
Abstract
The aim of this study was to isolate a novel esterase from a hypersaline lake by sequence-based metagenomics. The metagenomic DNA was isolated from the enriched hypersaline lake sediment. Degenerate primers targeting the conserved regions of lipolytic enzymes of halophilic microorganisms were used for polymerase chain reaction (PCR) and a whole gene was identified by genome walking. The gene was composed of 783 bp, which corresponds to 260 amino acids with a molecular weight of 28.2 kDa. The deduced amino acid sequence best matched with the esterase from Halomonas gudaonensis with an identity of 91%. Recombinantly expressed enzyme exhibited maximum activity towards pNP-hexanoate with a kcat value of 12.30 s−1. The optimum pH and temperature of the enzyme were found as 9 and 30 °C, respectively. The effects of NaCl, solvents, metal ions, detergents and enzyme inhibitors were also studied. In conclusion, a novel enzyme, named as hypersaline lake “Acıgöl” esterase (hAGEst), was identified by sequence-based metagenomics. The high expression level, the ability to maintain activity at cold temperatures and tolerance to DMSO and metal ions are the most outstanding properties of the hAGEst.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Esterases (EC 3.1.1.1) are ubiquitous enzymes which hydrolyze ester bonds, acting on the partly water-soluble small molecules. Along with lipases and other hydrolases, they constitute the 70% of enzyme sales in the market, which is expected to reach 10.7 billion US dollars in 2024 (Ramnath et al. 2017). They are capable of catalyzing not only hydrolysis but also ester synthesis, acidolysis, interesterification and alcoholysis (Villeneuve et al. 2000). Their high chemo-, regio- and enantio-selectivity make them attractive for many applications, especially for the production of optically pure compounds.
Lipolytic enzymes have some characteristic amino acid residues that are conserved. They show α/β hydrolase fold and constitute two conserved regions. One of them is the active site which is constituted of a nucleophilic amino acid (serine, cysteine or aspartic acid), an acidic amino acid and histidine. The other conserved site, called oxyanion hole, stabilizes the tetrahedral form during hydrolysis (Pleiss et al. 2000). These proteins are separated into three classes, namely GX, GGGX and Y type, according to the sequence and structure of their oxyanion holes (Borrelli and Trono, 2015). Two of the residues forming the oxyanion hole donate their backbone amide protons, so the stabilization occurs (Fischer and Pleiss, 2003). Although the sequences around amino acid residues that form oxyanion hole are not so strictly conserved, the oxyanion regions are identified with the help of the crystal structure of known lipases and multiple alignments of homologous proteins (Bell et al. 2002).
Up to date, many lipolytic enzymes from different organisms have been isolated, characterized and some of them were industrially produced. More active and robust enzymes were obtained by applying protein engineering techniques or using extremophilic organisms. However, the use of lipases with new specificities is still a need (Hasan et al. 2009; Borrelli and Trono 2015). When a large quantity of enzymes is required, the production costs become a major challenge. In addition, when used for esterification, inhibition by short-chain alcohols, glycerol and other impurities limits the use of these catalysts (Ribeiro et al. 2011). De Godoy et al. (2015) studied the history of lipases extensively, which starts with the first discovery of them in the 1930s. They reviewed research publications, patents and related industrial status of lipases. Using the concept of technology life cycle, they evaluated the need for new lipases. According to the analysis results, it is concluded that the industry is not satisfied with the current status and still open for new lipolytic enzymes.
Metagenomic studies have enabled researchers to identify novel enzymes from environmental samples, especially from extreme environments, whose microflora cannot be cultivated by traditional microbiological methods. Generally, metagenomic studies can be accomplished by either function- or sequence-based approaches. In the function-based approach, a metagenomic library is constructed and screened for the desired activity. However, in the sequence-based approach, the target genes are detected using the previously known sequence information. Within the scope of this study, we are focused on gene-targeted metagenomics. In this strategy, using the advantage of the presence of the conserved domains, novel enzymes can be identified from environmental samples. First, genes encoding for the specific enzymes in the same class are retrieved from databases and aligned. Then degenerate primers targeting the conserved regions are designed. Although there are alternatives such as GeneFisher (Giegerich et al. 1996), HYDEN (Linhart and Shamir 2002) and DePiCt (Wei et al. 2003), consensus-degenerate hybrid oligonucleotide primer (CODEHOP) (Rose et al. 1998) strategy is widely used for this purpose. Once the partial sequence of the gene is amplified by PCR, new gene-specific primers are designed and unknown 3′ or 5′ ends are identified by genome walking (Kotik 2009). Genome walking can be accomplished by many ways but the most widely used strategy is based on the digestion of metagenomic DNA, followed by adaptor ligation and further amplification using the adaptor and gene-specific primer pairs as described in Morris et al. (1995).
There are various reports in the literature that used this strategy to identify novel enzymes from environmental samples. Bell et al. (2002) isolated a lipase from a hot spring that was expressed in E. coli. In a similar study, two novel lipases from environmental DNA were isolated and characterized (Jiang et al. 2006). The same strategy was applied to isolate a thermostable lipase from a hot spring (Kumar and Kashmir 2012). Iwai et al. (2010) applied gene-targeted metagenomics and pyrosequencing on the metagenome of a polychlorinated biphenyl-contaminated soil. The study resulted in an extensive diversity of aromatic dioxygenases. Nitrile hydratase (Liebeton and Eck 2004), epoxide hydrolase (Zhao et al. 2004), cyclomaltodextrinase (Tang et al. 2006), and starch-modifying enzymes (Labes et al. 2008) were also isolated from environmental samples, using a combination of degenerate primers and genome walking strategies with other molecular biological techniques.
Halophilic organisms are found in three main domains of life (Bacteria, Archaea and Eucarya) (Ma et al. 2010). They are classified according to the salinity that they can live in: extreme halophiles (best growth in 2.5–5.2 M salt), borderline extreme halophiles (best growth in 1.5–4.0 M salt), moderate halophiles (best growth in 0.5–2.5 M salt) and halotolerant microorganisms that do not require salt for growth but can tolerate high salt concentrations (Oren 2008). Halophilic habitats are interesting research environments for metagenomics and there are many interesting reports in the literature (Ferrer et al. 2005; Ghai et al. 2011; Narasingarao et al. 2012; Fernández et al. 2014; Cowan et al. 2015). Halophilic enzymes are usually exploitable in organic solvents and receive the attention of researchers due to their ability to maintain the activity in low-water-content medium.
