
Abstract
Six organometallic compounds coming from a basic Mo-based complex were analyzed from the perspective of the dual descriptor in order to detect subtle influences that a substituent group could exert on the reactive core at a long range. Since the aforementioned complexes are open-shell systems, the used operational formula for the dual descriptor is that one defined for those aforementioned systems, which was then compared with spin density. In addition, dual descriptor was decomposed into two terms, each of which was also applied on every molecular system. The obtained results indicated that components of dual descriptor could become more useful than the operational formula of dual descriptor because differences exerted by the substituents at the para position were better detected by components of dual descriptor rather than the dual descriptor by itself.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The importance of finding new molybdenum–Oxo complexes
There has been a growing interest to find alternative fuels for the replacement of oil, mainly influenced by climate change concerns. Several efforts are being carried out in order to decrease carbon dioxide emissions into the atmosphere coming from the massive use of sources of energy based on carbon such as fossil fuels [1,2,3,4]. Among sources of renewable energy, molecular hydrogen [5,6,7,8,9,10,11] has been considered a possible candidate to be used as a sustainable fuel, thus leading to an effervescent search at present in achieving this goal from different materials, mainly because hydrogen is characterized by its almost omnipresence on Earth, since water is constituted from this element [12].
In particular, the chemical conversion H2O(l)→ H2(g)+ 1/2O2 (g) has an enthalpy of 285.8 kJ mol− 1 (= 68.32 kcal mol− 1= 2.96 eV at t = 25 °C and P = 1 atm) [13, 14]. The high energetic cost of splitting water into its constituent parts, dihydrogen and dioxygen, can be carried out using electrochemical means. However, the high potential for this process (\(\thicksim \)− 1.23 Volts at pH = 0; t = 25°C and P = 1 atm) [14] is one of the key factors in achieving this goal, so that finding new substances that are able to easily release molecular hydrogen is desirable in the short term. The difficulty thus lies in the fact that the conversion from water into molecular hydrogen and oxygen implies a significant cost in energy [6, 15]. Consequently, substances should be catalysts easily synthesizable from one or more abundant elements on Earth with the aim of decreasing its cost of production.
On the other hand, Mo is a transition metal that is considered by specialists to be appropriate for applications in leading-edge technology and in the metallurgical and chemical industries. Several uses of Mo have been detected, starting from its use as a metal for alloys and also its used as a metal by itself in structural mechanical pieces. This metal is also involved in high-technology compounds and in making some substances for specific uses, for instance lubricants, precursors for some types of synthesis and catalysts; the development of Mo-based catalysts to yield other products is a good example of capabilities offered by this metal and recent research gathered from scientific literature indicates a promising future for Mo in H2 production from water [16, 17].
Accidentally, during certain investigations performed about the second-row transition-metal chemistry employing the ligand 2,6-bis[1,1-bis(2-pyridil)ethyl]-pyridine, simply PY5Me2 [18], Karunadasa et al. [19] detected an organometallic Mo-based compound, whose molecular formula is [(PY5Me2)MoO(CF3SO3)]+ and which may exchange the (CF3SO3)− ligand by one water molecule. Once the water molecule is coordinated onto the metal center, the release of molecular hydrogen becomes easier than release it from water molecule embedded in liquid water due to several steps involved during the entire process [19].
One of those steps involves the transference of a hydrogen atom from the bound water toward the molybdenum atom followed by the molecular hydrogen release (MHR) according to an environmental friendly catalytic cycle that involves an organometallic-basic structure being responsible of the MHR: the 2,6-bis[1,1-bis(2-pyridyl)ethyl]-pyridine oxo-molybdenum complex monocation and its possible derivatized structures arising when a substituent group is attached at specific positions of pyridine rings so favoring or unfavoring the MHR process.
Karunadasa et al. explored the use of fluorine and methyl solely as substituent groups at all p a r a positions of pyridine rings [20]. The systematic analysis would shed light on how the substituent group influences the reactivity of the Mo-based complex and in consequence on the dihydrogen production.
