
Abstract
Six nitramines (N1–6) were designed with all possible arrangements of N–NO2 groups on a cyclic skeleton and structural optimization was performed using the density functional theory (DFT). We observed that all nitramines have high positive heats of formation proportionate to the number of N−NO2 groups in their molecular structure. Among the designed nitramines, N5 and N6 have crystal densities of 1.77 and 1.81 g cm−3, respectively, which lead to reasonable respective detonation velocities (D = 8.70 and 9.07 km s−1) and detonation pressures (P = 33.23 and 36.57 GPa) comparable to those of RDX. To understand the relationship between sensitivity and molecular structure, bond dissociation energies, impact sensitivities (h 50), free space in crystal lattice, imbalance between the positive and negative surface potentials and heats of detonation (Q) were investigated. The comparable performance of N5 and N6 with RDX highlights the potential application of these nitramine derivatives as high energy materials and also supports the advantage of N–N bonds in the backbone and substitution of N–NO2 groups.

Electrostatic potential on the 0.001 electron/bohr3 molecular surface of N6
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
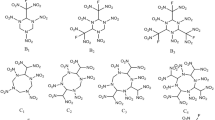
The high stored energy and potential of energetic materials makes them well suited to propellant and explosive applications [1–3]. The search for high performance energetic materials with better stability and lower sensitivity is the main objective of energetic materials research and is a never-ending process [4–7]. Nitramine (N–NO2) compounds are an important class of organic explosive, and are known to combine oxygen balance, density, performance and stability. In addition, previous reports have shown that introduction of a nitramine group into a molecule can increase its density and, thus, detonation performance [8–10]. 1,3,5-Trinitro-1,3,5-triazacyclohexane (RDX), 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane (HMX), and 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) are the most significant members of this class of compounds. Most nitramines have higher shattering capability and chemical resistance compared to their nitrate ester analogs [11]. RDX and HMX are the most extensively used military explosives; however, they have the risk of quick detonation and catastrophic explosion if used in high caliber guns and in the magazines of ships [11]. Similarly, CL-20 is superior in detonation performance but has limited explosive application due to its high sensitivity. In this article, we present six new nitramine molecules containing an octahydro[1, 2, 4, 5]tetrazino[1,2-a][1,2,4,5]tetrazine (OHTT) backbone, which were designed to achieve the high performance characteristics derived from cyclic structure and nitramine groups. The designed molecular structures are shown in Fig. 1. The OHTT ring has been reported in the literature (see Fig. S1 in Supporting Information) [12]; however, its nitramine derivatives have not been cited in experimental or theoretical studies. Owing to the difficulties in synthesis of the hypothetical molecules, a known strategy for the preparation of OHTT ring with four N–H sites also increases the chances for synthesis of designed nitramine derivatives. We believe that systematic computational investigation of their energetic properties and details of the structure–property relationship before experimental studies will help to screen potential molecules in this class of compounds.

Structures of designed nitramine derivatives
Methods
All the calculations were carried out using the Gaussian 03 suite of programs [13]. Geometric structure optimization and frequency analyses were performed using B3PW91/6-31G(d,p) level of theory and characterized to true local energy minima on the potential energy surfaces showing no imaginary frequencies. Isodesmic reactions were designed to predict the gas phase heat of formation (HOFgas) and are given in the Supporting Information. The usage of HOFgas in the calculation of detonation properties slightly overestimates the values of detonation velocity and detonation pressure; solid phase HOF (HOFsolid) can effectively reduce such errors. HOFsolid is calculated as the difference between HOFgas and heat of sublimation (HOFsub) as,
HOFsub depends on the molecular surface properties and is calculated using Eq. (2) proposed by Politzer et al. [14],
where A represent the surface area of the 0.001 electrons bohr−3 isosurface of electronic density, v denotes the degree of balance between the positive and negative surface potentials, and \( {\sigma}_{tot}^2 \) is the electrostatic potential variance. The molecular surface properties were obtained using the Multiwfn program [15].
The density (ρ) was calculated using Eq. (3), as developed by Politzer et al. [16] for CHNO energetic compounds,
where M is the molecular mass in g mol−1, V m is the volume enclosed by the 0.001 a.u. contour of the molecule’s electronic density, and \( v{\upsigma}_{\mathrm{tot}}^2 \) is an electrostatic interaction index.
Kamlet and Jacobs equations [17] were used to calculate the detonation velocity (D, km s−1) and detonation pressure (P, GPa),
N is the number of moles of gaseous detonation products per gram of explosive, M is their average molecular mass in g mol−1, and Q is the chemical energy of detonation given as calories per gram of explosive.
