
Abstract
Platelets play central roles for maintaining the homeostasis of the blood coagulation. As they are also involved in immune responses and host defenses, increasing evidences have suggested that platelets exert other roles beyond their well-recognized function in preventing bleeding. This review is focused on inflammation, allergy and immune modulations of platelets. Platelets conduct immunoregulation through secretion of functional mediators, interaction with various immune cells, endothelial cells and beneficial for the leukocyte infiltration to inflamed/allergic tissues. In these regulations, the leukocytes are influenced by and receiving the signals from platelets. In contrast, rare attentions were focused on platelet regulations by immune system. An intriguingly example in the intravenous immunoglobulin (IVIg) treatment is discussed, in which dendritic cells exert anti-inflammatory effect through platelets. This further suggests that coagulant and immune systems are tightly associated rather than separate entities. The cross-talks between these two systems implicate that platelet therapy may have application beyond thrombosis, and immune interventions may have potentials to treat thrombosis diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Not much more than a century ago, people had not yet identified which blood elements prevent bleeding. In 1882, Italian doctor Giulio Bizzozero first described a novel small element in the blood that has a role in hemostasis. The element was named a “platelet” because of its morphology similar to a small plate (Mazzarello et al. 2001; Michelson 2007; Ribatti and Crivellato 2007). The platelet’s structure and related functions were continually studied by many research groups thereafter, and it became known that various platelet factors were involved in blood coagulation. In addition to the critical role in blood clotting, increasing literatures have implied that platelets exert other functional roles in immunoregulation (Michelson 2007; Semple et al. 2011).
The average lifespan of a platelet is approximately 5–9 days in humans. Derived from cellular fragments of the bone marrow precursor megakaryocytes, platelets are small anucleate cells with a diameter normally around 1–3 μm and can reach to 6 μm if they are fully activated (Chang and Lo 1998). They patrol in the circulation system and are activated when bound to the collagen substratum or other extra-cellular matrix (ECM) proteins on the injured vessel wall (Joseph 1999). On the site of injury, collagen exposed from the subendothelial layer of the vessel wall would be allowed binding to the plasma ultra large von Willebrand factor (vWF). Immobilized vWF will then cause conformational changes and recruit circulating platelet via the interaction with platelet glycoprotein (GP) Ib–V–IX receptor complex under the high shear stress (Kumar et al. 2003). This allows platelets to form stable adhesion and transduce signals through two major collagen receptors GPVI and integrin α2β1 on their surfaces (Pugh et al. 2010; Varga-Szabo et al. 2008). During activation and aggregation, surface integrins and mainly the αIIbβ3 are transformed into high affinity structures. This allows platelet binding to some of the most notable ligands, fibrinogen, fibronectin, and vWF (Varga-Szabo et al. 2008) and such integrin–ligand interactions are primarily mediated through the cellular adhesion–receptor integrins and the Arg-Gly-Asp (RGD) tripeptide-motif of matrix proteins (Chang et al. 1993, 1997, 1998, 1999, 2001, 2002, 2005; Lo and Chang 2005). These interactions could transform platelet morphology (Fig. 1) and elicit sophisticated signal transductions in platelets (Michelson 2007; Sun et al. 2005a, b), by which it is critical for subsequent coagulation activation and the formation of blood clots (Davis 1998; Vargaftig et al. 1979). A lack of functional platelets may be fatal in various thrombocytopenic and hemorrhagic conditions (Kau et al. 2005, 2010; Sun et al. 2007).

Scanning electron microscope (SEM) images of platelets adhered on substrates coated with rhodostomin (an Arg-Gly-Asp containing peptide) (a) and laminin (b). Relatively more lamellipodia were induced after platelets were adhered on a rhodostomin substrate (a) as compared to more filopodia structures on a laminin substrate (b). Representative images are selected from three independent experiments. Scale bar 5 μm
At the same time, platelets are a natural source of proteins having biological functions other than hemostasis (Funa and Ahgren 1997; Koenen and Weber 2010; Leslie 2010; Nojima 1991). At the site of injury, proteins and cytokines released by platelets could limit the survival and dissemination of invading microbes and recruit leukocytes to the wounds (Huang et al. 2010; Li 2008; Sun et al. 2009; Zander and Klinger 2009). This prevents infection and simultaneously promotes tissue repair (Klinger and Jelkmann 2002; Nurden 2011). A number of investigators have concluded that platelets may act as a bridge to link innate and adaptive immunity (Semple et al. 2011; Vieira-de-Abreu et al. 2011; Weyrich and Zimmerman 2004). Evidence in this line suggests that platelets are versatile cells that play additional roles beyond their primary role in hemostasis. Because platelets can respond to wounding and interact promptly with both endothelial and immune cells, it is reasonable to conclude that a number of vascular and immune diseases (e.g. arthrosclerosis, allergy, and autoimmunity) may be associated with platelet functional disorders. This review is focused on platelet-related immunoregulation, in which the roles of platelets in modulating inflammation, allergy, innate and adaptive immune responses are discussed. In contrast to these platelet-to-leukocyte regulations, the leukocyte-to-platelet regulations are also discussed using an example of dendritic cell-mediated modulation of platelets in intravenous immunoglobulin (IVIg) treatments (Huang et al. 2010; Lazarus 2010).
