
Abstract
Electroless deposition of Pd is studied comparatively on glassy carbon (GCE) and spectral graphite (SGE) electrodes modified with electrodeposited poly(3,4-ethylene-dioxythiophene) (PEDOT) layers. Computed microtomography is used to explore the bulk structure of both types of carbon electrodes. It is found that SGE has high (27%) open porosity in contrast to GCE, the latter characterized by a dense and homogeneous structure with no porosity. Despite the structural difference of the electrodes, the intrinsic electrochemical activity of PEDOT is found to remain unaffected by the type of the underlying carbon substrate used for PEDOT electrochemical deposition. Electroless deposition of Pd is carried out on pre-reduced PEDOT-coated electrodes, in the absence of additional reductants in the electrolyte solution and expected to occur at the expense of PEDOT oxidation. It is found that at constant polymerization charge of PEDOT, the amount of metal deposited on PEDOT/SGE is several times higher in comparison to PEDOT/GCE. The high effectiveness for electroless metal deposition on PEDOT/SGE is explained by the involvement of pre-reduced hydrogen embedded in the highly porous structure of SGE that acts further as additional (apart from PEDOT) reductant contributing markedly to the amount of reduced Pd ions.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
It is known that the type and surface state of the electrode substrate affects the kinetics of electrodeposition. This effect is usually commented in terms of different types and numbers of “active sites” for nucleation on the electrode surface [1, 2]. The active sites may have different natures, for instance, physical defects (e.g., crystalline steps, edges, dislocations, grain boundaries, micropores, and nanopores) or chemical reaction sites (e.g., intentionally induced specific chemical bonding centers or, simply, chemical composition inhomogeneities). In the general case of electrodeposition, the proper nature of the active sites for nucleation is not known and the choice of the substrate is dictated mostly by practical reasons (appropriate potential window for given electrochemical reactions, good adhesion between depositing layer and substrate, opportunity for involvement in large-scale practical applications, etc.). The particular role of the substrate is rarely commented for processes involving bulky layers, once it does not induce specific properties in the electrodeposited layer, e.g., pre-designed chemical bonding or epitaxial growth.
The electrochemical formation of conducting polymer layers is intensively investigated over the last three decades, and different substrates are used in order to extract diverse information, e.g., ITO for spectroelectrochemical studies, Au-coated quartz crystals for microgravimetric studies, and mostly carbon electrodes for electrocatalytic or electroanalytic studies. It is known that the polymerization kinetics depends on a number of factors [3,4,5,6,7] (e.g., applied potential, concentration of monomer, type of solvent and available doping ions, electrochemical procedure for deposition). Despite the numerous studies, it is difficult to isolate the role of the substrate for the formation and properties of the obtained conducting polymer layers. In the present work, we address this point by showing results obtained with two types of carbon substrates—glassy carbon and spectral graphite electrodes. Glassy carbon electrodes (GCE) are very often involved in laboratory research as robust and inert (with respect to hydrogen- and oxygen-based reactions) electrodes. Spectrally clean graphite electrodes (SGE) although composed by carbon again are easily prepared, cheaper, and close to applications-oriented work. In the present study, these two types of electrodes are used for polymerization of 3,4-ethylenedioxythiphene (EDOT) and further for electroless deposition of Pd.
The electroless deposition process is carried out at the expense of the oxidation of the pre-reduced PEDOT-coated electrodes, in the absence of additional reductants in the electrolyte solution. It is known that this is a self-limited process depending on the available oxidation charge of the polymer that can be consumed for metal ion reduction [8,9,10]. This process is effective for the deposition of noble metals with equilibrium potentials far from the potential of the main redox transition of conducting polymer materials and was studied for electrosynthesized thin layers of polyaniline [11,12,13,14,15,16], polypyrrole [17,18,19], polythiophene [20,21,22,23,24,25,26,27,28], and polycarbazole [29, 30]. The prevailing number of these investigations [11, 14,15,16,17, 19,20,21,22, 24,25,26,27] is devoted to electroless deposition of Pd due to the involvement of this metal in a great variety of electrocatalytic and sensing applications [31].