Since there are multiple possibilities of substitution, a good starting point implies to study the most subtle influence that can be exerted by just one substituent group localized at the farthest position with respect to the reactive core (Mo and O), thus meaning the p a r a position at the axial pyridine ring where a general substituent group is represented by the capital letter G, as depicted by Fig. 1. Such a model suggests that the substituent group can exert an influence on the reaction mechanism, specifically on the kinetics and/or the thermodynamics of the process [20]. According to the latter, before performing a synthesis of a new compound based on this metal, a computational study should allow to understand the importance of including an electron-withdrawing or electron-donating substituent group and for that reason, previously to any quantification of the substituent effect, a visual analysis of this effect through the use of one type of scalar field could give us an insight about which regions of compounds like these are more affected by the respective substituent group. The first scalar field that could help us to understand the influence of the substituent groups is the spin density defined as the arithmetic difference between the alpha electron and beta electron densities [21], \(\rho _{{~}_{\mathrm {S}}}(\textbf {r})=\rho _{{~}_{\alpha }}(\textbf {r})-\rho _{{~}_{\beta }}(\textbf {r})\), but if it does not reveal the electron-withdrawing or electron-donating nature of substituent groups under study, a complementary tool called dual descriptor (DD) can be used. A brief definition of DD will be explained in the next subsection.

Molecular hydrogen release reaction: [(p-G-Py)Mo(H)(OH)]+ (a q)→ H2 (g) ↑ +[(p-G-Py)MoO]+ (a q). The organometallic product of this reaction model is of our interest in the present work
Dual descriptor
There are many scalar fields to estimate reactivity on molecules from quantum chemical calculations. In particular, from the framework of the density functional theory [22, 23] (DFT), chemists can find a plethora of reactivity descriptors [24] so giving rise to the conceptual DFT [21, 25] under the hypothesis that the total energy E of a system depends upon the total number of electrons N and the external potential υ(r), so that the application of successive ordinary and functional derivatives yields a wide range of reactivity descriptors, which are divided into three types: global, local, and non-local, all of them based on two essential physical observable quantities: energy, electronic density, or both. Local reactivity descriptors are scalar fields, thus meaning they depend on the vector position r in space surrounding a molecule.
Among the endless types of local reactivity descriptors that are possible to conceive, there is one defined at a third order [26] that is becoming more and more popular among some researchers interested in measuring local reactivities; this alluded descriptor is the dual descriptor [27,28,29] that has demonstrated to be a more robust tool than Fukui functions [30,31,32]. However, for open-shell systems, an operational formula has been proposed in terms of electron densities of frontier molecular orbitals for each spin symmetry in agreement with the strict definition of dual descriptor where N S (the spin number) and B(r) (external magnetic field in the z direction) must be kept constant:
The use of Eq. 1 implies to assume that local reactivity of a molecule is driven by frontier molecular orbitals, which is a good first approach to get visual insight into a family of molecules.
However, as the reader can see, Eq. 1 being an average could turn out to a loss of information, thus meaning that scalar fields would look very similar among them in spite of considering different substituent groups. As an alternative to the former operational formula of dual descriptor, a new operational formula was proposed [33] to be applied on open-shell systems, which is given by Eq. 2:
where the subscript Δ N S < 0 indicates that this operational formula is pertinent to use when considering a local reactivity that leads to decrease the spin number [33] (on the contrary of the operational formula given by Eq. 1 where the local reactivity is described by taking into account that the spin number is kept constant). Rigorously speaking, the operational formula given by Eq. 2 is not constrained to keep N S constant and therefore it can be considered as a component of Eq. 1, meaning that Eq. 2 is contained in Eq. 1 and then a comparison between these two operational formulae could be of interest in order to elucidate which one reveals more differences among substituent groups.
Furthermore, a counterpart of Eq. 2 is inferred as follows:
According to the latter, the Eq. 1 can be written as the sum of two components where each does not constrain the spin number to be kept constant:
As the f (2)(r) within the spin-polarized density functional theory [34,35,36] requires a constant spin number, the expression given by Eq. 4 allows us to analyze the working equation of dual descriptor by means of its components, which are free of such a restriction over the spin number so that we would be able to detect some influences exerted by substituent groups when the total expression defined by Eq. 1 is incapable of detecting expected differences due to the electron-donating and electron-withdrawing nature of those groups.
