risk factor. Furthermore, the presence of chronic inflammation in IA can further exacerbate lipid abnormalities and promote endothelial dysfunction, oxidative stress, and plaque formation. The dysregulated lipid metabolism in IA is not only linked to cardiovascular risk but also influences disease activity and progression. Lipid mediators, such as pro-inflammatory cytokines and eicosanoids derived from arachidonic acid, play a role in the inflammatory processes underlying IA. Dysfunctional lipid metabolism can perpetuate the inflammatory response, leading to joint damage and disease progression [86]. Understanding the interplay between dysregulated lipid metabolism and IA can provide valuable insights into the mechanisms driving both the joint pathology and cardiovascular complications associated with these conditions. Its impact on the EG has been investigated. Disturbed flow-induced changes to the EG components have been shown to correlate with heterogeneity in the cellular uptake of oxidized LDL and can initiate pro-atherosclerotic endothelial cell behavior. The lack of EG in cells containing cytoplasmic oxidized LDL indicates a link between EG degradation and the internalization of oxidized LDL. These findings suggest that the dysregulation of lipid metabolism and disturbed flow can contribute to GCX dysfunction, promoting the initiation and progression of atherosclerosis [87, 88]. Oxidized LDLs bind to lectin-like receptor (LOX-1) in endothelial cells, triggering signaling pathways involved in the synthesis of chemokines and cell adhesion molecules. Additionally, class B scavenger receptor CD36 mediates the uptake and degradation of oxidized LDL by macrophages, transforming them into foam cells, a hallmark of atherosclerotic plaque formation [89]. TNF has been shown to upregulate LOX-1 expression in endothelial cells, enhancing the uptake of oxidized LDL [90]. Understanding the intricate interplay between lipid metabolism, EG integrity, and atherosclerosis can provide valuable insights into the mechanisms underlying CVD and IA. Further research is needed to explore the precise mechanisms by which dysregulated lipid metabolism influences EG function and the progression of atherosclerosis.
Microparticles
Microparticles: origins, composition, and functions
Another key “participant” that could play a critical role in the pathogenetic process of atherosclerosis are microparticles (MPs) [91]. MPs are vesicle-like membrane fragments of the cell membrane, 0.1 to 1.0 μm in size, released after apoptosis or cellular activation of many cell types, including leukocytes, platelets, ECs, erythrocytes, and SMCs. MPs can be found in plasma, blood, and others. They express various molecules that provide information about the origin of their parent cells and may also express other markers of cellular activation [92].
The majority of circulating MPs originate from platelets and megakaryocytes, which have multiple receptors on their surface. The most expressed surface markers are CD41, CD42b, CD41a, CD61, CD62P, and AA. They influence inflammation, thrombosis, immunoregulation, and transmission of biological information, mainly due to the content of messenger RNA (mRNA), micro RNA (miRNA), and bioactive lipids through fusion or internalization with target cells and cytoplasmic and membrane protein from platelets [93]. MPs are capable of regulating a diverse series of events that result in cell proliferation, angiogenesis, immune response, and coagulation [94, 95].
Microparticles and atherosclerosis: implications for plaque formation
MPs have the ability to regulate cytokines and intercellular adhesion molecule-1 expression, which induces the migration of leukocytes to the vascular wall, which in turn leads to the initiation of atherosclerotic plaque formation [96]. In addition, MPs mediate inflammation by reducing on NO levels [97]. MPs contribute to platelet adhesion upon exposure to the subendothelial matrix [98]. Furthermore, they activate corneal division of smooth muscle and the activation of platelets and endothelial cells through the activation of bioactive lipids, which leads to the production of cytokines and tissue factors [99]. Another important aspect for the involvement of MPs in the atherosclerotic process is their proven presence in atherosclerotic plaques [100].
MPs also express target level markers, developing the markers vascular adhesion molecule-1 (VCAM-1), intertarget adhesion molecule-1 (ICAM-1), VE-cadherin (CD144), PECAM-1 (platelet-endothelial cell adhesion molecule 1/CD31), αv integrin, endoglin (CD105), melanoma endothelial adhesion molecule (MCAM) (CD146), VEGF (vascular endothelial growth factor) receptor 2, von Willebrand factor, E-selectin, and others. Many of these markers are both true soluble molecules and by expression of these different markers of target damage on the endothelial MPs may reflect the degree of endothelial dysfunction [101, 102]. Szotowski et al. have ascertain evidence of the link between the formation of reactive oxygen species (ROS) and the production of tissue factor familiar with EMP. Inhibition of ROS production was associated with lower expression of thrombogenic EMP, highlighting the positive relationship between ROS and the formation of thrombogenic EMP [103]. High levels of ROS can disrupt the redox balance of cells, causing oxidative stress and disruption of cell membrane organization, then releasing membrane microparticles initiating complete apoptosis [104].
Microparticles in IA: endothelial dysfunction and cardiovascular risk
MPs can be significantly elevated in patients with inflammatory rheumatic diseases and they have been considered as factors playing a role in the pathogenesis of some rheumatic diseases [105]. The positive correlation between the degree of RA activity and the levels of MPs has been investigated [106]. MPs in patient sera with IA can induce activation of endothelial cells, mainly those in the macrovasculature. This response is evidenced by an increase in the expression of the adhesion molecules CD54 and CD102; the production of inflammatory mediators, such as IL-6, CCL2, and CCL5; and by the adherence of monocytes to these cells. These vesicles also promote significant changes in the structure of endothelial monolayers, which reduces cell–cell adhesion, depolymerizes actin filaments, and induces cell death. All these changes can contribute to the increase in endothelial permeability observed in the response to MPs, which leads to the beginning of the formation of the atherosclerotic process [107]. MPs may represent a link between autoimmune responses and endothelial dysfunction by expressing TNF-α, altering endothelial apoptosis and autophagy [108].