Particularly, electroless deposition of Pd on PEDOT-modified electrodes is studied in several papers [20,21,22, 24,25,26,27] using polycrystalline Pt-carrying [20], ITO-carrying [22], or GCE-carrying [21, 24,25,26] substrates. No specific role of the electrode-substrate was assigned in any of these investigations. In a recent study [27], we have shown the possibility to obtain a high-density Pd nanocatalyst by using SGE as a carrying substrate for PEDOT electrodeposition. It was shown that a large amount of Pd becomes deposited and, besides, densely distributed nanoparticles with sizes in the few nanometer range cover homogeneously the entire polymer surface. The focus was set on the role of the doping ions used in the course of PEDOT synthesis for the process of Pd electroless deposition. The amount of deposited Pd exceeded largely the amount of Pd obtained using GCE under somewhat different conditions [26]. In order to elucidate these findings, here we extend our studies by revealing the role of the electrode substrate for the electroless deposition of Pd on PEDOT by using GCE and SGE under otherwise identical conditions.
Experimental
The electrochemical experiments were carried out in three electrode cells by using a platinum plate counter electrode and a mercury/mercury sulfate reference electrode (MSE) (Hg/Hg2SO4/0.5 mol cm−3 K2SO4). All potentials in the text are referred to as MSE, EMSE = 0.66 V vs. SHE. Spectral graphite rods (Ringsdorff-Werke GmbH) with diameter 0.3 cm and exposed front surface area, S = 0.071 cm2, were used as working electrodes (denoted as SGE). The exposed graphite surface was pre-treated with emery cloth (Buehler P5000) and rinsed with water before use. Alternatively, a homemade glassy carbon electrode (GCE) consisting of a glassy carbon disk with the same diameter (0.3 cm) sealed in epoxy resin and the same geometrical surface area was used. The exposed glassy carbon surface was pre-treated with emery cloth (Buehler P5000) and rinsed with water before use.
All electrolyte solutions were prepared with deionized water (ρ = 18.2 MΩ cm) obtained from the Millipore Synergy™ Ultrapure Water Purification System. Before starting electrochemical measurements, the electrolytes were de-aerated with argon for at least 20 min and kept under argon atmosphere thereafter. A computer-driven potentiostat/galvanostat (Autolab PGSTAT12, Eco Chemie, The Netherlands) was used for the electrochemical measurements.
Electrochemical polymerization of EDOT was carried out in aqueous solution consisting of 0.01 mol cm−3 EDOT and 0.034 mol cm−3 of sodium polystyrene sulfonate at constant anodic potential, Epoly = 0.36 V. After synthesis, the polymer-coated electrodes were transferred in supporting electrolyte (0.1 mol cm−3 LiClO4) for evaluation of their electroactivity. In the same electrolyte, the polymer layers were electrochemically reduced by keeping the working electrode at constant potential, E = − 1.4 V for 1200 s.
Electroless deposition of palladium was carried out by dipping the pre-reduced PEDOT layers in an aqueous solution of 0.002 mol cm−3 PdSO4 and 0.5 mol cm−3 H2SO4. The equilibrium potential of Pd2+/Pd0 in this solution is E0(Pd2+/Pd0) = 0.195 V. The electroless deposition process was monitored through the change in the open circuit potential (OCP) of the PEDOT-coated electrode. The process was stopped at EOCP = − 0.05 V. After this step, some of the specimens were used for SEM observation. Identical specimens were prepared and used to determine the mass of the deposited Pd by anodic voltammetric stripping completed in aqueous solution of 1.15 mol cm−3 hydrochloric acid.
A microscopic observation of the Pd/PEDOT-coated specimens was completed by means of a JSM-7800F (JEOL) device at JEOL Europe BV or JSM 6390 (JEOL) with INCA Oxford EDX analysis at the Institute of Physical Chemistry, Sofia. Imaging by the JSM-7800F (JEOL) device was accomplished at low accelerating voltage in beam deceleration mode.
Computed tomography studies of both types of electrodes were carried out in order to reveal their bulk structure. A SKYSCAN 1272 (Bruker) microtomograph was used at 50-kV voltage and 200-μA current of the X-ray tube. The size of the 3D pixel was 0.75 μm.