Evidently, the results expressed here are of a purely preliminary nature, and they will be compared in following articles with those results obtained by means of the operational formula written in terms of total electron densities [27,28,29]. In addition to this, the concept of so-called “state-specific dual descriptor (SSDD)” proposed by Tognetti et al. [37] will also be employed in the short term in order to assess how much the description of local reactivity improves when electronic densities of excited states are included in comparison with the rough (3) and (4) expressions that involve electronic densities of LUMO; this last pending task is relevant to be performed because according to those authors, the use of densities of the electronic excited states demonstrated its usefulness in those cases where attempts to explain local reactivity based on frontier molecular orbitals fails.
Computational methods
The BP86 (GGA-type) [38, 39] exchange-correlation functional along with the MWB28 [40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64] pseudopotential for the metal center and the standard Gaussian-type orbital 6-31+G(d,p) [65,66,67,68,69,70,71,72,73,74,75,76] basis set for non-metal atoms were employed to optimize the geometries of structures [77, 78]. This procedure was performed in the presence of an implicit solvent model based on the self-consistent reaction field method [79] through the integral equation formalism polarizable continuum model (IEFPCM) [80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100] with radii and non-electrostatic terms for Truhlar and coworkers’ SMD solvation model [101] using water as a solvent. Harmonic vibrational frequencies analysis was carried out at the BP86/MWB28/6-31+G(d,p) level of theory to confirm the character of minimum energy for all geometrically optimized monocationic structures. Single-point calculations were performed by means of the use of the M06L (meta-GGA-type) [102, 103] exchange-correlation functional along with Def2-TZVP [104] basis set complemented with density fitting basis sets [105, 106], this level of theory was applied on the studied structures with the aim of obtaining 3D pictures of spin density and dual descriptor. The organometallic [(p-G-Py)MoO]+ structures were generated through replacement of the G substituent group at the p a r a position in axial pyridine rings of Mo-based complexes where G ∈{NH2,CH3,H,F,CF3,CN}.
Results and discussion
As Fig. 1 indicates, the reactive core of these mono cations [(p-G-Py)MoO]+ is defined by the molybdenum and oxygen atoms, which concentrates the reactivity of these Mo–based complexes. The latter means that any influence of substituent group should be expressed surrounding those atoms through the use a proper local reactivity descriptor. Spin density, dual descriptor for open–shell systems and its components were tested in the present work.
Spin density, \(\protect \rho _{_{\mathrm {S}}}(\textbf {r})\)
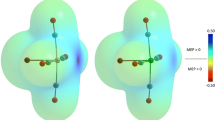
The spin density, according to Fig. 2, indicates a predominance of alpha electrons over beta electrons, however no more marked distinctions can be detected when \(\rho _{{~}_{\mathrm {S}}}(\textbf {r})\) is applied on this family of Mo-based complexes, thus indicating that the spin density is not a reliable parameter by itself to yield a 3D map of local reactivity able to reveal at least differences between electron-donating and electron-withdrawing substituent groups. Maybe the amino substituent is the only one able to concentrate the predominance of one type of electron surrounding the reactive core; however, it also shows a subtle presence of those electrons on the axial pyridine ring. Since the molecular hydrogen release occurs from the reactive core, local reactivity is expected to be focused on the Mo and O atoms. For this purposes, dual descriptor was applied to reveal more information about local reactivity, as will be described in the next subsections.

Spin density ρ: Isosurfaces are depicted at values of 0.001 a.u. Positive values of the spin density are indicated by blue-colored lobes, thus revealing a predominance of alpha electrons there; negative values of the spin density are indicated by green-colored lobes, thus revealing a predominance of beta electrons there
Dual descriptor, f (2)(r)
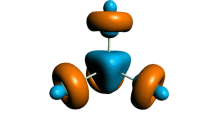
As displayed by Fig. 3, dual descriptor reveals some subtle differences between electron-donating and electron-withdrawing substituent groups (CF3 and CN). In the first place, contrary to spin density, dual descriptor reveals a reactivity that surrounds the reactive core noticeably. Secondly, electron-withdrawing substituent groups tend to yield 3D maps of dual descriptor that are localized around the reactive core and have no contributions from axial and equatorial pyridine rings. Meanwhile, electron-donating substituent groups (NH2, CH3, H and F) tend to yield 3D maps of dual descriptor that are less localized and does have contributions from the equatorial pyridine rings and subtle contributions from the axial pyridine ring.