Platelet and Inflammation
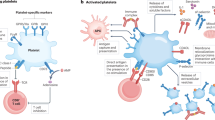
A prominent feature of inflammatory diseases is leukocyte recruitment to the inflamed tissue. This involves multistep processes including an initial interaction among platelets, leukocytes, and endothelial cells through adhesion receptors expressed on their surfaces (Michelson 2007; Passacquale et al. 2011). Among the various molecules involved in these interactions, P-selectin is an adhesion molecule that is essential for mediating leukocyte recruitment and subsequent inflammatory responses (Huo et al. 2003; McGregor et al. 2006; Yokoyama et al. 2005). P-selectin is a transmembrane protein of the selectin family of adhesion receptors, and is synthesized by megakaryocytes and endothelial cells and stored in their α-granules and Weibel–Palade bodies, respectively (Bonfanti et al. 1989; Larsen et al. 1989; Stenberg et al. 1985). Activated platelets and endothelial cells can translocate the P-selectin to their cell surfaces to mediate leukocyte–platelet and leukocyte–endothelial cells interactions, and initiate inflammatory reactions. Platelets are essential for leukocyte recruitment and subsequent firm adhesion to the endothelial cells at the site of inflammation; this consequently triggers autocrine and paracrine activation processes at the vascular wall (Bussolino and Camussi 1995; May et al. 2007). During severe inflammations, abnormal regulation of vascular anticoagulant systems may lead to hypercoagulation and thrombocytopenia (Chang et al. 2012; Sun et al. 2007). Increasing evidence has shown that platelet-induced chronic inflammatory processes at the vascular walls result in the development of atherosclerotic lesions (Gawaz et al. 2005; Huo et al. 2003; Ruggeri 2002). Interactions among platelets, leukocytes, and endothelial cells are now considered important steps leading to inflammation, thrombosis, and atherogenesis. Moreover, platelet–leukocyte interactions contribute to the exchange of signals between platelets and different types of leukocytes that bridge inflammatory immune reactions (Czapiga et al. 2004; Elzey et al. 2003, 2011). These interactions modulate a wide array of responses of both the innate and adaptive immune systems, thus contributing to the pathogenesis of inflammatory diseases and tissue damage (Totani and Evangelista 2010). For example, research has shown that platelet P-selectin is in part responsible for leukocyte–endothelial cell interactions, and responsible for the leukocyte exerting tissue damage in antigen-induced arthritis (Schmitt-Sody et al. 2007a, b). In arthritis, platelets amplify the inflammatory response through interleukin 1 (IL-1) pathway and collagen-dependent microparticle production. In contrast, removing the particles reduces the severity of the disease (Boilard et al. 2010). Platelet-derived P-selectin and RANTES have been detected in intestinal microcirculation, suggesting that activated platelets play a role in mediating leukocyte recruitment to an inflamed colon (Fagerstam et al. 2000). In addition, an increased level of soluble circulating CD40-ligand (CD40L, CD154) released from activated platelets was detected in patients with inflammatory bowel disease (IBD) (Danese et al. 2003). Since CD40–CD40L interactions have been suggested to be essential for angiogenesis in a mouse model of IBD, high levels of soluble CD40L released from activating platelets might play a role in chronic inflammatory processes during IBD formation (Danese and Fiocchi 2005).
Flow chamber and intravital microscopy analyses were used to investigate the molecular events involved in leukocyte recruitment during inflammation in a feline ischemia–reperfusion model (Gill et al. 2005). Evidence revealed that anti-inflammatory IVIg treatment exerted an inhibitory effect on the P-selectin dependent rolling that had led to reduced leukocyte recruitment and vascular dysfunction (Gill et al. 2005). Intriguingly, the in vitro flow-chamber analysis revealed a significant ameliorative effect of IVIg on leukocyte recruitment when whole blood was treated, but not when P-selectin-expressed endothelial cells were treated alone. The findings further suggested that IVIg amelioration of leukocyte rolling might be mediated through a platelet-P-selectin-dependent pathway. However, whether IVIg directly blocks the P-selectin mediating leukocyte interaction or subsequent inflammation requires further clarification.
Platelets and Allergic Inflammation
A different line of evidence also suggests that platelets exert significantly wider roles. Clinical data from patients suffering from allergic inflammatory reactions showed that the platelet counts and platelet activating markers in the plasma were significantly altered after exposure to an allergen (Kasperska-Zajac et al. 2008; Kemona-Chetnik et al. 2007; Kowal et al. 2006). Both intact platelets and platelet-released inflammatory mediators play crucial roles in the sustained inflammatory response of many allergic diseases. This type of allergy involves a change in platelet surface molecule expression, aggregation, adhesion, and arachidonic acid metabolism (Barnes 1987; Edenius et al. 1991; Pitchford et al. 2005). Platelet activation is necessary to transform arachidonic acid into lipid mediators, such as thromboxane A2 (TXA2), cysteinyl leukotrienes, and lipoxins, all of which exacerbates the allergic response (Barnes 1987; Edenius et al. 1991; Kantarci and Van Dyke 2003; Kasperska-Zajac and Rogala 2007). Thus, platelets are likely more directly involved in allergic initiation than simply exerting an accessory role to facilitate leukocyte rolling and adhesion. Research has shown that human platelets constitutively express functional receptors for the Fc fragment of IgE, both the low-affinity immunoglobulin E Fc receptor (FcεRII) (Capron et al. 1986) and the high-affinity receptor (FcεRI) (Hasegawa et al. 1999; Joseph et al. 1997). Upon coupling to the antigen–IgE complex, platelets are capable of releasing a variety of biologically active mediators and may thus participate in hypersensitive reactions, including anaphylaxis (Kasperska-Zajac and Rogala 2006; Pitchford 2007).