Within the scope of this study, we aimed to isolate a novel lipolytic enzyme from a hypersaline lake, Acıgöl, by sequence-based metagenomics approach. In the literature, there is a study about the isolation of lipolytic enzymes from various hypersaline lakes in Turkey (Salt Lake, Lake Acıgöl, Tuzla Lake) by traditional methods (Ozcan et al. 2009). The authors examined five different lipolytic enzymes produced by microorganisms isolated from these environments without specifying which isolate from which lake. However, here we introduce the production and characterization of recombinant esterase enzyme which was isolated as a result of the first metagenomic survey of Lake Acıgöl to search for biocatalytic potential. In this study, by applying PCR with degenerate primers and genome walking, a full-length esterase gene has been obtained. Then the recombinant expression and characterization studies of the novel esterase gene from Lake Acıgöl have been reported.
Materials and methods
Bacterial strains and plasmids
For cloning of the PCR-amplified inserts, pCR™4-TOPO® vector (Thermo Fisher Scientific, Waltham, USA) and OneShot® TOP 10 E. coli cells (Thermo Fisher Scientific, Waltham, USA) were used. For the expression studies, pET-28a(+) vector (Novagen, Madison, USA) and OverExpress™ C43 (DE3) strain of E. coli (Lucigen, Wisconsin, USA) were used.
Metagenomic DNA isolation from enrichment culture
Environmental samples were collected from a hypersaline lake in southwestern Turkey, Lake Acıgöl. The chemistry of the brine of the lake predominantly contains Na+, K+, Cl− and SO42− ions. Microbial mats, which are composed of halophilic microorganisms, around the lake usually cause carbonate precipitations. The salinity of the lake is approximately 7.5% (w/v) (Balci et al. 2016, 2018). Samples were stored at − 20 °C until handling. 10 g of sediment was inoculated into 100 ml of nutrient broth medium (Merck, Darmstadt, Germany) containing 10% of NaCl and incubated at 30 °C for 7 days. UltraClean Microbial DNA Isolation Kit (MOBIO, California, USA) was used to isolate metagenomic DNA (7.15 µg) from 5 ml of enriched culture.
Primer design and degenerate PCR
Halophilic microorganisms that were present in our environmental sample were determined by previous studies (Balci and Demirel 2016; Balci et al. 2016). For the design of primers, nucleotide sequences of lipolytic enzymes belonging to halophilic microorganisms present in our culture were retrieved from the NCBI database. Also, some sequences from other halophilic microorganisms which are not found in our samples, such as Chromohalobacter are also included in the groups to maximize the diversity. (NCBI accession numbers of retrieved sequences are WP_009724342.1; WP_009097462.1; WP_007114294.1; WP_011507233.1; CBV42568.1; WP_010628549.1 and WP_009287832.1). Degenerate primers were designed using Primer4Clades online tool (Contreras-Moreira et al. 2009) according to CODEHOP strategies (Rose et al. 1998). The quality parameters for the primers were evaluated by OligoAnalyzer (https://eu.idtdna.com/calc/analyzer, Integrated DNA Technologies). The list of degenerate primers used in this study is given in Table 1.
PCR was set using the 1xPCR buffer, 1.5 mM MgSO4, 0.2 mM dNTPs, 0.5 µM HCABAF and HCABUR primers, 1 U of Taq DNA polymerase (Roche Diagnostics, Mannheim, Germany) and 100 ng of metagenomic DNA. Amplification was performed in gradient thermocycler (Biometra, Gottingen, Germany) with an initial denaturation at 95 °C for 120 s, followed by 40 amplification cycles (denaturation at 95 °C for 2 min, annealing at 44–63 °C with 2–3 °C of intervals for 60 s, extension at 72 °C for 60 s) and a final extension at 72 °C for 5 min. After confirmation by agarose gel electrophoresis, amplicons were purified and cloned using Invitrogen TOPO® TA Cloning® Kit (Thermo Fisher Scientific, Waltham, USA). E. coli TOP10 cells were transformed, recombinant plasmids were isolated and sequence analysis was done by Genometri (Istanbul, Turkey) using M13 forward and reverse primers.
Genome walking
GenomeWalker™ Universal Kit (Clontech, Cambridge, USA) was used for genome walking experiments. Adaptors and primers used in genome walking are given in Table 1. Metagenomic DNA was digested with DraI, EcoRV, PvuII and StuI restriction enzymes. After adaptor ligation, PCR with adaptor primers and gene-specific primers were set up. Once amplicons were obtained, they were cloned and sequenced. The procedure was repeated for both 5′ and 3′ ends until the full-length gene was identified. Hereafter, the gene and the protein were designated as hagest and hAGEst, respectively.
Sequence and bioinformatic analyses
After the full-length gene was obtained, the similarity to known genes was examined using blastn tool on the NCBI website (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Putative amino acids were determined with the ExPASy translate tool (http://web.expasy.org/translate/). The multiple alignments of the hAGEst with the most similar five proteins were done by Jalview version 2.10.2b2 (Waterhouse et al. 2009). The representative sequences for previously defined lipolytic protein families were obtained and a bootstrapped phylogenetic tree (1000 replications) was built using the neighbor-joining method with the MEGA 6.0 (Tamura et al. 2013).
Cloning, expression and purification of the esterase
To amplify the targeted gene, PCR was set with 1 × PCR buffer, 50 µg of metagenomic DNA, 1.5 mM MgCl2, 0.2 mM dNTP, and 0.2 mM forward Abhf_F and reverse Abhf_R primers (see Table 1), and 1 U of Taq DNA polymerase. Amplification was performed by initial denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 45 s, annealing at 66 °C for 45 s and extension at 72 °C for 1 min, and final extension at 72 °C for 5 min. For cloning, the amplified product was digested with EcoRI and HindIII (Fermentas, Waltham, USA) and ligated to digested and dephosphorylated pET-28-a(+) plasmid (Novagen, Madison, USA) and transformed into E. coli C43 (DE3) (Lucigen, Wisconsin, USA) cells.
For the expression of the protein, LB medium (1000 ml) supplemented with kanamycin (40 µg/µl) was inoculated with 50 ml of an overnight starter culture. The culture was incubated at 37 °C until OD600 nm reached 0.8. The culture was induced by 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 6 h at 30 °C and then the cells were harvested by centrifugation (4000 g for 20 min). The pelleted cells were stored at − 20 °C. Purification of the six histidine-tagged recombinant proteins was performed with Ni–NTA agarose (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The purified protein was assayed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie brilliant blue G-250 staining (Laemmli 1970).