Dual descriptor: \(f^{(2)}(\textbf {r})\), total operational formula given by Eq. 1. Isosurfaces are depicted at values of 0.001 a.u. Positive values of the DD are indicated by dark-colored lobes, thus revealing a preference to undergo nucleophilic attacks there; negative values of the DD are indicated by white-colored lobes, thus revealing a preference to undergo electrophilic attacks in such a region
Dual descriptor: \(f^{(2)}_{{~}_{\varDelta N_{S} < 0}}(\textbf {r})\) component
Figure 4 shows 3D maps of the \(f^{(2)}_{{~}_{\varDelta N_{S} <0}}(\textbf {r})\) component of dual descriptor. It allows us to scrutinize in more detail some aspects of local reactivity revealed by dual descriptor and it indicates that the cyanide substituent decreases totally the reactivity of the reactive core. Since all of these mono-cation complexes are one of the products given by the MHR reaction as Fig. 1 depicts, if the local reactivity of the cyanided complex is zero, then we expect that the reaction involving such a type of complex presents the most exoenergetic change for this process.

Dual descriptor: \(f^{(2)}_{{~}_{\varDelta N_{S} <0}}(\textbf {r})\) component given by Eq. 2. Isosurfaces are depicted at values of 0.001 a.u. Positive values of the DD are indicated by dark-colored lobes, thus revealing a preference to undergo nucleophilic attacks there; negative values of the DD are indicated by white-colored lobes, thus revealing a preference to undergo electrophilic attacks in such a region
Dual descriptor: \(f^{(2)}_{{~}_{\varDelta N_{S} > 0}}(\textbf {r})\) component
This component of the dual descriptor reveals that the Mo-based complex with an amino substituent group yields the most locally reactive complex cation because, as observed in Fig. 5, such a complex cation exhibits a \(f^{(2)}_{{~}_{\varDelta N_{S} >0}}(\textbf {r})\) scalar field too much spread over the reactive core in comparison with the remaining complexes studied here, so that we could expect that the released energy during this process should be the least in comparison with the involved overall energy of the remaining processes of molecular hydrogen release that includes the other Mo-based complexes analyzed in the present work, according to these statements, a manuscript in preparation will be submitted soon where the energy involved in this MHR process will be analyzed in terms of the influence exerted by the substituent groups, thus the latter being a support for our hypothesis: the less reactive is the product, the more exoenergetic the MHR process is.

Dual descriptor: \(f^{(2)}_{{~}_{\varDelta N_{S} >0}}(\textbf {r})\) component given by Eq. 3. Isosurfaces are depicted at values of 0.001 a.u. Positive values of the DD are indicated by dark-colored lobes, thus revealing a preference to undergo nucleophilic attacks there; negative values of the DD are indicated by white-colored lobes, thus revealing a preference to undergo electrophilic attacks in such a region
Conclusions
-
Contrary to what one could expect, the influence generated by the presence of just one substituent group localized at the farthest position from the reactive core can be detected by means of the use of the dual descriptor and its components. The use of the spin density was not able to detect noticeable differences among the substituent groups.
-
Nevertheless, we have to take into account that we have used the approximated operational formula of dual descriptor that uses the electron densities of frontier molecular orbitals for open-shell systems. The same analysis should be performed in a short term involving total electron densities, as was done for closed-shell systems [107]. State-specific dual descriptor (SSDD) will also be applied on these systems [37].
-
Although the operational formula given by Eq. 1 of dual descriptor for open-shell systems, f (2)(r), is most abided to the strict definition required by the conceptual spin-polarized density functional theory, the use of its components \(f^{(2)}_{{~}_{\varDelta N_{S} <0}}(\textbf {r})\) and \(f^{(2)}_{{~}_{\varDelta N_{S} >0}}(\textbf {r})\), defined by Eqs. 2 and 3 respectively, has demonstrated to visually reveal in a better manner the effect exerted by a substituent group on the reactive core of the Mo-complexes studied here.