Emerging evidence underscores the pivotal role of TNF-α, expressed on the surface of microparticles, in modulating endothelial cell function in rheumatoid arthritis patients. MPs isolated from RA patients have been shown to exercise pathological effects on endothelial cells via surface-bound TNF-α. Remarkably, upon exposure to a TNF-α inhibitor, which likely binds to and blocks the action of surface-bound TNF-α, this detrimental impact on endothelial cells is significantly mitigated. This suggests the existence of a novel mechanism of endothelial injury that is mediated by MPs. The findings also reaffirm the protective benefits of anti-TNF therapy against endothelial damage in the context of RA patients. The interactions between MPs, endothelial cells, and the inflammatory milieu, particularly in conditions such as RA, constitute a critical area of investigation. Furthermore, these findings may have implications for the broader understanding of endothelial glycocalyx degradation, given the central role the glycocalyx plays in preserving endothelial integrity in the face of inflammatory assault [109].
Continuing the exploration of MPs role in IA, the study conducted by Sari et al. [110] provides further insight. The authors measured the levels of two specific types of MPs in men with AS, endothelial microparticles (EMPs) and platelet microparticles (PMPs). Elevated levels of both EMPs and PMPs were observed in the patients, indicating the potential involvement of these MPs in the pathogenesis of AS and possibly other IA. Endothelial microparticles, derived from endothelial cells, are particularly intriguing as they are known to carry various bioactive molecules and are considered as markers of endothelial dysfunction. Their presence in increased quantities could indicate ongoing endothelial damage or activation, which is a key event in the pathogenesis of atherosclerosis. On the other hand, PMPs, as products of activated or apoptotic platelets, are known to exhibit pro-coagulant properties and can contribute to a hypercoagulable state, further complicating the cardiovascular risk profile of IA patients. Interestingly, the study also noted a positive correlation between the levels of EMPs and disease activity in AS, suggesting that monitoring EMP levels could potentially serve as a marker for disease activity and cardiovascular risk in these patients [110].
The complex relationship between IA and cardiovascular pathology is significantly influenced by the release and activity of MPs, particularly PMPs and EMPs. Successful treatment with anti-TNF-α significantly reduced circulating MPs, including PMPs and EMPs, in patients with severe psoriasis [111]. Elevated PMP levels were reported in psoriasis patients without concurrent cardiovascular disease. These levels were directly proportional to the degree of inflammation and disease severity, providing a potential link between systemic inflammation seen in IA and the increased cardiovascular risk [112]. Interestingly, PMPs were also found to negatively impact the endothelial glycocalyx area and downregulate the expression of glypican-1 and occludin, crucial players in maintaining vascular homeostasis [113].
MPs, bearing bioactive molecules on their surface, play a critical role in the intercellular communication network, influencing endothelial function in the context of IA. In light of these findings, the study of MPs presents promising opportunities for unravelling the intricate pathways contributing to cardiovascular pathology, and for the potential development of novel therapeutic strategies.
Clinical implications
Risk prediction models that account for traditional CVD risk factors suggest that the systemic inflammation inherent to IA, rather than the specific type of arthritis, may be the primary driver of the increased CVD risk in these patients [5]. However, it is important to note that the manifestation of CVD can vary among different types of IA, likely due to their unique pathophysiological characteristics. PsA patients often present with metabolic syndrome, a known risk factor for CVD, which is less commonly observed in RA or AS patients [114]. Additionally, the concomitant hyperuricemia observed in PsA patients may have significant implications for cardiovascular health. In an animal model, uric acid has been found to induce endothelial-to-mesenchymal transition, a process often associated with endothelial dysfunction and cardiovascular disease. Concurrently, uric acid also prompts the shedding of the EG, mediated by MMPs. These mechanisms collectively suggest a potential pathway by which hyperuricemia could exacerbate the cardiovascular risk associated with PsA [115]. In PsA, an intriguing hypothesis known as the Koebner phenomenon has been proposed, which suggests that post-traumatic events can lead to the new onset of skin psoriasis or arthritis. This hypothesis can be extended to vascular trauma, where an initial injury could potentially exacerbate, through an immune hyper-reaction, into an atherosclerotic lesion. This process could lead to endothelial dysfunction and calciphylaxis. However, this remains speculative and further research is needed to confirm this hypothesis and to understand its potential implications for cardiovascular health in PsA patients [116,117,118].
Uveitis, an intraocular inflammatory condition commonly associated with SpA, has been identified as a potential predictor of atherosclerosis-related CVD. A systematic review and meta-analysis found that uveitis was linked to a 1.49-fold increase in atherosclerosis-related CVD in AS patients [119]. The relationship between AS, and large-vessel vasculitis (LVV) such as aortitis, is a topic of ongoing research. Aortitis, a form of LVV, involves inflammation of the aorta and can lead to serious complications such as aneurysm or aortic dissection. In the context of AS, this association is particularly noteworthy as it suggests a potential overlap in the pathophysiological mechanisms of these conditions. Patients with coexisting SpA and LVV were younger and had higher CRP levels at presentation, indicating a more severe inflammatory response [120]. While the association between IA and CVD is well-established, the specific pathophysiological changes underlying this association in different types of IA remain to be fully elucidated. The observed variations in cardiovascular manifestations among different types of IA suggest that unique pathophysiological mechanisms may be at play in each condition. Further research is needed to fully understand these mechanisms and to develop targeted strategies for cardiovascular risk reduction in patients with different types of IA.