Results and discussion
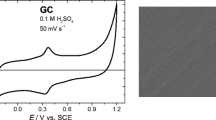
Figure 1 shows the voltammetric curves of both electrodes measured in 0.1 M LiClO4. The large difference in the currents (GCE currents are multiplied by a factor of 50 in the plot) shows that a large surface roughness should be expected for the SGE electrode. Furthermore, there is also a difference with respect to the hydrogen reduction reaction with SGE supporting easily hydrogen evolution and showing a reversible behavior. The porosity of both types of electrodes was studied by 3D computed tomography (CT) (Fig. 2). According to the CT analysis of the image histogram, the porosity of SGE is 27.5%, whereas 3D analysis by means of image segmentation results in а porosity of 21.7% with only 0.2% closed porosity. The mean size of the pores is 13 ± 5 μm, and the mean size between them is 6 ± 3 μm. In contrast, GCE has a dense, homogeneous, and isotropic structure with no porosity. There are only few defects with negligible total volume fraction amounting to 0.0013%.

Cyclic voltammetric curves measured at SGE (solid line) and GCE (dashed line) in supporting electrolyte (0.1 mol cm−3 LiClO4). (The current measured at GCE is multiplied by 50)

Cross sections in the bulk of SGE (a) and GCE (b) obtained by computed microtomography
The polymerization of EDOT on both types of electrodes reveals different kinetics of PEDOT formation especially in the initial stage (Fig. 3). A delayed initiation of the polymerization is observed on GCE whereas a diffusion-limited process seems to operate from the very beginning on SGE. This effect should originate from the much higher effective surface area of SGE. SEM examination of both types of PEDOT-coated electrodes has shown complete coverage of the surface by the polymer layer in the two cases with a rougher polymer structure observed in the PEDOT/SGE case.

Polymerization transients obtained at E = 0.36 V vs. MSE in aqueous solution of 0.01 mol cm−3 EDOT and 0.034 mol cm−3 NaPSS at SGE (solid line) and GCE (dashed line). Polymerization charges: Qpoly = 5.6 mC
A further question arising in our studies was whether the difference in the kinetics of polymerization results also in a difference in the extent of oxidation of PEDOT obtained on both types of electrodes. To answer this question, a series of polymerization transients was obtained at both SGE and GCE using different polymerization times. After synthesis, all PEDOT layers were characterized by cyclic voltammetry in the supporting electrolyte. An illustrative measurement is shown in Fig. 4. The comparison between both types of electrodes reveals a seemingly larger redox activity for PEDOT/SGE in the pseudocapacitive potential region but also a much larger permeability for hydrogen and promotion of the hydrogen evolution reaction. Bearing this in mind, the effective redox charge of PEDOT deposited on SGE was obtained by subtracting the capacitive charge measured on the bare electrode from the charge measured on the PEDOT-coated electrodes. The effective PEDOT redox charge thus obtained is further denoted by Q*redox. A summary of the data for Q*redox corresponding to PEDOT layers with different polymerization charges, Qpoly, is shown in Fig. 5. The comparison of results obtained for PEDOT/GCE and PEDOT/SGE shows that although the two carbon electrodes differ in their physical structure and promote polymerization in markedly different ways, the Q*redox to Qpoly ratio remains the same. The latter should be considered as an indication for the preserved extent of oxidation and thus identical molecular structure of the polymer chains.

Cyclic voltammetric curves measured in 0.1 mol cm−3 LiClO4 on bare (gray lines) and PEDOT-coated (black lines) GCE and SGE. (Qpoly = 5.6 mC)

Dependence of Q*redox on Qpoly for PEDOT layers obtained at GCE (filled circle) and SGE (filled square)
Electroless deposition of Pd was further studied at pre-reduced PEDOT/PSS layers deposited on SGE and GCE. Figure 6 shows OCP transients measured in the course of the electroless process at PEDOT-coated GCE and SGE with equal polymerization charges of the deposited PEDOT. For comparison, the OCP transient measured in the absence of palladium ions in the solution is also shown. For both types of electrodes, an increase of OCP to more positive potentials is observed in the Pd plating solution which indicates the deposition of the metal phase. Nevertheless, a substantial difference is found in the OCP kinetics for both electrode substrates. The process is markedly delayed when using the highly porous SGE. The trend observed in Fig. 6 can be further followed in the series of OCP transients measured in the course of electroless Pd deposition at pre-reduced PEDOT layers with different polymerization charges (Fig. 7). A steep OCP increase for GCE (Fig. 7a) in contrast to a much slower and gradual OCP change for SGE (Fig. 7b) is observed for all transients. This could be an indication of either different morphology of the PEDOT layers at the supramolecular level originating from the very different rates of polymerization or a special role of the SGE structure involving slow diffusion processes inside the micropores.