References
Mastral AM, Callén MS, García T (2000) Energy Fuels 14(2):275–281
McKellar JM, Bergerson JA, MacLean HL (2010) Energy Fuels 24(3):1687–1695
Hashi M, Tezel FH, Thibault J (2010) Energy Fuels 24(9):4628–4637
Eide I, Neverdal G (2014) Energy Fuels 28(4):2617–2623
Winkler M, Hemschemeier A, Gotor C, Melis A, Happe T (2002) Int J Hydrog Energy 27:1431–1439. BIOHYDROGEN
Mazloomi K, Sulaiman NB, Moayedi H (2012) Int J Electrochem Sci 7:3314–3326
Márquez-Reyes LA, del Pilar Sánchez-Saavedra M, Valdez-Vazquez I (2015) Int J Hydrog Energy 40 (23):7291–7300
Liu H, Wang H, Qin H (2016) Int J Hydrog Energy 41(48):22786–22792
Tan SC, Gui H, Yang X, Yuan B, Zhan SH, Liu J (2016) Int J Hydrog Energy 41(48):22663–22667
Wang S, Gao X, Hang X, Zhu X, Han H, Liao W, Chen W (2016) J Amer Chem Soc 138 (50):16236–16239
Ortiz AL, Zaragoza MM, Collins-Martínez V (2016) Int J Hydrog Energy 41(48):23363–23379
Punshon S, Moore RM (2008) Mar Chem 108(3-4):215–220
Himmelblau DM, Riggs JB (2012) Basic Principles and Calculations in Chemical Engineering, 8th edn. Prentice Hall International Series in the Physical and Chemical Engineering Sciences, New York
Lide DR (ed) (2005) CRC handbook of chemistry and physics. CRC Press, Boca Raton
Sun Y, Bigi JP, Piro NA, Tang ML, Long JR, Chang CJ (2011) J Amer Chem Soc 133 (24):9212–9215
Li G, Zhang D, Qiao Q, Yu Y, Peterson D, Zafar A, Kumar R, Curtarolo S, Hunte F, Shannon S, Zhu Y, Yang W, Cao L (2016) J Amer Chem Soc 138(51):16632–16638
Garrett BR, Click KA, Durr CB, Hadad CM, Wu Y (2016) J Amer Chem Soc 138(41):13726–13731
Bechlars B, D’Alessandro DM, Jenkins DM, Iavarone AT, Glover SD, Kubiak CP, Long JR (2010) Nat Chem 2(5):362–368
Karunadasa HI, Chang CJ, Long JR (2010) Nature 464:1329–1333
Sundstrom EJ, Yang X, Thoi VS, Karunadasa HI, Chang CJ, Long JR, Head-Gordon M (2012) J Amer Chem Soc 134(11):5233–5242
Parr R, Yang W (1989) Density–functional theory of atoms and molecules. Oxford University Press, New York
Hohenberg P, Kohn W (1964) Phys Rev 136:B864–B871
Kohn W, Sham LJ (1965) Phys Rev 140:A1133–A1138
Chermette H (1999) J Comput Chem 20:129–154
Geerlings P, De Proft F, Langenaeker W (2003) Chem Rev 103:1793–1874
Geerlings P, De Proft F (2008) Phys Chem Chem Phys 10:3028–3042
Morell C, Grand A, Toro-Labbé A (2005) J Phys Chem A 109:205–212
Fuentealba P, Parr R (1991) J Chem Phys 94(8):5559–5564
Morell C, Grand A, Toro-Labbé A (2006) Chem Phys Lett 425:342–346
Morell C, Hocquet A, Grand A, Jamart-Grégoire B (2008) J Mol Struct: THEOCHEM 849:46–51
Morell C, Ayers P, Grand A, Gutiérrez-Oliva S, Toro-Labbé A (2008) Phys Chem Chem Phys 10:7239–7246
Martínez-Araya JI (2015) J Math Chem 53:451–465
Martínez-Araya JI (2011) Chem Phys Lett 506(1):104–111
Galván M, Vela A, Gázquez JL (1988) J Phys Chem 92:6470–6474
Chamorro E, Pérez P, Duque M, De Proft F, Geerlings P (2008) J Chem Phys 129:064117