The assessment of CVD risk is a crucial strategy in managing patients with IA. Conventional algorithms such as Framingham and SCORE often underestimate the risk in many RA patients, providing a suboptimal risk stratification and consequently limiting proper clinical management, especially in those categorized as having a low to intermediate risk. Several CV imaging techniques have been reported as being useful in assessing CVD involvement, both for screening, diagnosis, and follow-up. However, it is still unclear which methods should be used to evaluate the CV risk in IA patients in clinical practice, taking into account costs and availability [121].
Conclusion
This review has underlined the intricate interactions between IA and CVD, demonstrating the impact of IA-induced chronic inflammation not only on local joint pathology but also on systemic vascular health. The EG, suffering degradation under chronic inflammation, emerges as a crucial element in this scenario, contributing to endothelial dysfunction and thus paving the way for CVD development and progression. The molecular pathways at play, involving enzymatic actions of heparanase and hyaluronidase, chemotaxis, and various immune cells, present both enlightening insights into disease pathogenesis and potential therapeutic targets. Furthermore, the advent of novel tools enabling non-invasive, direct assessment of EG degradation marks a significant step forward in tracking disease progression and gauging the efficacy of treatment modalities. Hence, to further our therapeutic strategies and improve patient outcomes, it is imperative to deepen our understanding of the dynamic interactions between IA, EG degradation, and endothelial dysfunction. Future research should maintain its focus on elucidating these links, discovering novel therapeutic targets, and enhancing disease progression monitoring tools.
References
Bäck M, Yurdagul A Jr, Tabas I, Öörni K, Kovanen PT (2019) Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 16:389–406. https://doi.org/10.1038/s41569-019-0169-2
Vonderlin N, Siebermair J, Kaya E, Köhler M, Rassaf T, Wakili R (2019) Critical inflammatory mechanisms underlying arrhythmias. Herz 44:121–129. https://doi.org/10.1007/s00059-019-4788-5
Adamo L, Rocha-Resende C, Prabhu SD, Mann DL (2020) Reappraising the role of inflammation in heart failure. Nat Rev Cardiol 17:269–285. https://doi.org/10.1038/s41569-019-0315-x
Agca R, Heslinga SC, Rollefstad S et al (2017) EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis 76(1):17–28. https://doi.org/10.1136/annrheumdis-2016-209775
Lauper K, Courvoisier DS, Chevallier P, Finckh A, Gabay C (2018) Incidence and prevalence of major adverse cardiovascular events in rheumatoid arthritis, psoriatic arthritis, and axial spondyloarthritis. Arthritis Care Res (Hoboken) 70(12):1756–1763. https://doi.org/10.1002/acr.23567
Moltó A, Nikiphorou E (2018) Comorbidities in spondyloarthritis. Front Med (Lausanne) 5:62. https://doi.org/10.3389/fmed.2018.00062
Liew JW, Ramiro S, Gensler LS (2018) Cardiovascular morbidity and mortality in ankylosing spondylitis and psoriatic arthritis. Best Pract Res Clin Rheumatol 32:369–389. https://doi.org/10.1016/j.berh.2019.01.002
Polachek A, Touma Z, Anderson M, Eder L (2017) Risk of cardiovascular morbidity in patients with psoriatic arthritis: a meta-analysis of observational studies. Arthritis Care Res 69:67–74. https://doi.org/10.1002/acr.22926
Zimba O, Gasparyan AY (2023) Cardiovascular issues in rheumatic diseases. Clin Rheumatol. https://doi.org/10.1007/s10067-023-06656-y
Ross R (1999) Atherosclerosis—an inflammatory disease. N Engl J Med 340:115–126. https://doi.org/10.1056/NEJM199901143400207
Libby P, Hansson GK (2015) Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res 116:307–311. https://doi.org/10.1161/CIRCRESAHA.116.301313
Hedar AM, Stradner MH, Roessler A, Goswami N (2021) Autoimmune rheumatic diseases and vascular function: the concept of autoimmune atherosclerosis. J Clin Med 10:4427. https://doi.org/10.3390/jcm10194427
Sima P, Vannucci L, Vetvicka V (2018) Atherosclerosis as autoimmune disease. Ann Transl Med 6(7):116. https://doi.org/10.21037/atm.2018.02.02
Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352(16):1685–1695. https://doi.org/10.1056/NEJMra043430
Peters MJ, van Halm VP, Voskuyl AE, Smulders YM, Boers M, Lems WF et al (2009) Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study Arthritis Rheum 61(11):1571–1579. https://doi.org/10.1002/art.24836
Libby P, Ridker PM, Hansson GK (2011) Progress and challenges in translating the biology of atherosclerosis. Nature 473:317–325. https://doi.org/10.1038/nature10146
Tabas I, García-Cardeña G, Owens GK (2015) Recent insights into the cellular biology of atherosclerosis. J Cell Biol 209(1):13–22. https://doi.org/10.1083/jcb.201412052
Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S (2016) Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res 118(4):535–546. https://doi.org/10.1161/CIRCRESAHA.115.307611
Schött U, Solomon C, Fries D, Bentzer P (2016) The endothelial glycocalyx and its disruption, protection and regeneration: a narrative review. Scand J Trauma Resusc Emerg Med 24:48. https://doi.org/10.1186/s13049-016-0239-y
Deyab G, Reine TM, Vuong TT, Jenssen T, Hjeltnes G, Agewall S et al (2021) Antirheumatic treatment is associated with reduced serum Syndecan-1 in rheumatoid arthritis. PLoS One. 16(7):e0253247. https://doi.org/10.1371/journal.pone.0253247
Agere SA, Kim EY, Akhtar N, Ahmed S (2018) Syndecans in chronic inflammatory and autoimmune diseases: pathological insights and therapeutic opportunities. J Cell Physiol 233:6346–6358. https://doi.org/10.1002/jcp.26388
Zhang X, Sun D, Song JW, Zullo J, Lipphardt M, Coneh-Gould L, Goligorsky MS (2018) Endothelial cell dysfunction and glycocalyx — a vicious circle. Matrix Biol 71–72:421–431. https://doi.org/10.1016/j.matbio.2018.01.026
Qi F, Zhou H, Gu P, Tang ZH, Zhu BF, Chen JR et al (2021) Endothelial glycocalyx degradation is associated with early organ impairment in polytrauma patients. BMC Emerg Med 21(1):52. https://doi.org/10.1186/s12873-021-00446-y
Gasparyan AY, Ayvazyan L, Blackmore H, Kitas GD (2011) Writing a narrative biomedical review: considerations for authors, peer reviewers, and editors. Rheumatol Int 31(11):1409–1417. https://doi.org/10.1007/s00296-011-1999-3
Rueda-Gotor J, Genre F, Corrales A, Blanco R, Fuentevilla P, Portilla V et al (2018) Detection of high cardiovascular risk patients with ankylosing spondylitis based on the assessment of abdominal aortic calcium as compared to carotid ultrasound. Arthritis Res Ther 20(1):195. https://doi.org/10.1186/s13075-018-1684-y
Tinggaard AB, Hjuler KF, Andersen IT, Winther S, Iversen L, Bøttcher M (2021) Prevalence and severity of coronary artery disease linked to prognosis in psoriasis and psoriatic arthritis patients: a multi-centre cohort study. J Intern Med 290(3):693–703. https://doi.org/10.1111/joim.13311
Crowson CS, Liao KP, Davis JM 3rd, Solomon DH, Matteson EL, Knutson KL et al (2013) Rheumatoid arthritis and cardiovascular disease. Am Heart J 166(4):622-628.e1. https://doi.org/10.1016/j.ahj.2013.07.010
Tekaya AB, Mehmli T, Mrad IB, Fendri A, Boukriba S, Bouden S et al (2022) Increased epicardial adipose tissue thickness correlates with endothelial dysfunction in spondyloarthritis. Clin Rheumatol 41(10):3017–3025. https://doi.org/10.1007/s10067-022-06261-5
Dehghan P, Rajaei A, Moeineddin R, Alizadeh AM (2015) Prevalence of atherosclerosis in patients with inactive rheumatoid arthritis. Clin Rheumatol 34:1363–1366. https://doi.org/10.1007/s10067-015-2996-9
Westerlind H, Rönnelid J, Hansson M, Alfredsson L, Mathsson-Alm L, Serre G et al (2020) Anti-citrullinated protein antibody specificities, rheumatoid factor isotypes, and incident cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheumatol 72(10):1658–1667. https://doi.org/10.1002/art.41381
Linton MF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, Doran AC, Vickers KC (2019) The role of lipids and lipoproteins in atherosclerosis. In: Feingold KR, Anawalt B, Blackman MR et al (eds) Endotext. MDText.com Inc, South Dartmouth (MA). https://www.ncbi.nlm.nih.gov/books/NBK343489/. Accessed 30 Jun 2023
Schwartz DM, Parel P, Li H, Sorokin AV, Berg AR, Chen M et al (2022) PET/CT-based characterization of 18F-FDG uptake in various tissues reveals novel potential contributions to coronary artery disease in psoriatic arthritis. Front Immunol 13:909760. https://doi.org/10.3389/fimmu.2022.909760
Gerganov G, Georgiev T, Dimova M, Shivacheva T (2023) Vascular effects of biologic and targeted synthetic antirheumatic drugs approved for rheumatoid arthritis: a systematic review. Clin Rheumatol. https://doi.org/10.1007/s10067-023-06587-8
Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M (2010) Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res 87:300–310. https://doi.org/10.1093/cvr/cvq137
Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L et al (2012) The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 18(8):1217–1223. https://doi.org/10.1038/nm.2843
Chappell D, Dörfler N, Jacob M, Rehm M, Welsch U, Conzen P, Becker BF (2010) Glycocalyx protection reduces leukocyte adhesion after ischemia/reperfusion. Shock 34:133–139. https://doi.org/10.1097/SHK.0b013e3181cdc363
Petrey AC, de la Motte CA (2014) Hyaluronan, a crucial regulator of inflammation. Front Immunol 5:101. https://doi.org/10.3389/fimmu.2014.00101
Thathiah A, Blobel CP, Carson DD (2002) Tumor necrosis factor-alpha converting enzyme/ADAM 17 mediates MUC1 shedding. J Biol Chem 278:3386–3394. https://doi.org/10.1074/jbc.M208326200
Tarbell JM, Cancel LM (2016) The glycocalyx and its significance in human medicine. J Intern Med 280(1):97–113. https://doi.org/10.1111/joim.12465
