
Abstract
The Antarctic soil microbial community has a crucial role in the growth and stabilization of higher organisms, such as vascular plants. Analysis of the soil microbiota composition in that extreme environmental condition is crucial to understand the ecological importance and biotechnological potential. We evaluated the efficiency of isolation and abundance of strict anaerobes in the vascular plant Deschampsia antarctica rhizosphere collected in the Antarctic’s Admiralty Bay and associated biodiversity to metabolic perspective and enzymatic activity. Using anaerobic cultivation methods, we identified and isolated a range of microbial taxa whose abundance was associated with Plant Growth-Promoting Bacteria (PGPB) and presences were exclusively endemic to the Antarctic continent. Firmicutes was the most abundant phylum (73 %), with the genus Clostridium found as the most isolated taxa. Here, we describe two soil treatments (oxygen gradient and heat shock) and 27 physicochemical culture conditions were able to increase the diversity of anaerobic bacteria isolates. Heat shock treatment allowed to isolate a high percentage of new species (63.63 %), as well as isolation of species with high enzymatic activity (80.77 %), which would have potential industry application. Our findings contribute to the understanding of the role of anaerobic microbes regarding ecology, evolutionary, and biotechnological features essential to the Antarctic ecosystem.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microbial life in the Antarctic continent represents a unique source for ecological and evolutionary studies of life on Earth. Moreover, the intrinsic potential for the discovery of new species with biotechnological applications is tantalizing. Antarctic biogeography is mainly characterized by extreme physicochemical conditions, including temperatures as low as −90 °C. According to the continental drift theory, isolation of Antarctica dates to around 120 million years ago (Rogers 2007; Clarke and Crame 1992). Studies based on molecular approaches about Antarctic vascular plants rhizosphere reveal a markedly different microbial phylogenetic profile compared with the rest of the planet, suggesting that this environment is a unique source of conserved microbial life from an intact era of microbial life evolution (Teixeira et al. 2010). Studies on microbial diversity based on culture-dependent and independent techniques were conducted in different regions of the Antarctic continent to measure the abundance and diversity of microbial communities. These studies unveiled an intriguing relationship between microbial community structure and presence of vegetation, latitude, and soil composition (Yergeau et al. 2009; Wynn-Williams 1996; Pointing et al. 2009; Harris and Tibbles 1997). In temperate climate environments, the soil microbiota is directly influenced by the vegetation, which modifies microbiota composition favoring certain groups of microorganisms, offering protection from low temperatures and harsh conditions; thus allowing for a more heterogeneous microbiota composition when compared soil without vegetation cover (Kowalchuk et al. 2002).
Worldwide, the phylum of bacteria most often detected in soil rhizosphere are Proteobacteria, Actinobacteria, Acidobacteria, Chloroflexi, Verrucomicrobia, Bacteroidetes, Planctomycetes, Gemmatimonadetes, and Firmicutes, as described by Janssen (2006), but anaerobic bacteria drove special attention when Teixeira et al. (2010) demonstrated a peculiar microbial community pattern in rhizosphere soil from Antarctic vascular plants of Admiralty Bay. This study revealed that Firmicutes were the most abundant phyla, followed by Actinobacteria and Proteobacteria phylum. Both Firmicutes and Actinobacteria phylum showed predominance of anaerobic bacteria orders (Clostridiales and Bifidobacteriales, respectively). This unusual profile contradicts studies on microbial diversity in Antarctica based on cultivation-dependent techniques. In general, cultivation-dependent studies have suggested a restricted microbial community, mainly aerobic or facultative anaerobic, with very few reports of anaerobic bacteria isolates (Zengler et al. 2002; Tindall 2004; Smith et al. 2006; Wagner and Wiegel 2008).