Enzyme assays
Standard activity assay
The release of pNP due to the hydrolysis of pNP-octanoate was measured according to Winkler and Stuckmann (1979) with some modifications. The final concentrations of the pNP-octanoate and the enzyme in the reaction mixture were set as 1 mM and 1 µM, respectively. First, 10 mg of gum arabic and 40 µL of Triton X-100 were dissolved in 9 ml of 50 mM glycine–NaOH, pH 9. The substrate was dissolved in 1 ml of isopropanol, to give a final concentration of 1 mM in the reaction. These two solutions were mixed to form substrate solution. 180 µL of substrate solution was mixed with 20 µL of enzyme and the reaction mixture was incubated at 30 °C for 20 min. At the end of the incubation, microplates were immediately transferred into ice and absorbance at 410 nm was measured using Benchmark Plus Microplate Reader (Bio-Rad, California, USA). One unit of activity was defined as the amount of enzyme that releases 1 µmol of product per minute.
Enzyme assay to determine the substrate specificity
To determine the substrate specificity of the enzyme, para-nitrophenyl esters with different carbon chain lengths (pNP-butyrate, pNP-hexanoate, pNP-octanoate, pNP-decanoate, pNP-dodecanoate and pNP-hexadecanoate) were used. Reactions were set with varying substrate concentrations of 0.031, 0.0625, 0.125, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 2.5 and 3 mM. The assay was set as described in standard activity assay with one exception. For the enzymatic assays with pNP-butyrate and pNP-hexanoate, 50 mM Tris–HCl (pH 8) was used as a reaction buffer to eliminate autohydrolysis. For other substrates, 50 mM glycine–NaOH (pH 9) buffer was used as described above. The kinetic parameters were calculated using GraFit 7.0 software.
Enzyme assay to determine the effect of pH
To determine the optimum pH of the enzyme, the reaction was set up with different buffers (50 mM citrate/phosphate buffer, pH 6; 50 mM Tris–HCl buffer, pH 7, 8, and 8.5; 50 mM glycine–NaOH buffer, pH 9, 9.5 and 10). To determine the pH stability of the enzyme, 10 µM of enzyme was prepared in buffers with different pH values (citrate/phosphate buffer, pH 4, 5, 6; Tris–HCl buffer, pH 7, 8, and 8.5; glycine–NaOH buffer, pH 9, 9.5 and 10) and stored at 4 °C for 24 h. Relative and residual activities were measured according to standard assay.
Enzyme assay to determine the effect of temperature
To determine the optimum temperature of the enzyme, the reaction was set up on ice and transferred to the incubator with varying temperatures (5 °C–70 °C with 5 °C increments). For thermostability assay, 10 µM of enzyme was incubated at varying temperatures (25 °C–80 °C with 5 °C increments) for 20 min. Relative and residual activities were measured according to the standard assay.
Enzyme assay to determine the effect of NaCl
To determine the effect of NaCl on the activity of the enzyme, reactions were set up as to include 5, 10, 15, 20 and 25% NaCl. To determine the stability of the enzyme in the presence of NaCl, 100 µM of the enzyme was prepared in Tris–HCl buffer (pH 8) containing 10, 15, 20 and 25% NaCl and incubated at 4 °C for 24 h. Relative and residual activities were measured according to the standard assay.
Enzyme assay to determine the effect of different solvents
Similarly, the effect of different solvents [hexane, chloroform, acetone, dimethylformamide (DMF), dimethyl sulfoxide (DMSO), 2-propanol, butanol, 1-propanol, ethanol, methanol and glycerol] on the activity and the stability of the enzyme was evaluated. To determine the effect on the activity, solvents were added to the reaction mixture with a ratio of 10%. To determine the stability of the enzyme, 100 µM of the enzyme was incubated in buffer containing 15% and 30% solvents at 4 °C for 24 h. Relative and residual activities were measured according to standard assay.
Enzyme assay to determine the effect of metal salts, detergents and inhibitors
To determine the activity of the enzyme in the presence of metal salts [MgCl2, FeSO4, KCl, CaCl2, ZnSO4, Al2SO4, CuSO4, CoCl2, NiCl2, MnSO4, AgNO3], detergents [sodium dodecyl sulfate (SDS), cetrimonium bromide (CTAB), Triton X-100, Tween-80, Nonidet-P40] and inhibitors [phenylmethanesulfonyl fluoride (PMSF), dithiothreitol (DTT), ethylenediamine tetraacetic acid (EDTA) and ethyleneglycoltetraacetic acid (EGTA)], they were added to the reaction mixture with a final concentration of 1 mM and 5 mM. Relative activities were measured according to the standard assay.
Nucleotide sequence accession number
The sequence of hagest was deposited in GenBank under accession number KY906189.
Results
Primer design, degenerate PCR and genome walking
Considering the microbial diversity of the sample, which was previously determined (Balci et al. 2016), the nucleotide sequences of lipolytic enzymes of these microorganisms were retrieved from the NCBI database (Supplementary Fig. 1). After the amplification with the degenerate primers HCABAF and HCABUR (Table 1 and Supplementary Fig. 2), two different sequences were obtained which showed best similarities to alpha/beta hydrolases from Halomonas titanicae (WP_009287832.1) and H. gudaonensis (WP_089686035.1) with identities of 97.5% and 88.39%. To increase the chance that we obtain a novel enzyme, we continued the experiments with the second sequence (88.39% similarity), which shows a lower similarity to known proteins. Genome walking was applied to identify the unknown regions of the gene. The 5′ and 3′ ends of the gene were successfully amplified and sequenced. The Cap3 program was used to assemble the 5′ and 3′ end sequences with the partial gene obtained by degenerate PCR (Huang and Madan 1999). The assembled sequence was used to detect the putative open reading frame (Supplementary Fig. 3).
Sequence and bioinformatics analyses
The gene is composed of 783 nucleotides, which corresponds to 260 amino acids. It starts with start codon ATG and ends up with UGA stop codon. The molecular weight and the isoelectric point (pI) of the protein were theoretically calculated as 28,367.98 Da and 5.27, respectively, using the ExPASy tool. The nucleotide sequence was compared on the non-redundant database using BlastX tool. The most similar protein is an alpha/beta hydrolase from Halomonas gudaonensis (WP_089686035.1) with 91% identity. This entry from Halomonas gudaonensis is also identical with an esterase (SDJ79551.1) from the same organism. The multiple alignments of the hAGEst with the most similar five proteins are given in Fig. 1a.