OCP transients obtained in the course of Pd electroless deposition on PEDOT/SGE (full black line) and PEDOT/GCE (dashed black line) (Qpoly = 5.6 mC). Reference OCP curves obtained at PEDOT/SGE and PEDOT/GCE in the absence of Pd2+ ions (full gray lines)

OCP transients obtained in the course of Pd electroless deposition on PEDOT/GCE (a) and PEDOT/SGE (b) at different polymerization charges of the polymer coatings denoted by figures in millicoulombs. The gray line in b shows OCP transient obtained at bare SGE
Data for the amount of deposited Pd were obtained by dissolution of the metal phase in HCl. Fig. 8 presents dissolution curves obtained after Pd deposition on PEDOT/SGE and PEDOT/GCE. The currents measured at GCE (Fig. 8a) are multiplied by a factor of 10 in order to make them visible in the scale of the plot. It is obvious that the amount of deposited Pd is much larger on SGE in comparison to GCE. Thus, despite the results presented in Fig. 5 and the indication for the identical redox behavior of PEDOT, deposited on both SGE and GCE, there is a different amount of oxidation charge spent for the electroless metal deposition process depending on the substrate. A series of dissolution curves obtained after deposition of Pd on PEDOT/SGE at different polymerization charges of PEDOT (Fig. 8b) shows the expected dependence of the amount of deposited metal on the polymerization (and thus redox) charge, but even for very thin PEDOT layers large amounts of Pd are obtained with metal dissolution charges exceeding the polymerization charge of PEDOT. To clarify this result, a further experiment was carried out by using bare SGE and completing the same sequence of steps as with PEDOT-coated electrodes (i.e., reduction at constant potential, subsequently registering the OCP curve in a PdSO4-containing solution and finally cyclic voltammetry in HCl solution).The obtained OCP transient (gray line in Fig. 7) shows the already observed holdup (in the − 0.35 to − 0.2 V potential window) and thus indicates to an electrochemical process taking place in this potential interval. The voltammetric dissolution experiment gives evidence for deposition of metallic phase (gray line in Fig. 8b). Thus, it becomes obvious that the porous SGE supports metal electroless deposition even in the absence of PEDOT very probably at the expense of oxidation of previously reduced hydrogen atoms embedded in the porous structure. (The same reference experiment was attempted also with GCE, but the long-term reduction at − 1.4 V resulted in irreversible changes of the electrode and activation in the hydrogen reduction region without the possibility to recuperate its initial state.)

Dissolution curves of palladium deposited on a PEDOT/SGE and PEDOT/GCE at Qpoly = 5.6 mC and b PEDOT/SGE for different Qpoly denoted by figures in millicoulombs. The gray line in b shows dissolution of Pd when using bare SGE
Finally, SEM was used to reveal the characteristics of the metal particles deposited on PEDOT/GCE and PEDOT/SGE (Figs. 9 and 10). SEM on GCE (Fig. 9a) did not allow resolving individual particles although EDX analysis gives evidence for the presence of Pd (Fig. 9b). Pd/PEDOT/SGE samples were examined with high-resolution FE-SEM (Fig. 10). Pd particles are clearly visualized by using very low accelerating voltages. They are homogeneously distributed over the entire polymer surface and have sizes in the several nanometer range (3 to 8 nm).

SEM a and EDX analysis b at Pd/PEDOT/GCE

High-resolution FE-SEM at Pd/PEDOT/SGE at different magnifications a 100,000× and b 400,000×
Conclusions
The present work focuses on the specific intrinsic properties of the carbon electrode substrate that interfere with processes occurring at conducting polymer-coated electrodes. Despite the fact that the intrinsic electrochemical activity of PEDOT seems unaffected by the type of the underlying carbon substrate, it is demonstrated that electroless metal deposition proceeds in a completely different way on both electrodes. The high effectiveness for electroless metal deposition on PEDOT-coated SGE is seemingly due to the involvement of the porous structure of this substrate in the process by providing additional species that are easily oxidized at the expense of metal ion reduction. In fact, the overlaying polymer layer which presents also a fine porous structure seems to act as a reaction medium that provides the opportunity for redox interaction between entrapped hydrogen and metal ions. This specific situation results in very high metal deposition yields and highly homogeneous distribution of the metal particles that was not achieved before. The permeability of the polymer layer for both ionic species should play a crucial role for the amount and spatial distribution of the metal particles. That is why factors such as thickness (as shown here) and type of the doping ions (as demonstrated in [27]) are expected to affect markedly the metal electroless deposition.