Alterations in the oxygen concentration in the atmosphere mark one important moment in life evolution; before that, anaerobic bacteria were the most important conductors of the main biological functions on Earth (Jeltsch 2013). This intriguing predominance of anaerobes in the Antarctic rhizosphere revealed by deep-sequencing techniques draws attention to their ecological role and physiological properties. It also points to an extraordinary reservoir of genetic and metabolic properties important for ecological, evolution, and biotechnological studies, stressing the need for cultivation of their representatives. The cultivation of this microbial community is the challenge of this study. Only a small amount of the microbial species found in natural environments is easily isolated and grown in artificial culture media (Hugenholtz et al. 1998; Rondon et al. 1999; Hugenholtz 2002; Connon and Giovannoni 2002; Torsvik et al. 2002; Song et al. 2009). The goal of this study is to isolate and cultivate the anaerobe microbial community in Antarctic rhizosphere. Here, we describe an array of anaerobe culture conditions, such as temperature, redox potential (redox), salt concentration, and alkalinity, are able to isolate a large number of anaerobic bacteria species from the vascular plant Deschampsia antarctica rhizosphere. We identified a range of microbial taxa whose abundance was associated with Plant Growth-Promoting Bacteria (PGPB). The genus Clostridium was the most abundant isolated taxa, followed by Bacteroides, Propionibacterium, and Enterobacter species. Growing these species in vitro is essential for understanding their physiology and metabolic repertoire for forward research in ecology and biotechnology application. The basic prerogative was to evaluate the efficiency and abundance of strict anaerobic bacteria isolation, associating biodiversity to metabolic perspective and enzymes activity features.
Materials and methods
Sampling site and procedure
The study was carried out at the Brazilian Antarctic Station, Estação Antártica Comandante Ferraz (EACF: 621040S, 581210W), located in Martel Inlet, Admiralty Bay, King George Island, Antarctic Peninsula, which is part of the South Shetlands Archipelago in Maritime Antarctica, during the austral summer of 2011–2012. Deschampsia antarctica vascular plants were sampled in triplicate at three sites (A, B, and C). In these areas, the soil was previously characterized as loamy sand (Simas et al. 2008). The sediments were obtained with a manual sampler and sterile Whirl pack bags. From each site, 500 g of soil were collected from a depth of 25–50 cm. The upper layer of soil was discarded due to the high oxygen concentration present, as suggested by the previous works (Bach et al. 2005). The samples were sealed in anaerobic jars, and anaerobic atmosphere was achieved using GasPack (Oxoid). The jars were kept at −20 °C for transportation and processing.
Sampling processing
The methodology used for bacterial isolation was conducted to obtain a landscape of different strict anaerobic bacteria from the rhizosphere (D. antarctica) across physicochemical media parameters. Three sample treatment variations were tested from the standard procedure of cultivation, by adding pre-incubation in oxygen gradient, heat shock, and a combination of both. The samples were then inoculated in brain-heart infusion (BHI) (Difco, USA) agar broth with various combinations of pH, temperature, and salinity (pH 6, 7 and 8; NaCl 0.5, 1.5 and 2.5 % w/v; temperature 5, 15, and 25 °C, respectively).
The sediment was diluted 1:5 in pre-reduced and anaerobically sterilize (PRAS) Phosphate buffer (pH 7.4) containing 1 mg/ml of cysteine, under CO2 flux to avoid oxygen. The suspension was sonicated in a water bath at a frequency of 55,000 cycles/s for 5 min. For the heat shock treatment, the samples were incubated in a water bath for 30 min at 60 °C. The samples were serially diluted tenfold to 10−3, and 0.1 ml of this dilution was spread over solid media. For the oxygen gradient pre-incubation, the samples were loaded in a high column containing thioglycolate media (Difco, USA). All the steps henceforward were performed in oxygen-free atmosphere, in an anaerobic incubator (Coylabs, USA). For the isolation and cultivation procedure, BHI agar was supplemented with hemin (5 μg/ml, Sigma Co.), menadione (10 μg/ml, Sigma Co). The plates were prepared with different combinations of pH, NaCl concentration and incubated for 24–72 h, in 5, 25, and 37 °C. All the cultures were tested for respiratory metabolism and stored frozen at −80 °C in 20 % (v/v) glycerol.