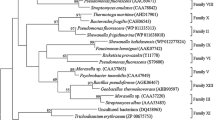
a The multiple alignments of hAGEst with most similar five proteins which show more than 76% identity. Sequences were found by BlastX search, and the alignment was done by Sequence Manipulation Suite (Stothard, 2000). The accession numbers and identities are as follows: WP_089686035.1 for Halomonas gudaonensis (91%), WP_104204164.1 for Halomonas saliphila (81%), WP_010628549.1 for Halomonas sp. KM-1 (77%), WP_102651855.1 for Halomonas endophytica (77%), WP_103967955.1 for Halomonas daqingensis (77%). Similar amino acids are shown with the same background colors: gray for Gly, Ala, Val, Leu, Ile; orange for Phe, Tyr, Trp; yellow for Cys, Met; green for Ser, Thr; red for Lys, Arg, His; blue for Asp, Glu; brown for Asn, Gln and pink for Pro. The residues which do not show consensus are not colored. b The phylogenetic tree of hAGEst and other lipolytic proteins. The phylogenetic tree was constructed using MEGA 6.0, with the neighbor-joining method and bootstrapping was applied with 1000 replicates. The scale bar indicates the number of amino acid substitutions per site
As shown in Fig. 1a, all six proteins share a common conserved site, Gly-His-Ser-Met-Gly, in which the catalytic serine is located at position 87. For the analysis of the phylogenetic relationship of hAGEst with other lipases, a tree was constructed using 36 sequences that represent 18 families (Arpigny and Jaeger 1999; Li et al. 2016; Samoylova et al. 2018). Representative protein sequences for each family, which were first defined by (1999), were retrieved from the protein database and aligned. According to the phylogenetic tree shown in Fig. 1b, the hAGEst was involved in the family V, along with lipolytic enzymes from Sulfolobus acidocaldarius and Haemophilus influenza. It is known that family V proteins are usually active towards short-chain substrates and do not display interfacial activation, unlike lipases (Ramnath et al. 2017).
Production and purification of the enzyme
After sequence analysis, the enzyme was successfully cloned and produced in E. coli C43 (DE3). The expression was optimum when the cell culture was induced by 0.1 mM IPTG for 6 h at 30 °C (data not shown). SDS-PAGE analysis showed that the targeted protein was successfully bound to Ni–NTA resin (Fig. 2a). Although some amount of protein was lost during the wash steps, expressed protein amount was satisfactory for further analysis (about 30 mg/L of cell culture). The molecular weight of the protein was calculated as 28.2 kDa, consistent with the calculated theoretical value.

a SDS-PAGE analysis of the samples obtained during His-tag purification. Lane FT: unbound proteins after Ni–NTA incubation; Lanes W1 and W3: wash samples, Lanes E1–E6: eluted proteins, Lane M: molecular weight marker (Pierce™, #26,610). b Enzyme solution in tubes after storage at 4 °C for 5 days in 1) Tris–HCl (pH 8) buffer containing 10% NaCl and 2) Tris–HCl (pH8) buffer without NaCl
All elution fractions were collected and concentrated by ultrafiltration. To assess the effect of the addition of NaCl in the storage buffer, enzymes were concentrated using 50 mM Tris–HCl, pH 8, or the same buffer containing 10% NaCl. The absence of NaCl in the storage buffer causes the protein to form aggregates, probably related to the loss of three-dimensional structure (Fig. 2b). In addition, when the activities of the enzymes stored in NaCl-free or 10% NaCl-included buffers were compared, it is observed that the activity was improved 1.55-fold with the addition of NaCl (data not shown). For further studies, elution samples were collected, buffer exchanged and concentrated using the Tris–HCl buffer containing 10% NaCl, pH 8.
The purification table (Table 2) shows that the protein was recovered with a yield of 59% and specific activity was increased by 4.72-fold when the enzyme was purified.
Substrate specificity
Lipolytic enzymes can catalyze many types of reactions by converting a wide variety of substrates to products. To determine the substrate specificity of the enzyme, enzymatic reactions were set up using para-nitrophenyl esters with 4, 6, 8, 10, 12 and 16 C chain length (para-nitrophenyl butyrate, para-nitrophenyl hexanoate, para-nitrophenyl octanoate, para-nitrophenyl decanoate, para-nitrophenyl dodecanoate and para-nitrophenyl hexadecanoate, respectively) as substrates.
As can be seen in Table 3 (and Supplementary Fig. 4), the enzyme has a significant activity towards short-chain fatty acid esters, pNP-butyrate, pNP-hexanoate and pNP-octanoate. The activity is dramatically low when substrates with a carbon chain length of ten or higher are used. While the catalytic turnover numbers for pNP-butyrate and pNP-octanoate are close to each other (6.03 s−1 and 6.36 s−1, respectively), the enzyme has a greater activity towards pNP-hexanoate with a turnover number of 12.30 s−1. The Km values were calculated as 0.95 mM, 0.15 mM and 0.19 mM for pNP-butyrate, pNP-hexanoate and pNP-octanoate, respectively. The kcat/Km values indicate that the enzyme is specific for pNP-hexanoate (82 s−1 mM−1). To differentiate the esterases and lipases in terms of their substrate specificity, ester chain length of ten carbon atoms is usually accepted as the set value (López-López et al. 2015). Due to the preference of this enzyme towards short-chain esters, we can conclude that the enzyme is an esterase.
Effect of pH and temperature
As shown in Fig. 3a, the enzyme showed higher activity in basic values than acidic values. Optimum pH of the enzyme was determined as 9. To assess the stability of the enzyme at different pH conditions, it was pre-incubated at different pH values. The enzyme displayed stability between pH 8 and 10. While incubation at neutral pH slightly decreased the activity of the enzyme, incubation at acidic buffers completely inhibited the enzyme (Fig. 3b).

a Effect of pH on the activity. The enzymatic reactions were set with respective buffers at different pH. b Effect of pH on the stability. 10 µM enzyme solution was incubated at respective buffers with different pH at 4 °C for 24 h. At the end of incubation, the activity was measured according to the standard assay. c Effect of temperature on the activity. The enzymatic reactions were set at respective temperatures. d Effect of temperature on the stability. 10 µM enzyme solution was incubated at different temperatures for 20 min. The residual activity was measured according to the standard assay
The optimum temperature was found as 30 °C, in accordance with the mesophilic nature of the environmental samples used in this study. However, the enzyme is cold-active which can show 47% of its maximum activity even at 5 °C, and 63% of its maximum activity at 15 °C. The activity of the enzyme rapidly decreased at a temperature above 40 °C. At 70 °C, the enzyme showed 8% of its maximum activity (Fig. 3c). The effect of temperature on the stability of the enzyme was also assessed (Fig. 3d). 10 µM of enzyme solution was incubated for 20 min at varying temperatures from 25 to 80 °C with 5 °C increments. The enzyme retained most of its activity (92%) until 55 °C but incubation at 60 °C caused a high decrease in the activity (27%).