Finally, the present study demonstrates the specific situation arising from the combination of two conducting porous media with complementary roles (i.e., providing additional oxidizing species and inhibiting fast diffusion of species in the electrolyte solution) that results further in most suitable conditions for a highly effective metal electroless deposition process.
References
Milchev A (2002) Electrocrystallization. Fundamentals of nucleation and growth. Kluwer, Boston
Scholz F (2011) Active sites of heterogeneous nucleation understood as chemical reaction sites. Electrochem Commun 13(9):932–933. https://doi.org/10.1016/j.elecom.2011.06.003
Choi SJ, Park SM (2002) Electrochemistry of conductive polymers. XXVI Effects of Electrolytes and Growth Methods on Polyaniline Morphology. J Electrochem Soc 149:E26–E34
Biallozor S, Kupniewska A (2005) Conducting polymers electrodeposited on active metals. Synth Met 155(3):443–449. https://doi.org/10.1016/j.synthmet.2005.09.002
Heinze J, Frontana-Uribe BA, Ludwigs S (2010) Electrochemistry of conducting polymers—persistent models and new concepts. Chem Rev 110(8):4724–4771. https://doi.org/10.1021/cr900226k
Inzelt G (2012) Conducting polymers: a new era in electrochemistry. In: Scholz F (ed) Monographs in electrochemistry. Springer, Heidelberg. https://doi.org/10.1007/978-3-642-27621-7
Inzelt G (2017) Recent advances in the field of conducting polymers. J Solid State Electrochem 21(7):1965–1975. https://doi.org/10.1007/s10008-017-3611-6
Tsakova V (2008) How to affect number, size and location of metal particles deposited in conducting polymer layers. J Solid State Electrochem 12(11):1421–1434. https://doi.org/10.1007/s10008-007-0494-y
Tsakova V (2010) In: Eftekhari A (ed) Nanostructured conductive polymers. Chichester, John Wiley & Sons
Kondratiev V, Malev V, Eliseeva S (2016) Composite electrode materials based on conducting polymers loaded with metal nanostructures. Russ Chem Rev 85:14–37
Abrantes LM, Correia JP (1995) Polymer films containing metal particles—nobel metals in polyaniline. Mater Sci Forum 191:235–240. https://doi.org/10.4028/www.scientific.net/MSF.191.235
Zang AQ, Cui CQ, Lee JY, Loh FC (1995) Interactions between polyaniline and silver cations. J Electrochem Soc 142(4):1097–1104. https://doi.org/10.1149/1.2044136
Ivanov S, Tsakova V (2005) Electroless versus electrodriven deposition of silver crystals in polyaniline: role of silver anion complexes. Electrochim Acta 50(28):5616–5623. https://doi.org/10.1016/j.electacta.2005.03.040
Mourato A, Wong SM, Siegenthaler H, Abrantes LM (2006) Polyaniline films containing palladium microparticles for electrocatalytic purpose. J Solid State Electrochem 10(3):140–147. https://doi.org/10.1007/s10008-005-0053-3
Mourato A, Viana AS, Correia JP, Siegenthaler H, Abrantes LM (2004) Polyaniline films containing electrolessly precipitated palladium. ElectrochimActa 49(14):2249–2257. https://doi.org/10.1016/j.electacta.2004.01.006
Lyutov V, Tsakova V (2011) Palladium-modified polysulfonic acid-doped polyaniline layers for hydrazine oxidation in neutral solutions. J Electroanal Chem 661(1):186–191. https://doi.org/10.1016/j.jelechem.2011.07.043
Lim VWL, Kang ET, Neoh KG (2001) Electroless plating of palladium and copper on polypyrrole films. Synth Met 123(1):107–115. https://doi.org/10.1016/S0379-6779(00)00592-0
Song FY, Shiu KK (2001) Preconcentration and electroanalysis of silver species at polypyrrole film modified glassy carbon electrodes. J Electronal Chem 498(1-2):161–170. https://doi.org/10.1016/S0022-0728(00)00360-0