One study compared the histology data for mice lungs regarding platelet chemotaxis, between ovalbumin-sensitized wild-type mice versus FcRγ−/− mutant mice that lacked the FcεRIγ response. These results revealed that for the wild type, but not the FcRγ−/− mice, isolated platelets migrated out of vessels and were localized beneath the airways after the mice had been allergen-challenged (Pitchford et al. 2008). Clarification of platelet chemotaxis further revealed that the binding of allergen–IgE complex to surface FcεRI allows platelets to participate directly in allergic tissue inflammation (Pitchford et al. 2008). This hypothesis has been supported by another line of evidence, which showed that chemotaxis platelets, and subsequently infiltrated leukocytes, might be equally important in generating allergic inflammation in the respiratory tract. For example, allergic asthma has been characterized by airway inflammation, normal platelet counts, but not to those thrombocytopenic animals (Coyle et al. 1990). This suggests that platelets are required for eosinophil infiltration. In a mouse model, allergen exposure appeared to induce platelet migration to the airways, a condition that was necessary for eosinophil recruitment and activation (Pitchford et al. 2005). Thus, platelets played an important role in airway eosinophilia and triggering of inflammatory reactions (Benton et al. 2010; Hogan 2007). Furthermore, platelets were found to be essential for tissue remodeling in a mouse model with chronic allergic airway inflammation (Pitchford et al. 2004, 2008). Platelet membranes were able to induce airway smooth-muscle cell proliferation in a mechanism dependent on 5-lipoxygenase (5-LOX) and reactive oxygen species (ROS) pathways (Svensson Holm et al. 2008, 2011). The consequence was asthma with persistent and chronic inflammation. Another study showed that the platelet activation maker, platelet factor 4 (PF-4), stimulated histamine release from mast cells in a dose-dependent manner (Suzuki et al. 2002). Circulating platelet factor 4 (PF-4) is an important factor in the development of seasonal allergic rhinitis and asthma (Kasperska-Zajac et al. 2008). The expression of the murine homologue of PF-4 was highly increased in the spleen of mice having atopic dermatitis (Watanabe et al. 1999), and the hyper-aggregability of platelets in the physiology of skin inflammation in atopic dermatitis has been demonstrated (Katoh 2009; Tamagawa-Mineoka et al. 2007). As a result, suppression of platelet function or interference of platelet-induced leukocyte recruitment to the site of inflammation might provide an alternative target in the alleviation of allergic diseases.
Platelets in Innate and Adaptive Immunity
Platelet responses in assisting allergy and inflammation have also been found to modulate the immune response. Platelets express toll-like receptors and may serve as sentinels in the circulatory system, with characteristics similar to those of an innate immune cell (Aslam et al. 2006). Innate immune cells can bind rapidly to pathogens, actively mediating innate immune responses and perhaps more importantly activating the mononuclear phagocyte system (MPS) (Johansson et al. 2011; Semple and Freedman 2010; Semple et al. 2011). Thus, platelets are actually potent effector cells of the innate immune system, capable of initiating an inflammatory response at the site of infection (Aslam et al. 2006; Clark et al. 2007; Semple and Freedman 2010). In addition to their roles in innate immunity, many evidences suggest that platelets act as mediators between innate and adaptive immune responses (Aslam et al. 2006; Elzey et al. 2011; Elzey et al. 2003; Semple et al. 2011). Among these, dendritic cells (DCs) are essential to link innate and adaptive immunity (Banchereau and Steinman 1998; Catani et al. 2006; Corrigall et al. 2009; Gallucci and Matzinger 2001; Medzhitov and Janeway 2000). Evidence suggests that platelets serve as early effectors of DC activation in tissue injury by displaying and releasing to DCs the “danger signals” of CD40L and IL-1β (Banchereau and Steinman 1998). Thrombin-stimulated murine platelets induced activation and maturation of primary mouse bone marrow DCs through a platelet-CD40L-dependent pathway (Elzey et al. 2003). At the same time, in a co-cultured system using M-CSF and IL-4, immobilized P-selectin enhanced the generation of CD14+CD16+ DC-like cells from primary human monocytes. These DC-like cells primed naive allogeneic CD4+ T cells to produce significantly less IFN-γ, and inhibited macrophage maturation from human peripheral blood monocytes (Li et al. 2003). Platelet-mediated leukocyte adhesion may also enhance lymphocyte trafficking during adaptive immunity and host defense (Diacovo et al. 1996, 1998; von Hundelshausen et al. 2007). Recent studies utilizing CD40L knockout mice found that platelet-derived CD40L modulates the adaptive immune response. This led to an enhanced antigen presentation, improved CD8+ T cell responses, and played a critical role in T dependent humoral immunity (Elzey et al. 2005; Sprague et al. 2007). Because CD40L served as part of the interface between platelets and adaptive immunity (Elzey et al. 2011), these findings may provide a basis to expand the current paradigm of B cell activation and germinal center formation (Sprague et al. 2007, 2008).