Effect of NaCl and solvents
The effect of NaCl on the activity of the enzyme was evaluated by adding NaCl into substrate emulsion to give a final concentration of 1–25%. The enzyme was added to the solution and the absorbance values were recorded at the end of 20 min. The results are given in Fig. 4a. Until 10% of NaCl (1.7 M), the enzyme retains 80% of its maximum activity. While the activity drops to 40% of its maximum at 20% NaCl (3.4 M) concentration, and it is totally lost at 25% NaCl (4.25 M). In contrast to the effect of NaCl on the activity, the enzyme was stable after incubation for 24 h in the Tris–HCl buffer containing 25% NaCl (Fig. 4b).

a Effect of NaCl on the activity. The reactions were set as to include NaCl at different concentrations. b Effect of NaCl on the stability. 100 µM of enzyme solution was incubated at buffers containing a different concentration of NaCl for 24 h at 4 °C. The residual activity was measured according to the standard assay. c Effect of solvents on the activity. The reactions were set as to include 10% solvents. d Effect of solvents on the stability. 100 µM of enzyme solution was incubated in buffers containing 15% (dark gray) and 30% (light gray) solvents for 24 h at 4 °C. The residual activity was measured according to the standard assay
The activity of the enzyme in the presence of 10% organic solvent was measured (Fig. 4c). No significant activity was observed in the presence of chloroform, butanol and 1-propanol (< 5%). The activity was lower than 50% of the maximum activity in the presence of hexane, acetone, DMF, isopropanol and ethanol. The presence of methanol and DMSO reduced the activity to 81% and 92%, respectively, while glycerol did not have a significant effect. Except for DMSO, apolar solvents strongly reduce the activity of the enzyme while more polar ones have a smaller effect. To test the stability of the enzyme, 100 µM of enzyme solution was incubated in 50 mM Tris–HCl buffer, pH 8, with 10% solvent for 24 h at 4 °C. The residual activity was measured with the standard assay (Fig. 4d). The minimum activity (86%) at 15% solvent concentration was obtained with 1-propanol. Hexane, acetone, DMF, DMSO, isopropanol, ethanol, methanol and glycerol slightly increased the activity, while chloroform and butanol slightly reduced the activity at 15% concentration. Comparison of the activities of the enzymes incubated with 30% solvent (acetone, isopropanol and 1-propanol) showed a reduction in activity to 85, 62 and 23% of its maximum value, respectively. No significant effect was observed for other solvents at 30% concentration.
Effect of metal ions, detergents and inhibitors on the activity of the enzyme
To assess the effect of metal ions on the activity of the enzyme, metal salts were added at 1 mM and 5 mM final concentrations to the reaction mixture (Fig. 5a). Addition of 1 mM Mg2+, Ca2+, and Mn2+ significantly enhanced the activity (approx. %30). Same metal ions increased the activity (4%–10%) when used in 5 mM concentration. While 1 mM Fe2+, Cu2+, Co2+ and Ni+2 improved the activity, increasing the concentration to 5 mM completely inhibited the activity. 1 mM K+ increased the activity by 28%, while 5 mM K+ did not significantly affect. Ag+ and Zn2+ had a great inhibitory effect on the enzyme. The enzyme showed 91% of its maximum activity with 1 mM Al3+ but increasing the concentration to 5 mM completely inhibited the activity.

a Effect of metal ions, b detergents and c inhibitors on the activity of the enzyme. The compounds were included in the reaction mixture at final concentrations of 1 mM (dark gray) and 5 mM (light gray)
The sensitivity of the enzyme to detergents was assessed by the addition of 1 mM and 5 mM reagents into the reaction mixture. As shown in Fig. 5b, the addition of 1 mM Triton-X100, Tween 80 and Nonidet P-40 did not have any effect. Enzyme still retained more than 80% of its activity when 5 mM Triton X-100, Tween-80 and Nonidet P-40 were present in the reaction. 1 mM SDS as an anionic detergent and CTAB as a cationic detergent lowered the activity of the enzyme by 14% and 9%, respectively. When the concentration was increased to 5 mM, the enzyme was inactivated by SDS and CTAB.
The effect of the inhibitors on the activity of the enzyme was investigated (Fig. 5c). Inhibitors PMSF, DTT, EDTA and EGTA were added at a final concentration of 1 mM and 5 mM. PMSF, which is a serine hydrolase inhibitor, sulfonylates the hydroxyl groups of the serine residues in a protein, which are crucial in catalysis in case of hydrolases (Hanson et al. 2005). As can be predicted from the amino acid sequence, the hAGEst is a serine hydrolase, so it is completely inhibited by 1 mM PMSF. DTT reduces the disulfide bonds in proteins. The protein is not expected to have any disulfide bond because there is only one cysteine residue in the primary sequence of the protein. However, the reduced activity in the presence of DTT (37% and 25% for 1 mM and 5 mM, respectively) can be associated with other factors. At the beginning of the reaction, the substrate in the reaction tube quickly became hydrolyzed, releasing a yellow color with high intensity. This shows that DTT acts on the substrate, changes the chemistry of the reaction and activity measurement became inapplicable.
The effects of chelating agents EDTA and EGTA on the activity were also investigated. In the presence of 1 mM EGTA, the activity was not changed while 5 mM EGTA inhibited the activity by 12%. EGTA especially acts on Ca2+ ions and the activity of the esterase was increased with Ca2+ (Fig. 5a). Although any calcium ion was not added into the reaction, the relatively small EGTA inhibition can be associated with the chelating of the trace amount of calcium, which may be bound to the enzyme. The activity was inhibited by EDTA in both 1 mM and 5 mM concentrations by 10% and 33%, respectively, which can be explained with the same situation as EGTA.
Discussion
Metagenomic approach has been widely used for the identification of novel enzymes from a wide variety of environments. In the present work, we identified, isolated and characterized an esterase, hAGEst, from a hypersaline lake, Acıgöl, by sequence-based metagenomics.