Pérez P, Chamorro E, Ayers P (2008) J Chem Phys 128:204108
Tognetti V, Morell C, Ayers PW, Joubert L, Chermette H (2013) Phys Chem Chem Phys 15:14465–14475
Becke AD (1988) Phys A 38:3098–3100
Perdew JP (1986) Phys B 33:8822–8824
Fuentealba P, Preuss H, Stoll H, Szentpály LV (1982) Chem Phys Lett 89(5):418–422
von Szentpály L, Fuentealba P, Preuss H, Stoll H (1982) Chem Phys Lett 93(6):555–559
Fuentealba P, Stoll H, von Szentpály L, Schwerdtfeger P, Preuss H (1983) J Phys B: Atom Mol Phys 16(11):L323
Stoll H, Fuentealba P, Schwerdtfeger P, Flad J.v., Szentpály L, Preuss H (1984) J Chem Phys 81(6):2732–2736
Igel G, Wedig U, Dolg M, Fuentealba P, Preuss H, Stoll H, Frey R (1984) J Chem Phys 81 (6):2737–2740
Fuentealba P, von Szentpály L, Preuss H, Stoll H. (1985) J Phys B: Atom Mol Phys 18(7):1287
Dolg M, Wedig U, Stoll H, Preuss H (1987) J Chem Phys 86(2):866–872
Igel-Mann G, Stoll H, Preuss H (1988) Mol Phys 65(6):1321–1328
Dolg M, Stoll H, Preuss H (1989) J Chem Phys 90(3):1730–1734
Schwerdtfeger P, Dolg M, Schwarz WHE, Bowmaker GA, Boyd PDW (1989) J Chem Phys 91 (3):1762–1774
Dolg M, Stoll H, Savin A, Preuss H (1989) Theor Chim Acta 75(3):173–194
Andrae D, Häußermann U, Dolg M, Stoll H, Preuß H (1990) Theor Chim Acta 77(2):123–141
Dolg M, Fulde P, Küchle W, Neumann CS, Stoll H (1991) J Chem Phys 94(4):3011–3017
Kaupp M.v.R, Schleyer P, Stoll H, Preuss H (1991) J Chem Phys 94(2):1360–1366
Küchle W, Dolg M, Stoll H, Preuss H (1991) Mol Phys 74(6):1245–1263
Dolg M, Stoll H, Flad HJ, Preuss H (1992) J Chem Phys 97(2):1162–1173
Bergner A, Dolg M, Küchle W, Stoll H, Preuss H (1993) Mol Phys 80(6):1431–1441
Dolg M, Stoll H, Preuss H (1993) Theor Chim Acta 85(6):441–450
Häussermann U, Dolg M, Stoll H, Preuss H, Schwerdtfeger P, Pitzer R (1993) Mol Phys 78 (5):1211–1224
Dolg M, Stoll H, Preuss H, Pitzer RM (1993) J Phys Chem 97(22):5852–5859
Küchle W, Dolg M, Stoll H, Preuss H (1994) J Chem Phys 100(10):7535–7542
Nicklass A, Dolg M, Stoll H, Preuss H (1995) J Chem Phys 102(22):8942–8952
Leininger T, Nicklass A, Stoll H, Dolg M, Schwerdtfeger P (1996) J Chem Phys 105(3):1052–1059
Cao X, Dolg M (2001) J Chem Phys 115(16):7348–7355
Cao X, Dolg M (2002) J Mol Struct: THEOCHEM 581(1–3):139–147
Ditchfield R, Hehre WJ, Pople JA (1971) J Chem Phys 54(2):724–728
Hehre WJ, Ditchfield R, Pople JA (1972) J Chem Phys 56(5):2257–2261
Hariharan P, Pople J (1973) Theor Chim Acta 28(3):213–222
Hariharan P, Pople J (1974) Mol Phys 27(1):209–214
Gordon MS (1980) Chem Phys Lett 76(1):163–168
Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA (1982) J Chem Phys 77(7):3654–3665
Binning RC, Curtiss LA (1990) J Comput Chem 11(10):1206–1216
Blaudeau JP, McGrath MP, Curtiss LA, Radom L (1997) J Chem Phys 107(13):5016–5021
Rassolov VA, Pople JA, Ratner MA, Windus TL (1998) J Chem Phys 109(4):1223–1229