Mourato A, Cabrita JF, Ferrari AM, Botelho do Rego AM, Abrantes LM (2010) Electrocatalytic activity of polypyrrole films incorporating palladium particles. Catal Today 158(1-2):2–11. https://doi.org/10.1016/j.cattod.2010.07.004
Ilieva M, Tsakova V, Erfurth W (2006) Electrochemical formation of bi-metal (copper-palladium) electrocatalyst supported on poly-3,4-ethylenedioxythiophene. ElectrochimActa 52(3):816–824. https://doi.org/10.1016/j.electacta.2006.06.015
Eliseeva SN, Malev VV, Kondratiev VV (2009) Electrochemical properties of composite films based on poly-3,4-ethylenedioxythiophene with inclusions of metallic palladium. Russ J Electrochem 45(9):1045–1051. https://doi.org/10.1134/S1023193509090109
Eliseeva S, Ubyivovk E, Bondarenko A, Vyvenko O, Kondratiev V (2010) Synthesis and structure of poly-3,4-ethylenedioxythiophene film with the inclusions of palladium nanoparticles. Russ J Gen Chem 80:1143–1148
Kondratiev V, Pogulaichenko N, Tolstopjatova E, Malev V (2011) Hydrogen peroxide electroreduction on composite PEDOT films with included gold nanoparticles. J Solid State Electrochem 15(11-12):2383–2393. https://doi.org/10.1007/s10008-011-1494-5
Kondratiev V, Babkova T, Eliseeva S (2012) Structure and electrochemical properties of composite films based on poly-3, 4-ethylenedioxythiophene with metallic palladium inclusions. Russ J Electrochem 48:205–211
Kondratiev V, Babkova T, Tolstopjatova E (2013) PEDOT-supported Pd nanoparticles as a catalyst for hydrazine oxidation. J Solid State Electrochem 17:1621–1630
Ilieva M, Nakova A, Tsakova V (2016) Pd-modified PEDOT layers obtained through electroless metal deposition—electrooxidation of glycerol. J Solid State Electrochem 20:3015–3023
Nakova A, Ilieva M, Boiadjieva-Scherzer T, Tsakova V (2017) High-density Pd nanoparticles distribution on PEDOT obtained through electroless metal deposition on pre-reduced polymer layers. Electrochim Acta 253:128–133
Karabozhikova V, Tsakova V (2017) Electroless deposition of silver on poly(3,4-ethylenedioxythiophene):role of the organic ions used in the course of electrochemical synthesis. Chem Papers 71(2):339–346. https://doi.org/10.1007/s11696-016-0076-5
Fedorczyk A, Ratajczak J, Czerwinski A, Skompska M (2014) Selective deposition of gold nanoparticles on the top or inside a thin conducting polymer film, by combination of electroless deposition and electrochemical reduction. Electrochim Acta 122:267–274. https://doi.org/10.1016/j.electacta.2013.08.035
Fedorczyk A, Ratajczak J, Kuzmych O, Skompska M (2015) Kinetic studies of catalytic reduction of 4-nitrophenol with NaBH4 by means of Au nanoparticles dispersed in a conducting polymer matrix. J Solid State Electrochem 19(9):2849–2858. https://doi.org/10.1007/s10008-015-2933-5
Chen A, Ostrom C (2015) Palladium-based nanomaterials: synthesis and electrochemical applications. Chem Rev 115(21):11999–12044. https://doi.org/10.1021/acs.chemrev.5b00324
Acknowledgements
Special thanks to JEOL Europe BV (Mr. K. Toshiyuki) for providing high-resolution SEM images and to Dr. D. Tachev (Institute of Physical Chemistry, Sofia) for computed microtomographic analysis. Financial support of CEST, Kompetenzzentrum für elektrochemische Oberflächentechnologie GmbH, Wiener Neustadt, Austria (PhD grant for A.N.), is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nakova, A., Ilieva, M., Boiadjieva-Scherzer, T. et al. Electroless deposition of palladium nanoparticles on poly(3,4-ethylene-dioxythiophene)—role of the electrode substrate. J Solid State Electrochem 22, 1901–1908 (2018). https://doi.org/10.1007/s10008-018-3900-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-018-3900-8