The amino acid composition of the hAGEst is consistent with the general features of proteins from halophilic microorganisms. It is statistically proven that halophilic proteins have a higher proportion of acidic amino acids and small hydrophobic amino acids, while they have a lower content of basic amino acids. Due to the complex interaction between amino acids, salt and water, the halo adaptation of proteins is achieved (Madern et al. 2000). The percentage of the basic and acidic amino acid residues in hAGEst is 13.8% and 14.3%, respectively. The total percentage of small hydrophobic residues (Gly, Ala and Val) is 30%, while Lys is present with a percentage of 0.8%, consistent with the amino acid compositions of halophilic proteins.
Lipolytic enzymes consist of lipases and esterases. The main criterion to differentiate them is their substrate specificity. Lipases usually prefer substrates with a long carbon hydrophobic chain while esterases can hydrolyze substrates with short carbon chain (López-López et al. 2015). hAGEst was found as the most active towards pNP-hexanoate, with a kcat/Km of 82.03 (s−1 mM−1) and a specific activity of 25.99 ± 0.42 U/mg (Table 3). Wang et al. studied a salt-tolerant esterase from Alkalibacterium sp. SL3 and found kinetic parameters as Km = 0.15 ± 0.01 mM and kcat = 307.69 s−1 when pNP-acetate was used as the substrate (Wang et al. 2016). Another halophilic esterase from an Antarctic soil sample was found to have a Km value of 0.87 ± 0.12 mM and a kcat value of 0.35 s−1 when para-methylumbelliferyl butyrate was used as a substrate (Castilla et al. 2017). Also, there is another report that lipolytic enzymes from halophilic strains were characterized and their Km values were in the range between 0.119 and 1.363 mM (Ozcan et al. 2009). The specific activity of an esterase from a deep-sea hydrothermal vent was found as 18.2 U/mg for pNP-butyrate and 1.9 U/mg for pNP-octanoate (Zhu et al. 2013). In another study, the specific activities of lipolytic enzymes from deep-sea sediments were reported as between 1.7 and 558.2 U/mg for different substrates (Lee et al. 2011). As we can conclude, kinetic parameters for different enzymes isolated from different habitats can vary. The outstanding features of the enzymes are usually revealed by detailed biochemical characterization studies.
Characterization of the hAGEst revealed that the enzyme shows the best activity at pH 9, which is an expected value for proteins from halophilic microorganisms. The habitat of the halophilic microorganisms is usually alkaline environment and enzymes from these organisms generally show maximum of their activities at slightly basic pH values. This feature usually makes halophilic enzymes applicable in detergent formulations (López-López et al. 2014). Li et al. (2012) isolated an extracellular esterase from Halobacillus sp. strain LY5 and found the optimum pH of the enzyme as 10. In another study, a halophilic lipase was isolated and optimum pH was found to be 9 using pNP-decanoate as a substrate (Esakkiraj et al. 2016). In a similar study, optimum pH of different isolates from halophilic archaeal strains was found to be between 8 and 8.5 (Ozcan et al. 2009). The enzyme retained nearly 90% of its maximum activity at pH 8.5. Increasing the pH to 9.5 did not dramatically affect the activity towards pNP-octanoate. As the chain length of the para-nitrophenyl esters decreases, the autohydrolysis occur at alkaline pH values (Glogauer et al. 2011), which reduces the difference between the absorbance values of blank and enzyme-added reactions. So, the activity measurements pNP-butyrate and pNP-hexanoate at pH 9 did not reveal reliable results.
The hAGEst showed maximum activity at 30 °C, consistent with the mesophilic nature of the environmental source (Fig. 3c). In a similar study, the optimum temperature of an esterase isolated from sea sediment was found as 35 °C and enzyme lost much of its activity at 40 °C (Zhang et al. 2017). A similar situation is observed in our case: the activity of the enzyme faces a significant activity loss when the temperature was above 40 °C. Interestingly, hAGEst retained about half of its activity at temperatures as low as 5 °C and 10 °C. This ability is especially important in the industry in terms of lowering energy costs. Some reactions require higher temperatures for catalysis but cold-active enzymes can effectively catalyze the same reaction at lower temperatures. These enzymes can find application in the production of detergent and food, bioremediation of soil or sea and biocatalysis reactions in which thermo-sensitive or highly volatile compounds are used (López-López et al. 2014).
Although the hAGEst could not show significant activity temperatures above 40 °C, thermostability assays showed that the enzyme is still stable after incubation at 55 °C for 20 min (Fig. 3d). This can be explained by the reversible or only partial denaturation at mid-level elevated temperatures.
Biotransformation in organic solvents has numerous advantages such as high solubility of hydrophobic reactants, a thermodynamic shift from hydrolysis to synthesis, and the prevention of unwanted by-products by the elimination of water molecules (Salihu and Alam 2015). So, enzymes applicable in organic solvents are of great interest in biotechnology. hAGEst showed significantly high activity in the presence of 10% DMSO, methanol and glycerol (Fig. 4c). The comparison of the effect of metal ions (1 mM) on the activity of hAGEst and other enzymes in the literature is summarized in Table 4. The enzymes to compare are selected according to their origin or salt tolerance properties: a metal-resistant esterase from Red Sea brine pool (Mohamed et al. 2013), a salt-tolerant esterase from Tibetan glacier (De Santi et al. 2015), a halophilic esterase from Antarctic soil sample (Castilla et al. 2017), and a salt-tolerant esterase from deep-sea sediment (Zhang et al. 2017). The activity of hAGEst is higher than other enzymes for many of the metal ions.
The effect of the presence of surfactants at 1 mM and 5 mM concentrations was evaluated (Fig. 5b). Detergents at 1 mM concentration did not significantly affect the activity but when the concentration was increased, the inhibitory effect of ionic detergents, SDS and CTAB, was observed. When we look at the literature, it is found that as ionic detergents, SDS (Mohamed et al. 2013; Wongwatanapaiboon et al. 2016; Castilla et al. 2017) and CTAB (Wongwatanapaiboon et al. 2016) dramatically reduce the activities of esterases and lipases. Other non-ionic detergents, Triton X-100, Tween-80 and Nonidet P-40 did not cause dramatic change at 5 mM concentration. The activity of a lipase from Aureobasidium melanogenum was completely inhibited by 1 mM Triton X-100 (Wongwatanapaiboon et al. 2016). In another study, esterase from Halobacillus sp. was isolated and 2 mM Triton X-100 reduced the activity of the enzyme to 90% of its maximum activity (Li et al. 2012). In our case, the enzyme retained 88% activity of its maximum value in the presence of 5 mM Triton X-100.