Woodcock TE, Woodcock TM (2012) Revised Starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. Br J Anaesth 108(3):384–394. https://doi.org/10.1093/bja/aer515
Lipowsky HH (2012) The endothelial glycocalyx as a barrier to leukocyte adhesion and its mediation by extracellular proteases. Ann Biomed Eng 40(4):840–848. https://doi.org/10.1007/s10439-011-0427-x
Holers VM, Banda NK (2018) Complement in the initiation and evolution of rheumatoid arthritis. Front Immunol 9:1057. https://doi.org/10.3389/fimmu.2018.01057
Conway EM (2015) Reincarnation of ancient links between coagulation and complement. J Thromb Haemost 13:S121–S132. https://doi.org/10.1111/jth.12950
Veraldi N, Vivès RR, Blanchard-Rohner G, L’Huillier AG, Wagner N, Rohr M, Beghetti M, De Agostini A, Grazioli S (2022) Endothelial glycocalyx degradation in multisystem inflammatory syndrome in children related to COVID-19. J Mol Med (Berl) 100(5):735–746. https://doi.org/10.1007/s00109-022-02190-7
Rangarajan S, Richter JR, Richter RP, Bandari SK, Tripathi K, Vlodavsky I, Sanderson RD (2020) Heparanase-enhanced shedding of syndecan-1 and its role in driving disease pathogenesis and progression. J Histochem Cytochem 68(12):823–840. https://doi.org/10.1369/0022155420937087
Behl T, Chadha S, Sehgal A, Singh S, Sharma N, Kaur R, Bhatia S, Al-Harrasi A, Chigurupati S, Alhowail A, Bungau S (2022) Exploring the role of cathepsin in rheumatoid arthritis. Saudi J Biol Sci 29(1):402–410. https://doi.org/10.1016/j.sjbs.2021.09.014
Becker BF, Jacob M, Leipert S, Salmon AH, Chappell D (2015) Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br J Clin Pharmacol 80:389–402. https://doi.org/10.1111/bcp.12629
Lee DH, Dane MJ, van den Berg BM, Boels MG, van Teeffelen JW, de Mutsert R, den Heijer M, Rosendaal FR, van der Vlag J, van Zonneveld AJ, Vink H, Rabelink TJ, NEO study group, (2014) Deeper penetration of erythrocytes into the endothelial glycocalyx is associated with impaired microvascular perfusion. PLoS One 9:e96477. https://doi.org/10.1371/journal.pone.0096477
Ikonomidis I, Pavlidis G, Katsimbri P, Lambadiari V, Parissis J, Andreadou I, Tsoumani M, Boumpas D, Kouretas D, Iliodromitis E (2020) Tocilizumab improves oxidative stress and endothelial glycocalyx: a mechanism that may explain the effects of biological treatment on COVID-19. Food Chem Toxicol 145:111694. https://doi.org/10.1016/j.fct.2020.111694
Garcin C, Straube A (2019) Microtubules in cell migration. Essays Biochem 63:509–520. https://doi.org/10.1042/EBC20190016
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936. https://doi.org/10.1038/nature04478
Tarbell JM, Simon SI, Curry FR (2014) Mechanosensing at the vascular interface. Annu Rev Biomed Eng 16:505–532. https://doi.org/10.1146/annurev-bioeng-071813-104908
Lawson CD, Burridge K (2014) The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 5:e27958. https://doi.org/10.4161/sgtp.27958
Richter RP, Ashtekar AR, Zheng L, Pretorius D, Kaushlendra T, Sanderson RD, Gaggar A, Richter JR (2022) Glycocalyx heparan sulfate cleavage promotes endothelial cell angiopoietin-2 expression by impairing shear stress-related AMPK/FoxO1 signaling. JCI Insight 7:e155010. https://doi.org/10.1172/jci.insight.155010
Akwii RG, Sajib MS, Zahra FT, Mikelis CM (2019) Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells 8471. https://doi.org/10.3390/cells8050471
Kruglov A, Drutskaya M, Schlienz D, Gorshkova E, Kurz K, Morawietz L, Nedospasov S (2020) Contrasting contributions of TNF from distinct cellular sources in arthritis. Ann Rheum Dis 79(11):1453–1459. https://doi.org/10.1136/annrheumdis-2019-216068
Urschel K, Cicha I (2015) TNF-α in the cardiovascular system: from physiology to therapy. Int J Interferon Cytokine Mediat Res 7:9–25. https://doi.org/10.2147/IJICMR.S64894
Chappell D, Hofmann-Kiefer K, Jacob M, Rehm M, Briegel J, Welsch U, Conzen P, Becker BF (2009) TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol 104:78–89. https://doi.org/10.1007/s00395-008-0749-5
Wang P, Li S, Liu LN, Lv TT, Li XM, Li XP, Pan HF (2017) Circulating osteoprotegerin levels are elevated in rheumatoid arthritis: a systematic review and meta-analysis. Clin Rheumatol 36:2193–2200. https://doi.org/10.1007/s10067-017-3747-x
Jadon DR, Sengupta R, Nightingale A, Lu H, Dunphy J, Green A, Elder JT, Nair RP, Korendowych E, Lindsay MA, McHugh NJ (2017) Serum bone-turnover biomarkers are associated with the occurrence of peripheral and axial arthritis in psoriatic disease: a prospective cross-sectional comparative study. Arthritis Res Ther 19:210. https://doi.org/10.1186/s13075-017-1417-7
Baud’huin M, Duplomb L, Teletchea S, Lamoureux F, Ruiz-Velasco C, Maillasson M, Redini F, Heymann MF, Heymann D (2013) Osteoprotegerin: multiple partners for multiple functions. Cytokine Growth Factor Rev 24:401–409. https://doi.org/10.1016/j.cytogfr.2013.06.001