The methodology proposed by Akhavan [18] was used to predict explosive power and power index using the heat of explosion and volume of gaseous explosion products. The heat of explosion under constant volume (Q v) denotes heat liberated under adiabatic conditions and calculated from the difference between the total energies of formation of the explosive components and the total energies of formation of the decomposition products. The total volume of gaseous (V) products liberated represents the amount of work done by the explosive. Modified Kistiakowsky-Wilson (K-W) rules were used to predict the decomposition products and the overall decomposition reactions are given in the Supporting Information.
Politzer et al. [19, 20] established the correlation between free space in the crystal lattice (ΔV) of energetic material and sensitivity. The free space per molecule was calculated by subtracting the effective volume per molecule (V eff) and the intrinsic gas phase molecular volume (V int) as given in Eq. (8),
In energetic nitramine materials, generally, N–NO2 is the weakest bond, which ruptures easily upon applying external stimuli. Hence, we calculated the bond dissociation energy (BDE) of the longest N–NO2 bond using following Eq. (9) at B3PW91/6-31G(d,p) level,
where E R1-R2, E R1 and E R2 are the total energies with zero point energy (ZPE) correction of the precursor and the corresponding radicals produced by bond dissociation.
The impact sensitivity (h 50) is an important parameter that characterizes the safety nature of the energetic material. Politzer’s group [21] correlated h 50 with statistically defined quantities like strength of positive surface potential (σ 2+ ) and an electrostatic balance parameter (v) using following equation,
Results and discussion
Heat of formation
The HOF is an important parameter used to measure the detonation parameters and energy stored in chemical structure. The total energies (E 0), ZPE, thermal corrections (H T), and the calculated HOF values of the nitramine derivatives are given in Table 1. Isodesmic reactions were designed and used to predict the HOFs of the nitramine derivatives (see Figs. S2 and S3 in the Supporting Information). For the nitramine derivatives (N1–6), HOFsolid lies in the range between 348 and 460 kJ mol−1. These high HOFsolid values can be ascribed to the presence of N−N bonds in the OHTT backbone and to N−NO2 groups. It is worthwhile to compare HOFsolid of N5 and N6 with the RDX (HOFsolid = 79 kJ mol−1), which shows the effects of fused ring and N−NO2 groups on the HOFs. N5 and N6 exhibit higher HOF values compared to those of RDX and HMX. To understand the effect of N−NO2 groups, HOFgas, HOFsub and HOFsolid values for N1, N2, N5 and N6 are plotted in Fig. 2, which clearly shows that HOFs goes up with the number of N−NO2 groups and represent a good linear trend between them. Introduction of each N−NO2 group in N1 to N2, N2 to N5 and N5 to N6 increases the HOFsolid by ∼40 kJ mol−1. Among the N2−4 isomers, repulsion in nitro groups and their position on the OHTT ring varies the HOFsolid values.

Effect of N−NO2 groups on gas phase HOF (HOFgas), heat of sublimation (HOFsub), and solid phase HOF (HOFsolid). In molecules N1, N2, N5 and N6, −NO2 group increases from 1 to 4, respectively
Density and oxygen balance
Generally, high crystal density strongly influences the electronic and detonation properties of energetic materials. Politzer’s approach [16] was used to calculate the crystal densities of our nitramine derivatives, which proved to be reliable and accounts well for the intermolecular interactions in CHNO-containing molecules. The computed densities of compounds N1−N6 are given in Table 2; these show clearly that the density goes up with introduction of nitro groups into the molecular structure. Previous reports also support that an increase in nitramine groups in the cyclic structure improves the density [23–25]. The densities of N5 (1.77 g cm−3) and N6 (1.81 g cm−3) were comparable to that of RDX (1.80 g cm−3). In the case of N2, N3 and N4 isomers, we observed that repulsion and steric hindrance between nitro groups reduces the density. Along with higher densities, oxygen balance (OB) is also an important factor that governs the detonation performance of energetic materials. In CHNO compounds, OB shows the amount of oxygen required for conversion of carbon to CO2 and hydrogen to H2O. A high concentration of gaseous detonation products also helps improve performance. In this work, all the designed nitramine derivatives are composed of CHNO and possess negative OB ranges from −97 to −20 % (see Table 2). The results show that the OB of N6 (−20 %) is the largest, and is slightly better than that of RDX (−22 %). Overall, the addition of –NO2 groups is a good substituent for increasing OB and designing potential energetic materials.