Dendritic Cell-to-Platelet Regulation: An Example in IVIg Treatments
Featured by increased platelet destruction and insufficient platelet production, the pathophysiology of immune thrombocytopenia (ITP) is characterized by the development of autoantibodies against platelet glycoproteins and subsequently the macrophage Fcγ receptor-mediated clearance in patients (Cines and McMilan 2005; Provan et al. 2010; Rodeghiero et al. 2009; Samuelsson et al. 2001). Intravenous immunoglobulin (IVIg) is an effective treatment to ameliorate autoimmune and inflammation diseases, such as ITP (Imbach and Morell 1989; Kazatchkine and Kaveri 2001). The IVIg is prepared from pooled immunoglobulin (Ig)G fractions taken from thousands of healthy blood donors (Ephrem et al. 2005). Despite its high efficiency, the ameliorative mechanism of IVIg remains unclear. Several possible mechanisms have been suggested. It might inhibit complement (Basta and Dalakas 1994), provide immunomodulation through idiotypic antibodies (Levy et al. 1998), modulate cytokine production (Braun-Moscovici and Furst 2003; Gonzalez et al. 2004; Pashov et al. 1997), enhance pathogenic antibody degradation through neonatal Fc receptors (Akilesh et al. 2004), block cell-surface receptors for IgG (FcγRs) (Clynes 2005), or modulate inhibitory Fcγ receptor expression on innate effector cells (Samuelsson et al. 2001).
In addition to these, a more complicated two-step modulation may be involved. This possibility is suggested by the finding that colony-stimulating factor 1 (CSF-1)-dependent sensor macrophages were required for IVIg to induce inhibition of effector macrophages (Bruhns et al. 2003). The two-step model was further supported by data from a study of adoptive cell transfer, in which IVIg-primed splenic DCs were shown to play an ameliorative role in recipient mice with ITP (Siragam et al. 2006). The regulatory role of platelets was rarely discussed in IVIg-mediated amelioration. To investigate the role of platelets in IVIg-mediated amelioration, a novel approach, which combined experimental ITP, adoptive DC transfer, and platelet-splenic phagocyte (PLT-SP) bind/engagement experiments, was established (Huang et al. 2010). As the level of in vitro PLT-SP engagement is associated with the platelet count, and thus reflecting the IVIg ameliorative effect, respective leukocyte and platelet activities could be analyzed through this approach (Huang et al. 2010). Splenic CD11c+ DCs have been shown to initiate the IVIg-induced ameliorative effects (Crow and Lazarus 2008; Siragam et al. 2006). Intriguingly, after adoptively transferred IVIg-primed DCs (IVIg-DCs) into recipient ITP mice, platelets rather than the phagocytes changed their PLT-SP engaging property (experimental outline is shown in Fig. 2) (Huang et al. 2010). As both inhibitory Fcγ receptor IIB (FcγRIIB) and activating FcγRIII receptors (encoded by Fcgr2b and Fcgr3, respectively) are involved in IVIg-mediated amelioration (Samuelsson et al. 2001; Siragam et al. 2006), these FcγR null mice were tested. The IVIg-DCs did not ameliorate ITP in Fcgr2b −/−, Fcgr3 −/−, nor Selp −/− (P-selectin null) mice, implicating the potential involvement of these pathways in IVIg action (Huang et al. 2010). These findings suggested that a P-selectin-mediated leukocyte–platelet crosstalk is involved in IVIg-induced amelioration of ITP, and that FcγRs plays a critical role in IVIg-induced amelioration in ITP. As platelets are a component of DC regulatory circuits, these findings may suggest there are potentials to treat coagulant and thrombosis diseases through immune interventions. Agonists and antagonists of P-selectin and FcγRs may provide more maneuverability during the treatments. Further mechanism and application studies are needed for such development.

Adoptive cell transfer mouse model and flow cytometry analyses for platelet-splenic phagocyte (PLT-SP) engagement. Schematic illustration of the experiment outline used in the adoptive transfer mice model (a). Open triangle the recipients, filled triangle third party naïve wild type (WT) mice. CD11c+ dendritic cells (DCs) were isolated from donor mice and then transferred to ITP recipient mice (filled triangle) after IVIg priming (a). Ameliorative effect of IVIg-primed DCs was analyzed through measuring platelet counts and the engaging properties of platelet (PLT) and splenic phagocyte (SP) in the recipient mice (flow cytometry analysis) (b). Leukocytes were first distinguished from platelets using cell size and granularity characteristics (left R1), and PLT-engaged SPs were then quantified by measuring the percentage of CD14+ (SP marker) CD41+ (PLT marker) double positive cells (right PLT/SPs)
Conclusion
Aforementioned evidences indicate that platelets play functional roles not only in hemostasis and thrombosis but also in inflammation and immune response. Platelets conduct immunoregulation through secretion of functional mediators, interaction with various immune cells, endothelial cells and even migration to inflamed/allergic tissues. These responses are critically associated with the disease outcomes. DC-to-platelet regulation in IVIg treatments further suggests that coagulant and immune systems are not distinctly regulated. Platelets conduct bidirectional regulations and serve as an important bridge to coordinate the coagulant and immune systems. These results suggest anti-platelet therapy may have application beyond thrombosis, and also shed light upon the potential treatments of coagulant diseases through immune interventions. Although further investigations are required to clarify the underlying mechanism, new findings of immune-coagulant system cross-talks are starting to revolutionize our understanding of platelet biology.