To our knowledge, this is the only report on metagenomic exploration of Lake Acıgöl. The most similar study was conducted by Ozcan et al. (2000) in which five halophilic archaeal isolates from various hypersaline lakes in Turkey were investigated for their lipolytic activity. Although we tried to make a few comparisons above, making a full comparison between the two studies is not possible, though the designation for the enzyme from Lake Acıgöl is not indicated and only crude culture supernatants were used for activity assays, rather than purified recombinant proteins.
Conclusion
In conclusion, a novel esterase gene was successfully isolated from environmental source using sequence-based metagenomic approach. The enzyme was not only cloned and purified, but also detailed biochemical characterization was performed. hAGEst is the first enzyme, which is recombinantly produced from the hypersaline lake, Acıgöl, by metagenomic approach. Although the enzyme does not show a significantly high specific activity when compared to similar proteins, it has outstanding features such as high activity at low temperatures, high tolerance to DMSO and metal ions. The possible utilization of this enzyme will be investigated by further studies. The ability of the enzyme to catalyze diverse reactions such as transesterification, interesterification and hydrolysis with different substrates will be evaluated. After detailed biochemical characterization of the enzyme, possible industrial application areas (detergent formulations, production of pharmaceuticals, other fine chemicals, etc.) will be searched.
References
Arpigny JL, Jaeger KE (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343:177–183
Balci N, Demirel C (2016) Formation of carbonate nanoglobules by a mixed natural culture under hypersaline conditions. Minerals 6:122
Balci N, Demirel C, Akcer S, Gültekin AH, Kurt MA (2018) Evaluating abiotic and microbial factors on carbonate precipitation in Lake Acigöl, a hypersaline lake in Southwestern Turkey. Quat Int 486:116–128
Balci N, Menekşe M, Karagüler NG, Sönmez MS, Meister P (2016) Reproducing authigenic carbonate precipitation in the hypersaline lake Acigöl (Turkey) with microbial cultures. Geomicrobiol J 33:758–773
Bell PJL, Sunna A, Gibbs MD, Curach NC, Nevalainen H, Bergquist PL (2002) Prospecting for novel lipase genes using PCR. Microbiol 148:2283–2291
Borrelli GM, Trono D (2015) Recombinant lipases and phospholipases and their use as biocatalysts for industrial applications. Int J Mol Sci 16:20774–20840
Castilla A, Panizza P, Rodríguez D, Bonino L, Díaz P, Irazoqui G, Rodríguez Giordano S (2017) A novel thermophilic and halophilic esterase from Janibacter sp. R02, the first member of a new lipase family (Family XVII). Enzyme Microbiol Technol 98:86–95
Contreras-Moreira B, Sachman-Ruiz B, Figueroa-Palacios I, Vinuesa P (2009) primers4clades: a web server that uses phylogenetic trees to design lineage-specific PCR primers for metagenomic and diversity studies. Nucleic Acids Res 37:95–100
Cowan DA, Ramond JB, Makhalanyane TP, De Maayer P (2015) Metagenomics of extreme environments. Curr Opin Microbiol 25:97–102
De Godoy Daiha K, Angeli R, De Oliveira SD, Almeida RV (2015) Are lipases still important biocatalysts? A study of scientific publications and patents for technological forecasting. PLoS ONE 10:1–20
De Santi C, Ambrosino L, Tedesco P, Zhai L, Zhou C, Xue Y, Ma Y, de Pascale D (2015) Identification and characterization of a novel salt-tolerant esterase from a Tibetan glacier metagenomic library. Biotechnol Progress 31:890–899
Esakkiraj P, Prabakaran G, Maruthiah T, Immanuel G, Palavesam A (2016) Purification and characterization of halophilic alkaline lipase from Halobacillus sp. Proc Natl Acad Sci India Sect B Biol Sci 86:309–314
Fernández AB, Ghai R, Martin-Cuadrado AB, Sánchez-Porro C, Rodriguez-Valera F, Ventosa A (2014) Prokaryotic taxonomic and metabolic diversity of an intermediate salinity hypersaline habitat assessed by metagenomics. FEMS Microbiol Ecol 88:623–635
Ferrer M, Golyshina OV, Chernikova TN, Khachane AN, dos Santos VAM, Yakimov MM, Timmis KN, Golyshin PN (2005) Microbial enzymes mined from the Urania deep-sea hypersaline anoxic basin. Chem Biol 12:895–904
Fischer M, Pleiss J (2003) The lipase engineering database: a navigation and analysis tool for protein families. Nucleic Acids Res 31:319–321
Ghai R, Pasic L, Fernandez AB, Martin-Cuadrado AB, Mizuno CM, McMahon KD, Papke RT, Stepanauskas R, Rodriguez-Brito B, Rohwer F, Sanchez-Porro C, Ventosa A, Rodriguez-Valera F (2011) New abundant microbial groups in aquatic hypersaline environments. Sci Rep 1:1–10
Giegerich R, Meyer F, Schleiermacher C (1996) GeneFisher–software support for the detection of postulated genes. Proc Int Conf Intell Syst Mol Biol 4:68–77
Glogauer A, Martini VP, Faoro H, Couto GH, Müller-Santos M, Monteiro RA, Mitchell DA, de Souza EM, Pedrosa FO, Krieger N (2011) Identification and characterization of a new true lipase isolated through metagenomic approach. Microb Cell Fact 10:1–15
Hanson CV, Nishiyama Y, Paul S (2005) Catalytic antibodies and their applications. Curr Opin Biotechnol 16:631–636
Hasan F, Shah AA, Hameed A (2009) Methods for detection and characterization of lipases: a comprehensive review. Biotechnol Adv 27:782–798
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9:868–877
Iwai S, Chai B, Sul WJ, Cole JR, Hashsham S, Tiedje JM (2010) Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J 4:279–285
Jiang Z, Wang H, Ma Y, Wei D (2006) Characterization of two novel lipase genes isolated directly from environmental sample. Appl Microbiol Biotechnol 70:327–332
Kotik M (2009) Novel genes retrieved from environmental DNA by polymerase chain reaction: current genome-walking techniques for future metagenome applications. J Biotechnol 144:75–82
Kumar P, Kashmir S (2012) Characterization of a thermostable lipase showing loss of secondary structure at ambient temperature. Mol Biol Rep 39:2795–2804