Dekker M, Waissi F, Silvis MJM, Bennekom JV, Schoneveld AH, de Winter RJ, Isgum I, Lessmann N, Velthuis BK, Pasterkamp G, Mosterd A, Timmers L, de Kleijn DPV (2021) High levels of osteoprotegerin are associated with coronary artery calcification in patients suspected of a chronic coronary syndrome. Sci Rep 11:18946. https://doi.org/10.1038/s41598-021-98177-4
Arida A, Nezos A, Papadaki I, Sfikakis PP, Mavragani CP (2022) Osteoprotegerin and MTHFR gene variations in rheumatoid arthritis: association with disease susceptibility and markers of subclinical atherosclerosis. Sci Rep 12:9534. https://doi.org/10.1038/s41598-022-13265-3
Shrivastava AK, Singh HV, Raizada A, Singh SK (2015) C-reactive protein, inflammation and coronary heart disease. Egypt Heart J 67(2):89–97. https://doi.org/10.1016/j.ehj.2014.11.005
Rhodes B, Fürnrohr BG, Vyse TJ (2011) C-reactive protein in rheumatology: biology and genetics. Nat Rev Rheumatol 7:282–289. https://doi.org/10.1038/nrrheum.2011.37
Devaraj S, Yun JM, Adamson G, Galvez J, Jialal I (2009) C-reactive protein impairs the endothelial glycocalyx resulting in endothelial dysfunction. Cardiovasc Res 84(3):479–484. https://doi.org/10.1093/cvr/cvp249
Köhler M, Kaufmann I, Briegel J, Jacob M, Goeschl J, Rachinger W, Thiel M, Rehm M (2011) The endothelial glycocalyx degenerates with increasing sepsis severity. Crit Care 15:P22. https://doi.org/10.1186/cc10391
Bian F, Yang XY, Xu G, Zheng T, Jin S (2019) CRP-induced NLRP3 inflammasome activation increases LDL transcytosis across endothelial cells. Front Pharmacol 10:40. https://doi.org/10.3389/fphar.2019.00040
Xiong Y, Cai M, Xu Y, Dong P, Chen H, He W, Zhang J (2022) Joint together: the etiology and pathogenesis of ankylosing spondylitis. Front Immunol 13:996103. https://doi.org/10.3389/fimmu.2022.996103
Yin H, Liu N, Sigdel KR, Duan L (2022) Role of NLRP3 inflammasome in rheumatoid arthritis. Front Immunol 13:931690. https://doi.org/10.3389/fimmu.2022.931690
Qu J, Cheng Y, Wu W, Yuan L, Liu X (2021) Glycocalyx impairment in vascular disease: focus on inflammation. Front Cell Dev Biol 9:730621. https://doi.org/10.3389/fcell.2021.730621
Stark K, Massberg S (2021) Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol 18:666–682. https://doi.org/10.1038/s41569-021-00552-1
Huang HS, Chang HH (2012) Platelets in inflammation and immune modulations: functions beyond hemostasis. Arch Immunol Ther Exp (Warsz) 60:443–451. https://doi.org/10.1007/s00005-012-0193-y
Nieuwdorp M, Meuwese MC, Vink H, Hoekstra JB, Kastelein JJ, Stroes ES (2005) The endothelial glycocalyx: a potential barrier between health and vascular disease. Curr Opin Lipidol 16:507–511. https://doi.org/10.1097/01.mol.0000181325.08926.9c
Barrett TJ (2020) Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol 40:20–33. https://doi.org/10.1161/ATVBAHA.119.312802
Udalova IA, Mantovani A, Feldmann M (2016) Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol 12:472–485. https://doi.org/10.1038/nrrheum.2016.91
Evangelatos G, Fragoulis GE, Koulouri V, Lambrou GI (2019) MicroRNAs in rheumatoid arthritis: from pathogenesis to clinical impact. Autoimmun Rev 18(11):102391. https://doi.org/10.1016/j.autrev.2019.102391
Motta F, Pederzani A, Carena MC, Ceribelli A, Wordsworth PB, De Santis M, Selmi C, Vecellio M (2021) MicroRNAs in axial spondylarthritis: an overview of the recent progresses in the field with a focus on ankylosing spondylitis and psoriatic arthritis. Curr Rheumatol Rep 23:59. https://doi.org/10.1007/s11926-021-01027-5
Romaine SP, Tomaszewski M, Condorelli G, Samani NJ (2015) MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart 101:921–928. https://doi.org/10.1136/heartjnl-2013-305402
O’Brien J, Hayder H, Zayed Y, Peng C (2018) Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne) 9:402. https://doi.org/10.3389/fendo.2018.00402
Palte MJ, Raines RT (2012) Interaction of nucleic acids with the glycocalyx. J Am Chem Soc 134:6218–6223. https://doi.org/10.1021/ja2106477
Tanase DM, Gosav EM, Petrov D, Teodorescu DS, Buliga-Finis ON, Ouatu A, Tudorancea I, Rezus E, Rezus C (2022) MicroRNAs (miRNAs) in cardiovascular complications of rheumatoid arthritis (RA): what is new? Int J Mol Sci 23:52–54. https://doi.org/10.3390/ijms23095254
Sileno S, Beji S, D’Agostino M, Carassiti A, Melillo G, Magenta A (2021) MicroRNAs involved in psoriasis and cardiovascular diseases. Vasc Biol 3(1):R49–R68. https://doi.org/10.1530/VB-21-0007
Gunter S, Michel FS, Fourie SS, Singh M, le Roux R, Manilall A, Mokotedi LP, Millen AME (2022) The effect of TNF-α inhibitor treatment on microRNAs and endothelial function in collagen induced arthritis. PLoS One. 17(2):e0264558. https://doi.org/10.1371/journal.pone.0264558
Schmitz B, Niehues H, Lenders M, Thorwesten L, Klose A, Krüger M, Brand E, Brand SM (2019) Effects of high-intensity interval training on microvascular glycocalyx and associated microRNAs. Am J Physiol Heart Circ Physiol 316:H1538–H1551. https://doi.org/10.1152/ajpheart.00751.2018