Molecular approach
Genomic DNA was obtained from all bacterial strains using the DNEasy Blood and Tissue kit (QIAGEN) according to manufacturer’s instructions. The integrity of the DNA was confirmed by electrophoresis in a 0.8 % agarose gel with 0.5 TBE buffer (450 mM Tris–borate, 10 mM EDTA (pH 8.0) and stained with Blue Green Loading Dye I (LGC Biotecnologia) under UV light (MiniBis Pro®, Dnr Bio-Imaging System). DNA samples were stored at −20 °C. BOX-PCR was initially performed using the oligonucleotide BOXA1R 5′CTACGCCAAGGCGACGCCTGACG3′, aiming to generate BOX profiles (Versalovic et al. 1994). The 50 μl reactions were composed of a mixture of buffer 1× Taq polymerase, 3.75 mM MgCl2, 0.2 mM dNTPs, 2.5 μl 100 % DMSO; primer-BOX 10ρ moles; and 1 μl and 1.5 U Taq DNA sample of each strain. The PCR program used comprises a cycle of denaturation of DNA strands for 7 min at 94 °C, followed by 35 cycles of 1 min at 94 °C, 1 min at 53 °C, 8 min at 65 °C, and extension at 65 °C for 16 min. After the PCR, an aliquot of 15 μL of the sample is subjected to electrophoresis at 80 Volts agarose gel (1.2 % w/v) in 1X TBE for 5 h. Aliquots of 15 µl of PCR products were loaded on 1.2 % agarose gels, submitted to electrophoresis run at 80 Volts for 3.5 h, stained with Blue Green Loading Dye I (LGC Biotecnologia), and visualized and photographed under UV light using the MiniBis Pro® (Dnr Bio-Imaging System).
Ribosomal RNA gene sequencing was performed for bacterial strains that presented different band patterns revealed by BOX-PCR. The primers used to amplify the whole 16S RNA gene were 27f 5′-AGA GTT TGA TCA TGG CTC AG-3′ and 1492 5′-GTT TAC CTT GTT ACG ACT T-3′. Amplification reactions were performed in a final volume of 50 μL containing 50 ng of DNA that was extracted from each sample, buffer 1× Taq polymerase, 2.5 mM MgCl2, 200 mol of each dNTP, 20 ρmol of each primer, 2.5 U Taq polymerase (Invitrogen), and sterile Milli-Q water to complete the volume. The PCR program used comprises a cycle of denaturation of DNA strands for 3 min at 94 °C followed by 35 cycles of 1 min at 94 °C, 1 min at 55 °C, and 1 min at 72 °C, and extension at 72 °C for 10 min. To verify the success of amplification, an aliquot of 5 µL of sample was subjected to electrophoresis at 80 Volts agarose gel (1.2 %) for 2 h. After the run, the gels were visualized with Blue Green I Dye Loading (LGC Biotechnology) and exposed to UV light, and then photographed in an image capture system (MiniBis Pro ®—Dnr Bio—Imaging System).
The rRNA 16 s gene sequencing of selected strains was performed using primers from the ends 27f 5′-AGA GTT TGA TCA TGG CTC AG-3′, 1492 5′-GTT TAC CTT GTT ACG ACT T-3′, intermediate primer 532 5′-CGT GCC AGC AGC CGC GGT AA-3′, and 907 5′-CCG TCA ATT CMT TTG AGT TT-3′. The amplified products were purified with the PCR Purification System kit following the manufacturer’s recommendations (Qiagen). Each sequencing reaction was performed by automatic sequencing megabase (Sanger, ABI 3100 Applied). The sequences obtained were analyzed using the Ribosomal Database Project (RDP) and GenBank to align with database sequences deposited.