Performance parameters
To evaluate the performance of energetic materials, detonation velocity (D) and detonation pressure (P) are the main parameters. Table 2 lists the predicted detonation properties of nitramine derivatives and RDX. Density (ρ) is the most important factor in computing D and P and, as can be seen from Table 2, the rise in density from N1 to N6 has significant influence on them. Reviewing the performance of compounds N1−N6, it is also clear that D and P improve with the increasing number of nitro groups in the OHTT backbone. This also supports the statement that introduction of more nitro groups into a structure helps to boost its detonation performance [23–25]. N1 possess the lowest D and P values of the designed nitramine derivatives due to its lower density and HOF. Considering the calculated detonation parameters, N6 shows superior performance over RDX and other nitramine derivatives, while N5 shows values close to that of RDX. The computed heats of detonation (Q) in Table 2 indicate that substitution of the –NO2 group increases the heat of detonation whereas the position of the –NO2 group in the N2, N3 and N4 isomers has little effect.
Detonation reactions liberate heat and hot gases. Depending on the OB, the detonation of CHNO energetic materials results mainly in the formation of decomposition products like water (H2O), carbon monoxide (CO), carbon dioxide (CO2), and carbon. Explosive power and power index of nitramines N1–N6 were calculated using Eqs. (6) and (7), as suggested by Akhavan [18] and given in Table 2. Explosive power and power index depend on the volume of gases released (V in dm3 g−1) and the heat of explosion (Qv in kJ kg−1) of corresponding explosive. The predicted results show that the explosive power of N5 and N6 are above 5000, which is higher than that of RDX (4568). Similarly, both compounds show a high power index (187 %) and are superior to RDX (169 %). Regardless of having a more negative OB, N2−N4 show a comparable power index to RDX due to their high positive HOFs. In view of the performance parameters, we conclude that N5 and N6 exhibit notable detonation performance and may be regarded as promising candidates for energetic materials.
Sensitivity correlations
Understanding the stability and sensitivity of energetic materials, and their practical applications, is gaining wide interest. Bond dissociation energy (BDE) is used widely to predict initiation and decomposition of newly designed molecules. In general, a smaller value of BDE denotes a weaker trigger bond in the chemical structure and the corresponding compound is more unstable or sensitive. In this work, the longest N−NO2 bond in the OHTT backbone was considered as the weakest bond to estimate the BDEs. The BDEs of the N−N bond in the OHTT ring were found to be above 120 kJ mol−1, showing more strength than N−NO2 bonds thus showing the skeleton to be more stable. It can be seen from Table 3 that all the designed nitramine derivatives have BDEs larger than 88 kJ mol−1 and possess good stability. Comparing the BDEs of the N−NO2 bonds in the N2, N3 and N4 isomers reveals that steric hindrance and repulsion in N2 reduces the BDE by ∼12 kJ mol−1. On the whole, BDEs of N–NO2 decrease with increasing number in the skeleton, with the introduction of each group reducing the BDE by 10–15 kJ mol−1.
For early safety understanding, the impact sensitivity was calculated according to Politzer’s approach [21], which is used widely for nitramines. Impact sensitivity is usually represented by the height (h 50) from which a standard weight dropped upon the compound has a 50 % possibility of an explosion. The h 50 values of all the nitramine molecules are listed in Table 3. These values show clearly shows that, with increasing N−NO2 groups, the h 50 values lower and sensitivity increases. Compared with the h 50 value of RDX, it was found that, except N6, all the compounds were insensitive to impact. It is also noteworthy that the results of h 50 were consistent with the BDEs.
The Politzer approach [21], which relates crystal lattice effects and sensitivity, was applied to understand the sensitivities of the designed nitramine derivatives. It was observed that a greater number of electronegative atoms and nitro groups present in the molecular structure creates an imbalance of positive and negative surface potentials. In general, the larger the imbalance (v) between the positive and negative surface potentials, the higher the sensitivity [21, 26]. The calculated values of v (in Table 3) shows that introduction of N−NO2 groups increases the imbalance in potential and thus the sensitivity. Comparison of v values reveals the order of sensitivity as N6 > N5 > N2 > N1. Similarly, free space in crystal lattice increases gradually with the number of N−NO2 groups. Overall, a higher value of free space in the crystal lattice (larger ΔV) denotes more sensitivity. From the ΔV values of the nitramine derivatives, it can be noted that N5 and N6 possess higher ΔV, and are more sensitive than RDX.
In recent reports, Politzer and Murray investigated the relationship between heat of detonation (Q max) and sensitivity [27], concluding that higher values of Q are undesirable and increase sensitivity. The heats of detonation (Q) are calculated using the equation suggested by Kamlet-Jacobs [17] and are listed in Table 2. Politzer reported that detonation parameters (D and P) increase as Q become higher (Eqs. 4, 5). However, the dependence of D and P on heat of detonation is lower compared to that of density. We can see that N1–N4 have lower Q values than RDX (1508 cal g−1), while N5 (1569 cal g−1) and N6 (1661 cal g−1) surpass RDX. The higher Q values of N5 and N6 are due to their high HOF and low/high ratio of hydrogen/oxygen in the chemical formula. According to Q values, N5 and N6 are more sensitive than N1–N4 and RDX. Considering the BDEs, h 50, v, ΔV and heats of detonation values, all nitramines follow a similar trend of sensitivity (see Fig. 3).