Labes A, Karlsson EN, Fridjonsson OH, Turner P, Hreggvidson GO, Kristjansson JK, Holst O, Schönheit P (2008) Novel members of glycoside hydrolase family 13 derived from environmental DNA. Appl Environ Microbiol 74:1914–1921
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lee JH, Jeon JH, Kim JT, Lee HS, Kim SJ, Kang SG, Choi SH (2011) Novel lipolytic enzymes identified from metagenomic library of deep-sea sediment. J Evid Based Complementary Altern Med 2011:1–9
Li X, Yu HY, Lin YF (2012) Purification and characterization of an extracellular esterase from a moderately halophilic bacterium, Halobacillus sp. strain LY5. Afr J Biotechnol 11:6327–6334
Li M, Yang LR, Xu G, Wu JP (2016) Cloning and characterization of a novel lipase from Stenotrophomonas maltophilia GS11: the first member of a new bacterial lipase family XVI. J Biotechnol 228:30–36
Liebeton K, Eck J (2004) Identification and expression in E. coli of novel nitrile hydratases from the metagenome. Eng Life Sci 4:557–562
Linhart C, Shamir R (2002) The degenerate primer design problem. Bioinformatics 18:S172–S181
López-López O, Cerdán ME, González Siso MI (2014) New extremophilic lipases and esterases from metagenomics. Curr Protein Pept Sci 15:445–455
López-López O, Cerdán ME, González-Siso MI (2015) Thermus thermophilus as a source of thermostable lipolytic enzymes. Microorganisms 3:792–808
Ma Y, Galinski EA, Grant WD, Oren A, Ventosa A (2010) Halophiles 2010: life in saline environments. Appl Environ Microbiol 76:6971–6981
Madern D, Ebel C, Zaccai G (2000) Halophilic adaptation of enzymes. Extremophiles 4:91–98
Mohamed YM, Ghazy MA, Sayed A, Ouf A, El-Dorry H, Siam R (2013) Isolation and characterization of a heavy metal-resistant, thermophilic esterase from a red sea brine pool. Sci Rep 3:1–8
Morris DD, Reeves R, Gibbs MD, Saul DJ, Bergquist P (1995) Correction of the b-mannanase domain of the celC pseudogene from Caldicellulosiruptor saccharolyticus and activity of the gene product on kraft pulp. Appl Environ Microbiol 61:2262–2269
Narasingarao P, Podell S, Ugalde JA, Brochier-Armanet C, Emerson JB, Brocks JJ, Heidelberg KB, Banfield JF, Allen EE (2012) De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J 6:81–93
Oren A (2008) Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Syst 4:2
Ozcan B, Ozyilmaz G, Cokmus C, Caliskan M (2009) Characterization of extracellular esterase and lipase activities from five halophilic archaeal strains. J Ind Microbiol Biotechnol 36:105–110
Pleiss J, Fischer M, Peiker M, Thiele C, Schmid RD (2000) Lipase engineering database: understanding and exploiting sequence-structure-function relationships. J Mol Catal B Enzym 10:491–508
Ramnath L, Sithole B, Govinden R (2017) Classification of lipolytic enzymes and their biotechnological applications in the pulping industry. Can J Microbiol 63:1–14
Ribeiro BD, Castro AM, Coelho MAZ, Freire DMG (2011) Production and use of lipases in bioenergy: a review from the feedstocks to biodiesel production. Enzyme Res 2011:1–16
Rose TM, Schultz ER, Henikoff JG, Pietrokovski S, McCallum CM, Henikoff S (1998) Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res 26:1628–1635
Salihu A, Alam MZ (2015) Solvent tolerant lipases: a review. Process Biochem 50:86–96
Samoylova YV, Sorokina KN, Romanenko MV, Parmon VN (2018) Cloning, expression and characterization of the esterase estUT1 from Ureibacillus thermosphaericus which belongs to a new lipase family XVIII. Extremophiles 22:271–285
Stothard P (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28:1102–1104
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evolution 30:2725–2729
Tang K, Utairungsee T, Kanokratana P, Sriprang R, Champreda V, Eurwilaichitr L, Tanapongpipat S (2006) Characterization of a novel cyclomaltodextrinase expressed from environmental DNA isolated from Bor Khleung hot spring in Thailand. FEMS Microbiol Lett 260:91–99
Villeneuve P, Muderhwa JM, Graille J, Haas MJ (2000) Customizing lipases for biocatalysis: a survey of chemical, physical and molecular biological approaches. J Mol Catal B Enzym 9:113–148
Wang G, Wang Q, Lin X, Bun NT, Yan R, Lin J, Ye X (2016) A novel cold-adapted and highly salt-tolerant esterase from Alkalibacterium sp. SL3 from the sediment of a soda lake. Sci Rep 6:1–10
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (2009) Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191
Wei X, Kuhn DN, Narasimhan G (2003) Degenerate primer design via clustering degenerate primer design via clustering. Proc IEEE Comput Soc Bioinform Conf 2003:75–83
Winkler UK, Stuckmann M (1979) Glycogen, hyaluronate, and some other polysaccharides greatly enhance the formation of exolipase by Serratia marcescens. J Bacteriol 138:663–670
Wongwatanapaiboon J, Klinbunga S, Ruangchainikom C, Thummadetsak G, Chulalaksananukul S, Marty A, Chulalaksananukul W (2016) Cloning, expression, and characterization of Aureobasidium melanogenum lipase in Pichia pastoris. Biosci Biotechnol Biochem 80:2231–2240
Zhang Y, Hao J, Zhang YQ, Chen XL, Xie BB, Shi M, Zhou BC, Zhang YZ, Li PY (2017) Identification and characterization of a novel salt-tolerant esterase from the deep-sea sediment of the south china sea. Front Microbiol 8:1–10
Zhao L, Han B, Huang Z (2004) Epoxide hydrolase-catalyzed enantioselective synthesis of chiral 1, 2-diols via desymmetrization of m eso-epoxides. J Am Chem Soc 126:11156–11157
Zhu Y, Li J, Cai H, Ni H, Xiao A, Hou L (2013) Characterization of a new and thermostable esterase from a metagenomic library. Microbiol Res 168:589–597
Funding
This study was funded by Istanbul Technical University (project no: 39574).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by M. Moracci.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tutuncu, H.E., Balci, N., Tuter, M. et al. Recombinant production and characterization of a novel esterase from a hypersaline lake, Acıgöl, by metagenomic approach. Extremophiles 23, 507–520 (2019). https://doi.org/10.1007/s00792-019-01103-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-019-01103-w