Abbreviations
- ITP:
-
Immune thrombocytopenia
- IVIg:
-
Intravenous immunoglobulin
- DCs:
-
Dendritic cells
- CD40L:
-
CD40-ligand
- FcεR:
-
Immunoglobulin E Fc receptor
- FcγR:
-
Immunoglobulin G Fc receptor
References
Akilesh S, Petkova S, Sproule TJ et al (2004) The MHC class I-like Fc receptor promotes humorally mediated autoimmune disease. J Clin Invest 113:1328–1333
Aslam R, Speck ER, Kim M et al (2006) Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 107:637–641
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392:245–252
Barnes PJ (1987) Inflammatory mediator receptors and asthma. Am Rev Respir Dis 135:S26–S31
Basta M, Dalakas MC (1994) High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J Clin Invest 94:1729–1735
Benton AS, Kumar N, Lerner J et al (2010) Airway platelet activation is associated with airway eosinophilic inflammation in asthma. J Investig Med 58:987–990
Boilard E, Nigrovic PA, Larabee K et al (2010) Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 327:580–583
Bonfanti R, Furie BC, Furie B et al (1989) PADGEM (GMP140) is a component of Weibel–Palade bodies of human endothelial cells. Blood 73:1109–1112
Braun-Moscovici Y, Furst DE (2003) Immunoglobulin for rheumatic diseases in the twenty-first century: take it or leave it? Curr Opin Rheumatol 15:237–245
Bruhns P, Samuelsson A, Pollard JW et al (2003) Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity 18:573–581
Bussolino F, Camussi G (1995) Platelet-activating factor produced by endothelial cells. A molecule with autocrine and paracrine properties. Eur J Biochem 229:327–337
Capron M, Jouault T, Prin L et al (1986) Functional study of a monoclonal antibody to IgE Fc receptor (Fc epsilon R2) of eosinophils, platelets, and macrophages. J Exp Med 164:72–89
Catani L, Fagioli ME, Tazzari PL et al (2006) Dendritic cells of immune thrombocytopenic purpura (ITP) show increased capacity to present apoptotic platelets to T lymphocytes. Exp Hematol 34:879–887
Chang HH, Lo SJ (1998) Full-spreading platelets induced by the recombinant rhodostomin are via binding to integrins and correlated with FAK phosphorylation. Toxicon 36:1087–1099
Chang, WK, Sun, DS, Chan, H et al. (2012) Visible light responsive core-shell structured In2O3@CaIn2O4 photocatalyst with superior bactericidal property and biocompatibility. Nanomedicine-NBM 8:609–617
Chang HH, Hu ST, Huang TF et al (1993) Rhodostomin, an RGD-containing peptide expressed from a synthetic gene in Escherichia coli, facilitates the attachment of human hepatoma cells. Biochem Biophys Res Commun 190:242–249
Chang HH, Tsai WJ, Lo SJ (1997) Glutathione S-transferase-rhodostomin fusion protein inhibits platelet aggregation and induces platelet shape change. Toxicon 35:195–204
Chang HH, Lin CH, Lo SJ (1999) Recombinant rhodostomin substrates induce transformation and active calcium oscillation in human platelets. Exp Cell Res 250:387–400
Chang CP, Chang JC, Chang HH et al (2001) Positional importance of Pro53 adjacent to the Arg49-Gly50-Asp51 sequence of rhodostomin in binding to integrin alphaIIbbeta3. Biochem J 357:57–64
Chang HH, Shyu HF, Wang YM et al (2002) Facilitation of cell adhesion by immobilized dengue viral nonstructural protein 1 (NS1): arginine-glycine-aspartic acid structural mimicry within the dengue viral NS1 antigen. J Infect Dis 186:743–751
Chang JC, Chang HH, Lin CT et al (2005) The integrin alpha6beta1 modulation of PI3 K and Cdc42 activities induces dynamic filopodium formation in human platelets. J Biomed Sci 12:867–884
Cines DB, McMillan R (2005) Management of adult idiopathic thrombocytopenic purpura. Annu Rev Med 56:425–442
Clark SR, Ma AC, Tavener SA et al (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13:463–469
Clynes R (2005) Immune complexes as therapy for autoimmunity. J Clin Invest 115:25–27
Corrigall VM, Vittecoq O, Panayi GS (2009) Binding immunoglobulin protein-treated peripheral blood monocyte-derived dendritic cells are refractory to maturation and induce regulatory T-cell development. Immunology 128:218–226
Coyle AJ, Page CP, Atkinson L et al (1990) The requirement for platelets in allergen-induced late asthmatic airway obstruction. Eosinophil infiltration and heightened airway responsiveness in allergic rabbits. Am Rev Respir Dis 142:587–593
Crow AR, Lazarus AH (2008) The mechanisms of action of intravenous immunoglobulin and polyclonal anti-d immunoglobulin in the amelioration of immune thrombocytopenic purpura: what do we really know? Transfus Med Rev 22:103–116