Correlation of sensitivity with balance parameters between surface potentials (v), h 50, and free space in crystal lattice (ΔV). In molecules N1, N2, N5 and N6, the number of −NO2 groups increases from 1 to 4, respectively
The disproportionation energy (E dis) was determined to predict the strength of the interactions between the –NO2 groups using isodesmic reactions (see Fig. S4 in the Supporting Information) and corresponding values are listed in Table 3. As for N5 and N6, the E dis values of N6 (108 kJ mol−1) indicate that the strong repulsive interactions between –NO2 groups is very high and that they play a major role in making this compound more unstable than N5 (69 kJ mol−1). As expected, E dis increases as the number of adjacent –NO2 groups increases due to their repulsive interactions.
Conclusions
A series of nitramine derivatives were designed by taking advantage of a synthesis route available for their basic skeleton and their energetic properties were studied. N5 and N6 possess good OB, high density and reasonable detonation properties. Sensitivity correlations established by considering BDEs, h 50, free space in crystal lattice, balance parameter between positive and negative surface potentials and heats of detonation (Q), indicate that N5 and N6 are more sensitive than RDX. The detonation properties of N5 and N6 are comparable to those of RDX, which suggests that they can be considered as new energetics.
References
Badgujar DM, Talawar MB, Asthana SN, Mahulikar PP (2008) J Hazard Mater 151:289–305
Sikder AK, Sikder NA (2004) J Hazard Mater 112:1–15
Singh RP, Verma RD, Meshri DT, Shreeve JM (2006) Angew Chem Int Ed 45:3584–3601
Zhang Q, Shreeve JM (2014) Chem Rev 114:10527–10574
Gao H, Shreeve JM (2011) Chem Rev 111:7377–7436
Lin QH, Li YC, Qi C, Liu W, Wang Y, Pang SP (2013) J Mater Chem A 1:6776–6785
Klapötke TM, Piercey DG, Stierstorfer J (2012) Eur J Inorg Chem 2012(34):5694–5700
Qiu L, Xiao H, Gong X, Ju X, Zhu W (2006) J Phys Chem A 110:3797–3807
Zhao GZ, Lu M (2012) J Mol Model 18:2443–2451
Wang F, Wang G, Du H, Zhang J, Gong X (2011) J Phys Chem A 115:13858–13864
Agrawal JP, Hodgson RD (2007) Organic chemistry of explosives. Wiley, Chichester
Shawali AS, Elsheikh SM (2001) J Heterocycl Chem 38:541–559
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T Jr, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PM, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian 03, Revision A.1. Gaussian Inc, Pittsburgh
Politzer P, Ma Y, Lane P, Concha MC (2005) Int J Quantum Chem 105:341–347
Lu T, Chen F (2012) J Comput Chem 33:580–592
Politzer P, Martinez J, Murray JS, Concha MC, Toro-Labbé A (2009) Mol Phys 107:2095–2101
Kamlet MJ, Jacobs SJ (1968) J Chem Phys 48:23–35
Akhavan J (2004) The chemistry of explosives, 2nd edn. Royal Society of Chemistry, London
Politzer P, Lane P, Murray JS (2013) Cent Eur J Energ Mater 10:305–323
Politzer P, Murray JS (2014) J Mol Model 20:2223
Pospíšil M, Vávra P, Concha MC, Murray JS, Politzer P (2010) J Mol Model 16:895–901
Politzer P, Murray JS (2011) Cent Eur J Energ Mater 8:209–220
Qiu L, Gong XD, Ju XH, Xiao HM (2008) Sci China Ser B-Chem 51:1231–1245
Lu M, Zhao G (2013) J Braz Chem Soc 24:1018–1026
Xu XJ, Xiao HM, Ju XH, Gong XD, Zhu WH (2006) J Phys Chem A 110:5929–5933
Murray JS, Concha MC, Politzer P (2009) Mol Phys 107:89–97
Politzer P, Murray JS (2015) J Mol Model 21:262
Acknowledgments
S.D. thanks the National Institute of Technology, Kurukshetra for financial support. V.D.G. thanks the DST-SERB for research grant (Young Scientists, No. SB/FT/CS-110/2014). D.S. thanks the ACRHEM and School of Chemistry, University of Hyderabad for providing computational facilities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Devi, A., Deswal, S., Dharavath, S. et al. Molecular design and screening of energetic nitramine derivatives. J Mol Model 21, 298 (2015). https://doi.org/10.1007/s00894-015-2846-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-015-2846-3