Wang Y, Yu H, He J (2020) Role of dyslipidemia in accelerating inflammation, autoimmunity, and atherosclerosis in systemic lupus erythematosus and other autoimmune diseases. Discov Med 30(159):49–56
Harding IC, Mitra R, Mensah SA et al (2019) Glycocalyx degradation induces a proinflammatory phenotype and increased leukocyte adhesion in cultured endothelial cells under flow. PLoS One 14:e0220567. https://doi.org/10.1371/journal.pone.0220567
Mensah SA, Cheng MJ, Homayoni H et al (2021) Disturbed flow induces a sustained, stochastic NF-κB activation which communicates substrate topography in endothelial cells. Sci Rep 11:16526. https://doi.org/10.1038/s41598-021-95953-y
Moore KJ, Sheedy FJ, Fisher EA (2013) Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13:709–721. https://doi.org/10.1038/nri3520
Arjuman A, Chandra NC (2015) Differential pro-inflammatory responses of TNF-α receptors (TNFR1 and TNFR2) on LOX-1 signalling. Mol Biol Rep 42:1039–1047. https://doi.org/10.1007/s11033-014-3841-y
Tan KT, Lip GY (2005) The potential role of platelet microparticles in atherosclerosis. Thromb Haemost 94:488–492. https://doi.org/10.1160/TH05-03-0201
Piccin A, Murphy WG, Smith OP (2007) Circulating microparticles: pathophysiology and clinical implications. Blood Rev 21:157–171. https://doi.org/10.1016/j.blre.2006.09.001
Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME (2010) Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 327:580–583. https://doi.org/10.1126/science.1181928
Curtis AM, Edelberg J, Jonas R, Rogers WT, Moore JS, Syed W, Mohler ER (2013) Endothelial microparticles: sophisticated vesicles modulating vascular function. Vasc Med 18(204):214. https://doi.org/10.1177/1358863X13499773
Chirinos JA, Zambrano JP, Virani SS, Jimenez JJ, Jy W, Ahn E, Horstman LL, Castellanos A, Myerburg RJ, Ahn YS (2005) Correlation between apoptotic endothelial microparticles and serum interleukin-6 and C-reactive protein in healthy men. Am J Cardiol 95:1258–1260. https://doi.org/10.1016/j.amjcard.2005.01.063
Loyer X, Vion AC, Tedgui A, Boulanger CM (2014) Microvesicles as cell-cell messengers in cardiovascular diseases. Circ Res 114(2):345–353. https://doi.org/10.1161/CIRCRESAHA.113.300858
Lukasik M, Rozalski M, Luzak B, Michalak M, Ambrosius W, Watala C, Kozubski W (2013) Enhanced platelet-derived microparticle formation is associated with carotid atherosclerosis in convalescent stroke patients. Platelets 24:63–70. https://doi.org/10.3109/09537104.2011.654292
Priou P, Gagnadoux F, Tesse A, Mastronardi ML, Agouni A, Meslier N et al (2010) Endothelial dysfunction and circulating microparticles from patients with obstructive sleep apnea. Am J Pathol 177(2):974–983. https://doi.org/10.2353/ajpath.2010.091252
Weber A, Köppen HO, Schrör K (2000) Platelet-derived microparticles stimulate coronary artery smooth muscle cell mitogenesis by a PDGF-independent mechanism. Thromb Res 98:461–466. https://doi.org/10.1016/s0049-3848(00)00192-4
Mallat Z, Hugel B, Ohan J, Lesèche G, Freyssinet JM, Tedgui A (1999) Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation 99:348–353. https://doi.org/10.1161/01.cir.99.3.348
Burger D, Schock S, Thompson CS, Montezano AC, Hakim AM, Touyz RM (2013) Microparticles: biomarkers and beyond. Clin Sci (Lond) 124:423–441. https://doi.org/10.1042/CS20120309
Curtis AM, Edelberg J, Jonas R, Rogers WT, Moore JS, Syed W (2013) Mohler EREndothelial microparticles: sophisticated vesicles modulating vascular function. Vasc Med. 18:204–214. https://doi.org/10.1177/1358863X13499773
Szotowski B, Antoniak S, Goldin-Lang P et al (2007) Antioxidative treatment inhibits the release of thrombogenic tissue factor from irradiation- and cytokine-induced endothelial cells. Cardiovasc Res 73(4):806–812. https://doi.org/10.1016/j.cardiores.2006.12.018
Vince RV, Chrismas B, Midgley AW, McNaughton LR, Madden LA (2009) Hypoxia mediated release of endothelial microparticles and increased association of S100A12 with circulating neutrophils. Oxid Med Cell Longev 2:2–6. https://doi.org/10.4161/oxim.2.1.7611
Krajewska-Włodarczyk M, Owczarczyk-Saczonek A, Żuber Z, Wojtkiewicz M, Wojtkiewicz J (2019) Role of microparticles in the pathogenesis of inflammatory joint diseases. Int J Mol Sci 20:5453. https://doi.org/10.3390/ijms20215453
Knijff-Dutmer EA, Koerts J, Nieuwland R, Kalsbeek-Batenburg EM, van de Laar MA (2002) Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheum 46:1498–1503. https://doi.org/10.1002/art.10312
Atehortúa L, Rojas M, Vásquez G, Muñoz-Vahos CH, Vanegas-García A, Posada-Duque RA, Castaño D (2019) Endothelial activation and injury by microparticles in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res Ther 21:34. https://doi.org/10.1186/s13075-018-1796-4
Pamuk GE, Vural O, Turgut B, Demir M, Pamuk ON, Cakir N (2008) Increased platelet activation markers in rheumatoid arthritis: are they related with subclinical atherosclerosis? Platelets 19:146–154