Czapiga M, Kirk AD, Lekstrom-Himes J (2004) Platelets deliver costimulatory signals to antigen-presenting cells: a potential bridge between injury and immune activation. Exp Hematol 32:135–139
Danese S, Fiocchi C (2005) Platelet activation and the CD40/CD40 ligand pathway: mechanisms and implications for human disease. Crit Rev Immunol 25:103–121
Danese S, de la Motte C, Sturm A et al (2003) Platelets trigger a CD40-dependent inflammatory response in the microvasculature of inflammatory bowel disease patients. Gastroenterology 124:1249–1264
Davis GL (1998) Introduction to platelet update. Clin Lab Sci 11:355
Diacovo TG, Puri KD, Warnock RA et al (1996) Platelet-mediated lymphocyte delivery to high endothelial venules. Science 273:252–255
Diacovo TG, Catalina MD, Siegelman MH et al (1998) Circulating activated platelets reconstitute lymphocyte homing and immunity in L-selectin-deficient mice. J Exp Med 187:197–204
Edenius C, Stenke L, Lindgren JA (1991) On the mechanism of transcellular lipoxin formation in human platelets and granulocytes. Eur J Biochem 199:401–409
Elzey BD, Tian J, Jensen RJ et al (2003) Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity 19:9–19
Elzey BD, Grant JF, Sinn HW et al (2005) Cooperation between platelet-derived CD154 and CD4+ T cells for enhanced germinal center formation. J Leukoc Biol 78:80–84
Elzey BD, Ratliff TL, Sowa JM et al (2011) Platelet CD40L at the interface of adaptive immunity. Thromb Res 127:180–183
Ephrem A, Misra N, Hassan G et al (2005) Immunomodulation of autoimmune and inflammatory diseases with intravenous immunoglobulin. Clin Exp Med 5:135–140
Fagerstam JP, Whiss PA, Strom M et al (2000) Expression of platelet P-selectin and detection of soluble P-selectin, NPY and RANTES in patients with inflammatory bowel disease. Inflamm Res 49:466–472
Funa K, Ahgren A (1997) Characterization of platelet-derived growth factor (PDGF) action on a mouse neuroblastoma cell line, NB41, by introduction of an antisense PDGF beta-receptor RNA. Cell Growth Differ 8:861–869
Gallucci S, Matzinger P (2001) Danger signals: SOS to the immune system. Curr Opin Immunol 13:114–119
Gawaz M, Langer H, May AE (2005) Platelets in inflammation and atherogenesis. J Clin Invest 115:3378–3384
Gill V, Doig C, Knight D et al (2005) Targeting adhesion molecules as a potential mechanism of action for intravenous immunoglobulin. Circulation 112:2031–2039
Gonzalez H, Khademi M, Andersson M et al (2004) Prior poliomyelitis-IVIg treatment reduces proinflammatory cytokine production. J Neuroimmunol 150:139–144
Hasegawa S, Pawankar R, Suzuki K et al (1999) Functional expression of the high affinity receptor for IgE (FcepsilonRI) in human platelets and its’ intracellular expression in human megakaryocytes. Blood 93:2543–2551
Hogan SP (2007) Recent advances in eosinophil biology. Int Arch Allergy Immunol 143(Suppl 1):3–14
Huang HS, Sun DS, Lien TS et al (2010) Dendritic cells modulate platelet activity in IVIg-mediated amelioration of ITP in mice. Blood 116:5002–5009
Huo Y, Schober A, Forlow SB et al (2003) Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med 9:61–67
Imbach P, Morell A (1989) Idiopathic thrombocytopenic purpura (ITP): immunomodulation by intravenous immunoglobulin (IVIg). Int Rev Immunol 5:181–188
Johansson D, Shannon O, Rasmussen M (2011) Platelet and neutrophil responses to gram positive pathogens in patients with bacteremic infection. PLoS ONE 6:e26928
Joseph A (1999) The role of platelets and platelet glycoprotein IIb/IIIa receptor inhibition in acute coronary syndromes: an educational supplement. Introduction. J Emerg Med 17:565
Joseph M, Gounni AS, Kusnierz JP et al (1997) Expression and functions of the high-affinity IgE receptor on human platelets and megakaryocyte precursors. Eur J Immunol 27:2212–2218
Kantarci A, Van Dyke TE (2003) Lipoxins in chronic inflammation. Crit Rev Oral Biol Med 14:4–12
Kasperska-Zajac A, Rogala B (2006) Platelet function in anaphylaxis. J Investig Allergol Clin Immunol 16:1–4
Kasperska-Zajac A, Rogala B (2007) Platelet activation during allergic inflammation. Inflammation 30:161–166
Kasperska-Zajac A, Brzoza Z, Rogala B (2008) Seasonal changes in platelet activity in pollen-induced seasonal allergic rhinitis and asthma. J Asthma 45:485–487
Katoh N (2009) Platelets as versatile regulators of cutaneous inflammation. J Dermatol Sci 53:89–95