For the metabolic profile, we used the software Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt). This software allowed us to generate the metabolic profile of the isolates by functional gene content predictions based on 16S rRNA gene data present in the Greengenes and KEGG databases (Langille et al. 2013).
DNA sequences were assigned as OTUs (Operational Taxonomic Unit) and each OUT was aligned with their closer species obtained from the reference database GenBank and the Ribosomal Database Project (RDP), and Mothur, Bioedit, and MEGA 5 software subsequently analyzed these sequences. The trees were generated by the neighbor-joining method, while the model was Jukes–Cantor with a value equal to 1000 bootstrap and the species of the Archaea domain Aquifex pyrophilus was used as root. All sequences with coverage in the range of 1200–1644 bps and high-quality sequencing were allocated to the tree for a better comparison of closeness or dissimilarity.
Enzymatic activities screening
The ratio of halo diameter substrate degradation and colony was measured to obtain the enzymatic activity Index (I)2 in the medium (Hankin et al. 1971; Hankin and Anagnostakis 1975). Were considered as potential producers of enzymes strains that showed enzymatic index greater than or equal to 2.0 (Lealem and Gashe 1994). For proteolytic activity on casein agar (milk powder), 1 % w/v. strains were inoculated in triplicate in spots and incubated for 48–72 h at 25 °C. For proteolytic activity on gelatin, 0.4 % of gelatin was used. The reading was made by adding the solution of HgCl2 (1.5 % HgCl2, mL 20 % HCl). A clear halo in contrast with the opaque background was observed and measured with a caliper. For lipase activity, direct visualization in the petri dish for the presence of halos that form after growth on media containing the respective culture media with 1 % of the substrate tributyrin and 2 % agar was used. After growth (48 h and 1 week at 25 °C), the dye Victoria blue B was added to detect acidification of the medium (Hasan et al. 2009). The VITEK® automated system (Biomerieux) was used to perform a series of identification tests and biochemical profiles of the isolates.
Diversity assessment and distribution analysis
Eight complete 16S strain sequences were used for an assessment of the anaerobic diversity in the region. Their closest matches were searched for on GeneBank searches and an alignment (ClustalW) of the compiled sequences was generated (Fernandez-Carazo et al. 2011). All sequences were clustered with Mothur using the average neighbor method generating operational taxonomic units (OTUs). Each OTU contains complete 16S sequences with at least 97 % of similarity. GeneBank also allowed for an evaluation of the geographical distribution of the OTUs by looking at the isolation origin of the strains closest matches.
Statistical analysis
Statistical analysis (unpaired Student’s t test) was done using the Prism® software (GraphPad Software, Inc., San Diego, CA, USA); results were considered statistically significant if p < 0.05.
Results
To isolate and cultivate the anaerobe bacteria species from the rhizosphere of Deschampsia Antarctica, we tested three sample treatment variations from the standard procedure of cultivation, by adding pre-incubation in oxygen gradient (O2Gd), heat shock (HS), and a combination of both. Since spores are both heat resistant and activated to germinate by heat, the HS sample treatment stimulates bacteria spores to germinate and produce vegetative cells. In addition, oxygen gradient treatment was performed to stimulate fastidious bacteria to grow. Treated soil samples were then inoculated in brain-heart infusion (BHI) agar broth with various combinations of pH, temperature, and salinity (pH 6, 7 and 8; NaCl 0.5, 1.5 and 2.5 % w/v; Temperature 5, 15, and 25 °C, respectively).
The methodology for bacteria isolation conducted resulted in the isolation of 577 colonies. Initially, these colonies were tested for oxygen susceptibility growth and analyzed in BOX-PCR to obtain a comparative profile to identify dissimilarities between different bacterial isolates. As a result, 185 colonies represent strict anaerobic strains, and from those, 91 isolates showed differences in DNA fragments profile in BOX-PCR.