Barbati C, Vomero M, Colasanti T, Diociaiuti M, Ceccarelli F, Ferrigno S, Finucci A, Miranda F, Novelli L, Perricone C, Spinelli FR, Truglia S, Conti F, Valesini G, Alessandri C (2018) TNFα expressed on the surface of microparticles modulates endothelial cell fate in rheumatoid arthritis. Arthritis Res Ther 20:273. https://doi.org/10.1186/s13075-018-1768-8
Sari I, Bozkaya G, Kirbiyik H, Alacacioglu A, Ates H, Sop G, Can G, Taylan A, Piskin O, Yildiz Y, Akkoc N (2012) Evaluation of circulating endothelial and platelet microparticles in men with ankylosing spondylitis. J Rheumatol 39:594–599. https://doi.org/10.3899/jrheum.111073
Pelletier F, Garnache-Ottou F, Biichlé S, Vivot A, Humbert P, Saas P, Seillès E, Aubin F (2014) Effects of anti-TNF-α agents on circulating endothelial-derived and platelet-derived microparticles in psoriasis. Exp Dermatol 23:924–925. https://doi.org/10.1111/exd.12551
Papadavid E, Diamanti K, Spathis A, Varoudi M, Andreadou I, Gravanis K, Theodoropoulos K, Karakitsos P, Lekakis J, Rigopoulos D, Ikonomidis I (2016) Increased levels of circulating platelet-derived microparticles in psoriasis: possible implications for the associated cardiovascular risk. World J Cardiol 8:667–675. https://doi.org/10.4330/wjc.v8.i11.667
Wang GH, Ma KL, Zhang Y, Hu ZB, Liu L, Lu J, Chen PP, Lu CC, Ruan XZ, Liu BC (2019) Platelet microparticles contribute to aortic vascular endothelial injury in diabetes via the mTORC1 pathway. Acta Pharmacol Sin 40:468–476. https://doi.org/10.1038/s41401-018-0186-4
Labitigan M, Bahče-Altuntas A, Kremer JM, Reed G, Greenberg JD, Jordan N, Putterman C, Broder A (2014) Higher rates and clustering of abnormal lipids, obesity, and diabetes mellitus in psoriatic arthritis compared with rheumatoid arthritis. Arthritis Care Res 66:600–607. https://doi.org/10.1002/acr.22185
Ko J, Kang HJ, Kim DA, Kim MJ, Ryu ES, Lee S, Ryu JH, Roncal C, Johnson RJ, Kang DH (2019) Uric acid induced the phenotype transition of vascular endothelial cells via induction of oxidative stress and glycocalyx shedding. FASEB J 33:13334–13345. https://doi.org/10.1096/fj.201901148R
Thorarensen SM, Lu N, Ogdie A, Gelfand JM, Choi HK, Love TJ (2017) Physical trauma recorded in primary care is associated with the onset of psoriatic arthritis among patients with psoriasis. Ann Rheum Dis 76:521–525. https://doi.org/10.1136/annrheumdis-2016-209334
Puig L, Costanzo A, Muñoz-Elías EJ, Jazra M, Wegner S, Paul CF, Conrad C (2022) The biological basis of disease recurrence in psoriasis: a historical perspective and current models. Br J Dermatol 186:773–781. https://doi.org/10.1111/bjd.20963
Gabel CK, Chakrala T, Dobry AS, Garza-Mayers AC, Ko LN, Nguyen ED, Shah R, St John J, Nigwekar SU, Kroshinsky D (2021) The Koebner phenomenon may contribute to the development of calciphylaxis: a case series. JAAD Case Rep 13:57–61. https://doi.org/10.1016/j.jdcr.2021.04.016
Gao X, Lv T, Li G, Tse G, Liu T (2022) Association between atherosclerosis-related cardiovascular disease and uveitis: a systematic review and meta-analysis. Diagnostics (Basel) 12:3178. https://doi.org/10.3390/diagnostics12123178
Ernst D, Baerlecken NT, Schmidt RE, Witte T (2014) Large vessel vasculitis and spondyloarthritis: coincidence or associated diseases? Scand J Rheumatol 43:246–248. https://doi.org/10.3109/03009742.2013.850737
Atzeni F, AlciatiA, (2023) Cardiovascular risk in systemic inflammatory arthritis. J Clin Med 12:2779. https://doi.org/10.3390/jcm12082779
Acknowledgements
Figures were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
All authors have played a substantial role in shaping the study’s conception and design. They have actively engaged in acquiring and analyzing the data, as well as interpreting the results. Furthermore, the authors have collectively contributed to both the initial draft and subsequent critical revisions of the manuscript, providing their invaluable insights. Each author has granted their final approval for the manuscript’s submission to this journal, demonstrating their commitment to taking responsibility for all aspects of the work and being accountable for any potential concerns that may arise. Lastly, all contributing authors have meticulously reviewed and endorsed the final version of the manuscript and take full responsibility for the integrity and accuracy of all aspects of the work.
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Cardiovascular Issues in Rheumatic Diseases
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Angelov, A.K., Markov, M., Ivanova, M. et al. The genesis of cardiovascular risk in inflammatory arthritis: insights into glycocalyx shedding, endothelial dysfunction, and atherosclerosis initiation. Clin Rheumatol 42, 2541–2555 (2023). https://doi.org/10.1007/s10067-023-06738-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-023-06738-x