Kau JH, Sun DS, Tsai WJ et al (2005) Antiplatelet activities of anthrax lethal toxin are associated with suppressed p42/44 and p38 mitogen-activated protein kinase pathways in the platelets. J Infect Dis 192:1465–1474
Kau JH, Sun DS, Huang HS et al (2010) Sublethal doses of anthrax lethal toxin on the suppression of macrophage phagocytosis. PLoS ONE 5:e14289
Kazatchkine MD, Kaveri SV (2001) Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med 345:747–755
Kemona-Chetnik I, Bodzenta-Lukaszyk A, Butkiewicz A et al (2007) Thrombocytopoesis in allergic asthma. Pol Arch Med Wewn 117:9–13
Klinger MH, Jelkmann W (2002) Role of blood platelets in infection and inflammation. J Interferon Cytokine Res 22:913–922
Koenen RR, Weber C (2010) Platelet-derived chemokines in vascular remodeling and atherosclerosis. Semin Thromb Hemost 36:163–169
Kowal K, Pampuch A, Kowal-Bielecka O et al (2006) Platelet activation in allergic asthma patients during allergen challenge with Dermatophagoides pteronyssinus. Clin Exp Allergy 36:426–432
Kumar RA, Dong JF, Thaggard JA et al (2003) Kinetics of GPIbalpha-vWF-A1 tether bond under flow: effect of GPIbalpha mutations on the association and dissociation rates. Biophys J 85:4099–4109
Larsen E, Celi A, Gilbert GE et al (1989) PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 59:305–312
Lazarus AH (2010) IVIg conducts DC-platelet nuptials. Blood 116:4740–4741
Leslie M (2010) Cell biology. Beyond clotting: the powers of platelets. Science 328:562–564
Levy Y, Sherer Y, Ahmed A et al (1998) Autoantibody level modification in adult patients with idiopathic thrombocytopenic purpura following intravenous immunoglobulin treatment. Nat Immun 16:207–214
Li N (2008) Platelet-lymphocyte cross-talk. J Leukoc Biol 83:1069–1078
Li G, Kim YJ, Mantel C et al (2003) P-selectin enhances generation of CD14+ CD16+ dendritic-like cells and inhibits macrophage maturation from human peripheral blood monocytes. J Immunol 171:669–677
Lo SJ, Chang HH (2005) Recombinant snake disintegrins used for mammalian integrin study. Toxin Rev 24:95–111
May AE, Langer H, Seizer P et al (2007) Platelet–leukocyte interactions in inflammation and atherothrombosis. Semin Thromb Hemost 33:123–127
Mazzarello P, Calligaro AL, Calligaro A (2001) Giulio Bizzozero: a pioneer of cell biology. Nat Rev Mol Cell Biol 2:776–781
McGregor L, Martin J, McGregor JL (2006) Platelet-leukocyte aggregates and derived microparticles in inflammation, vascular remodelling and thrombosis. Front Biosci 11:830–837
Medzhitov R, Janeway C Jr (2000) Innate immunity. N Engl J Med 343:338–344
Michelson AD (2007) Platelets. California Academic Press, New York
Nojima S (1991) Platelet-activating factor (PAF): an introduction. Lipids 26:965–966
Nurden AT (2011) Platelets, inflammation and tissue regeneration. Thromb Haemost 105(Suppl 1):S13–S33
Pashov A, Bellon B, Kaveri SV et al (1997) A shift in encephalitogenic T cell cytokine pattern is associated with suppression of EAE by intravenous immunoglobulins (IVIg). Mult Scler 3:153–156
Passacquale G, Vamadevan P, Pereira L et al (2011) Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS ONE 6:e25595
Pitchford SC (2007) Defining a role for platelets in allergic inflammation. Biochem Soc Trans 35:1104–1108
Pitchford SC, Riffo-Vasquez Y, Sousa A et al (2004) Platelets are necessary for airway wall remodeling in a murine model of chronic allergic inflammation. Blood 103:639–647
Pitchford SC, Momi S, Giannini S et al (2005) Platelet P-selectin is required for pulmonary eosinophil and lymphocyte recruitment in a murine model of allergic inflammation. Blood 105:2074–2081
Pitchford SC, Momi S, Baglioni S et al (2008) Allergen induces the migration of platelets to lung tissue in allergic asthma. Am J Respir Crit Care Med 177:604–612
Provan D, Stasi R, Newland AC et al (2010) International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 115:168–186
Pugh N, Simpson AM, Smethurst PA et al (2010) Synergism between platelet collagen receptors defined using receptor-specific collagen-mimetic peptide substrata in flowing blood. Blood 115:5069–5079
Ribatti D, Crivellato E (2007) Giulio Bizzozero and the discovery of platelets. Leuk Res 31:1339–1341
Rodeghiero F, Stasi R, Gernsheimer T et al (2009) Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 113:2386–2393
Ruggeri ZM (2002) Platelets in atherothrombosis. Nat Med 8:1227–1234
Samuelsson A, Towers TL, Ravetch JV (2001) Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science 291:484–486