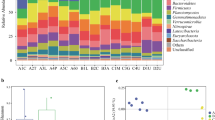
All 91 anaerobic isolated strains were identified by the sequencing of rrs phylogenetic marker gene. The composition of a cultivated anaerobic bacteria community present in the rhizosphere of the vascular plant D. antarctica was analyzed using Ribosomal Database Project (RDP) and its similarity indices were compared with sequences deposited in the GenBank database. The relative abundance of the isolates was demonstrated by phyla, order, and genera (Fig. 1), detailing the phylogenetic groups associated with the rhizosphere.

Distribution of partial sequences of bacterial isolates 16S rRNA genes from the rhizosphere of the Antarctic species D. antarctica, classified using Ribosomal Database Project (RDP) at genus, order, and phylum levels
The results indicate a significant prevalence of the Firmicutes phylum (73 %). Actinobacteria (10 %), Bacteroidetes (15 %), and Proteobacteria (2 %) phyla were also isolated and cultured, however, in smaller proportions. Analyzing the relative abundance in the order level, we observe prevalence of Clostridiales, Bacteroidales, and Actinomycetales within the Firmicutes, Bacteroidetes, and Actinobacteria phyla, respectively. The genus distribution within the Firmicutes phylum showed prevalence of the genus Clostridium spp. (46 %) (Fig. 1). Among the other bacteria isolates, we were able to identify the presence of taxa within the plant growth-promoting bacteria (PGPB) group, well-known to be important in nitrogen fixation and tryptophan metabolism, such as Enterobacter spp. (10 %), Tissierella spp. (5 %), and Granulicatella spp. (2 %) (Fig. 1).
However, what draws the most attention is that 24 % of the bacteria isolates could not be classified at species level, indicating a high prevalence of new bacteria species and potential new genus category isolated from this extreme ecosystem (Fig. 1), in the rrs gene similarity analysis, those sequences hit less than 97 % similarity to entries in RDP or GenBank databases and with good coverage sequencing (more than 1200pb) of the rrs gene were considered as potential new species candidates. Among the 91 isolates sequenced, 22 presented as candidates for new species (Clostridium sp. ISOL106, Cryobacterium sp. ISOL110, Bacillus sp. ISOL12A, Bacteroides sp. ISOL21, Tissierella sp. ISOL25A, Bacteroides sp. ISOL32, Clostridium sp. ISOL34A, Propionibacterium sp. ISOL38A, Clostridium sp. ISOL49, Clostridium sp. ISOL53, Clostridium sp. ISOL5A, Bacteroides sp ISOL68, Clostridium sp. ISOL70, Clostridium sp. ISOL76, Clostridium sp. ISOL78, Clostridium sp. ISOL85, Clostridium sp. ISOL87, Clostridium sp. ISOL8A, Clostridium sp. ISOL9, Clostridium sp. ISOLG1, Clostridium sp. ISOL45A, and Clostridium sp. ISOL59), with 15 of these candidates related to the Clostridium genus. The vast majority of isolates identified belong to the Firmicutes phylum and Clostridium genus, a major representative of anaerobes, which is widely distributed in nature.
A phylogenetic tree comparing the isolates obtained in this work with the most closely related previously described species and their type strains found in RDP database was generated based on the rrs sequencing results. All sequences with good coverage and high-quality sequencing could be allocated to the tree for a better comparison of closeness or dissimilarity (Supplementary Figure S1). The isolates identity, as verified in the tree distribution, allowed us to fit them within four major phyla: Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria. In the Firmicutes phylum, five main branches closely allocated were observed. However, in some cases, it is possible to observe that the isolates are closer to each other than to their closest related strains in database (ISOL78, ISOL49, ISOL59, ISOL21A, and ISOL112), suggesting that they may be classified as a new genus.