Rassolov VA, Ratner MA, Pople JA, Redfern PC, Curtiss LA (2001) J Comput Chem 22 (9):976–984
Frisch MJ, Pople JA, Binkley JS (1984) J Chem Phys 80(7):3265–3269
Clark T, Chandrasekhar J, Spitznagel GW, Schleyer PVR (1983) J Comput Chem 4(3):294–301
Peng C, Bernhard Schlegel H (1993) Israel J Chem 33(4):449–454
Peng C, Ayala PY, Schlegel HB, Frisch MJ (1996) J Comput Chem 17(1):49–56
Tapia O, Goscinski O (1975) Mol Phys 29(6):1653–1661
Miertuš S, Scrocco E, Tomasi J (1981) Chem Phys 55(1):117–129
Miertuš S, Tomasi J (1982) Chem Phys 65(2):239–245
Pascual-ahuir JL, Silla E, Tuñon I (1994) J Comput Chem 15(10):1127–1138
Cossi M, Barone V, Cammi R, Tomasi J (1996) Chem Phys Lett 255(4–6):327–335
Barone V, Cossi M, Tomasi J (1997) J Chem Phys 107(8):3210–3221
Cancès E, Mennucci B, Tomasi J (1997) J Chem Phys 107(8):3032–3041
Mennucci B, Tomasi J (1997) J Chem Phys 106(12):5151–5158
Mennucci B, Cancès E, Tomasi J (1997) J Phys Chem B 101(49):10506–10517
Barone V, Cossi M (1998) J Phys Chem A 102(11):1995–2001
Cossi M, Barone V, Mennucci B, Tomasi J (1998) Chem Phys Lett 286(3-4):253–260
Barone V, Cossi M, Tomasi J (1998) J Comput Chem 19(4):404–417
Cammi R, Mennucci B, Tomasi J (1999) J Phys Chem A 103(45):9100–9108
Cossi M, Barone V, Robb MA (1999) J Chem Phys 111(12):5295–5302
Tomasi J, Mennucci B, Cancès E (1999) J Mol Struct: THEOCHEM 464(1–3):211–226
Cammi R, Mennucci B, Tomasi J (2000) J Phys Chem A 104(23):5631–5637
Cossi M, Barone V (2000) J Chem Phys 112(5):2427–2435
Cossi M, Barone V (2001) J Chem Phys 115(10):4708–4717
Cossi M, Rega N, Scalmani G, Barone V (2001) J Chem Phys 114(13):5691–5701
Cossi M, Scalmani G, Rega N, Barone V (2002) J Chem Phys 117(1):43–54
Cossi M, Rega N, Scalmani G, Barone V (2003) J Comput Chem 24(6):669–681
Tomasi J, Mennucci B, Cammi R (2005) Chem Rev 105(8):2999–3094. PMID: 16092826
Marenich AV, Cramer CJ, Truhlar DG (2009) J Phys Chem B 113(18):6378–6396
Zhao Y, Truhlar DG (2006) J Chem Phys 125:194101
Zhao Y, Truhlar DG (2008) Theor Chem Accounts 120:215–241
Weigend F, Ahlrichs R (2005) Phys Chem Chem Phys 7:3297–3305
Dunlap BI (1983) J Chem Phys 78(6):3140–3142
Dunlap B (2000) J Mol Struct: THEOCHEM 529(1):37–40
Martínez-Araya JI (2016) J Comput Chem 37(25):2279–2303
Acknowledgements
The authors acknowledge the financial support provided by FONDECYT grants N° 1140289, 3150249, 1140340, and ICM, Millennium Nucleus Chemical Processes and Catalysis (CPC) grant N° 120082.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper belongs to Topical Collection P. Politzer 80th Birthday Festschrift
Rights and permissions
About this article

Cite this article
Martínez-Araya, J.I., Yepes, D. & Jaque, P. A 3D visualization of the substituent effect. J Mol Model 24, 31 (2018). https://doi.org/10.1007/s00894-017-3565-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-017-3565-8