Schmitt-Sody M, Gottschalk O, Metz P et al (2007a) Endothelial iNOS versus platelet iNOS: responsibility for the platelet/leukocyte endothelial cell interaction in murine antigen induced arthritis in vivo. Inflamm Res 56:262–268
Schmitt-Sody M, Metz P, Gottschalk O et al (2007b) Platelet P-selectin is significantly involved in leukocyte–endothelial cell interaction in murine antigen-induced arthritis. Platelets 18:365–372
Semple JW, Freedman J (2010) Platelets and innate immunity. Cell Mol Life Sci 67:499–511
Semple JW, Italiano JE Jr, Freedman J (2011) Platelets and the immune continuum. Nat Rev Immunol 11:264–274
Siragam V, Crow AR, Brinc D et al (2006) Intravenous immunoglobulin ameliorates ITP via activating Fc gamma receptors on dendritic cells. Nat Med 12:688–692
Sprague DL, Sowa JM, Elzey BD et al (2007) The role of platelet CD154 in the modulation in adaptive immunity. Immunol Res 39:185–193
Sprague DL, Elzey BD, Crist SA et al (2008) Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood 111:5028–5036
Stenberg PE, McEver RP, Shuman MA et al (1985) A platelet alpha-granule membrane protein (GMP-140) is expressed on the plasma membrane after activation. J Cell Biol 101:880–886
Sun DS, Lo SJ, Lin CH et al (2005a) Calcium oscillation and phosphatidylinositol 3-kinase positively regulate integrin alpha(IIb)beta3-mediated outside-in signaling. J Biomed Sci 12:321–333
Sun DS, Lo SJ, Tsai WJ et al (2005b) PI3-kinase is essential for ADP-stimulated integrin alpha(IIb)beta3-mediated platelet calcium oscillation, implications for P2Y receptor pathways in integrin alpha(IIb)beta3-initiated signaling cross-talks. J Biomed Sci 12:937–948
Sun DS, King CC, Huang HS et al (2007) Antiplatelet autoantibodies elicited by dengue virus non-structural protein 1 cause thrombocytopenia and mortality in mice. J Thromb Haemost 5:2291–2299
Sun H, Wang X, Degen JL et al (2009) Reduced thrombin generation increases host susceptibility to group A streptococcal infection. Blood 113:1358–1364
Suzuki R, Kimura T, Kitaichi K et al (2002) Platelet factor 4 fragment induces histamine release from rat peritoneal mast cells. Peptides 23:1713–1717
Svensson Holm AC, Bengtsson T, Grenegard M et al (2008) Platelets stimulate airway smooth muscle cell proliferation through mechanisms involving 5-lipoxygenase and reactive oxygen species. Platelets 19:528–536
Svensson Holm AC, Bengtsson T, Grenegard M et al (2011) Platelet membranes induce airway smooth muscle cell proliferation. Platelets 22:43–53
Tamagawa-Mineoka R, Katoh N, Ueda E et al (2007) The role of platelets in leukocyte recruitment in chronic contact hypersensitivity induced by repeated elicitation. Am J Pathol 170:2019–2029
Totani L, Evangelista V (2010) Platelet-leukocyte interactions in cardiovascular disease and beyond. Arterioscler Thromb Vasc Biol 30:2357–2361
Vargaftig BB, Conard J, Samama M (1979) Blood coagulation and platelet function: introduction. Pharmacol Ther B 5:225–227
Varga-Szabo D, Pleines I, Nieswandt B (2008) Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol 28:403–412
Vieira-de-Abreu, A, Campbell, RA, Weyrich, AS et al. (2011) Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol
von Hundelshausen P, Weber C (2007) Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res 100:27–40
Watanabe O, Natori K, Tamari M et al (1999) Significantly elevated expression of PF4 (platelet factor 4) and eotaxin in the NOA mouse, a model for atopic dermatitis. J Hum Genet 44:173–176
Weyrich AS, Zimmerman GA (2004) Platelets: signaling cells in the immune continuum. Trends Immunol 25:489–495
Yokoyama S, Ikeda H, Haramaki N et al (2005) Platelet P-selectin plays an important role in arterial thrombogenesis by forming large stable platelet-leukocyte aggregates. J Am Coll Cardiol 45:1280–1286
Zander DM, Klinger M (2009) The blood platelets contribution to innate host defense—what they have learned from their big brothers. Biotechnol J 4:914–926
Acknowledgments
This work was supported by National Science Council of Taiwan ROC under Grant No. 98-2320-B-320-004 MY3, and Tzu-Chi University under Grant No. TCIRP 95002-02, TCIRP 96004-01, TCIRP 98001-01, TCRPP 99020 and TCRPP100003.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Huang, HS., Chang, HH. Platelets in Inflammation and Immune Modulations: Functions Beyond Hemostasis. Arch. Immunol. Ther. Exp. 60, 443–451 (2012). https://doi.org/10.1007/s00005-012-0193-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-012-0193-y