The large number of new species candidates was obtained due to the broad conditions and treatments used for isolation. Among the different isolation conditions used, we observed that HS and O2Gd treatments have heightened the abundance and phylogeny diversity of the anaerobes strains isolated. It is important to note that 63.63 % of the new species candidates were obtained from HS treatment, indicating the importance of sample treatment in the isolation and culture of extremophile anaerobes.
The use of HS favors the isolation of Clostridiaceae, Propionibacteriaceae, and Bacillaceae families, regardless of the physicochemical conditions used in the culture medium. On the other hand, the use of O2Gd supports Bacteroidaceae family members with the cost of lower abundance of Clostridiaceae representatives (Fig. 2a–c). Although allowing higher frequency of isolation at mesophile conditions (0.5 % NaCl, 25 °C, and pH 7), O2Gd treatment did not show an increase in number of isolates, in general (Supplementary Figure 2Sa–c).

Relative proportion of taxonomic distribution by treatments from total of 91 isolates. a Taxonomic distribution of cultured bacteria (family level) from total isolates. b, c Taxonomic distribution of cultured bacteria (family level) by treatments, O2 gradient and heat shock, respectively
These results indicate that pre-treatment of the soil sample is extremely important to increase the phylogenetic range during isolation of anaerobe bacteria of extreme environments. Despite decreasing the absolute number of species isolated is some cases (Supplementary Figure 2d–i), O2Gd and HS treatment enable the isolation of members of rare families, such as Tissierellaceae, Peptostreptococcaceae, Microbacteriaceae, Flammeovirgaceae, and Enterobacteriaceae (Fig. 2b, c).
It is important to highlight that changes from neutral pH and low salinity (pH 7 and 0.5 % NaCl) in the culture medium decreased the number of anaerobic isolates drastically, in most of cases. The single exception was the culture condition 15 °C/pH 8/0.5 % NaCl. The two most important isolation procedures are presented in Supplementary Figures 2S A–C and 3S A–C, showing that the absence of treatment is important when physicochemical variations are used.
Those procedures were crucial to achieve a broad numbers of species, leading us to the next question; what is the role of those strains in the rhizosphere and their biotechnological features. PICRUSt algorithms allowed us to predict, from the 16S rRNA gene (16S), some functional capabilities as a community. The total anaerobe isolates as well as its subgroups divided by family profiles and treatments were analyzed. In general, we observed a functional representative community in comparison to microbial community described from the previous work when normalized by OTUs number (data not shown). From all available KEGG pathways, we elect five metabolism functions related to PGPB (Fig. 3a, b). We conducted the analysis with a cut-off distinction between the strains at family level to verify their functions in a rhizosphere environment. Gene pathways of the anaerobic strains from Propionibacteriaceae and Enterobacteriales family were related to the metabolism of inositol phosphate to solubilization and mineralization of phosphorus for plants. The strains from Bacteroidaceae showed a significant association with nitrogen fixation process when compared with others. Flammeovirgaceae, Microbacteriaceae, and Tissierella families were associated to the metabolism of tryptophan—Indole-3-Acetic Acid (IAA), which is related to the biosynthesis of hormones for plants. However, for propionate metabolism, no distinctions were found between families. Another important role associated with microorganisms is the resilience of the microbial community against xenobiotics. In this sense, the most prominent families were the Bacillales, Clostridiaceae, Microbacteriaceae, and Peptostreptococcaceae, which also have the greatest number of bacteria candidates for new species. The analysis comparing treatments for PGPB metabolism did not show any distinction (data not shown).

Metabolic distribution of the main functions associated with symbiosis between rhizosphere and plant. The relative proportions were calculated between the total numbers of genes indicated by PICRUSt algorithms for five of principal PGPB function sorted by: a association in relation to the level of family of the individual and b association in relation to the treatment used
Analyzing the isolates from a biogeographical distribution perspective, the Antarctic continent consists of a unique environment rich in endemic organisms. Our sequences have shown a geographical distribution restricted to the sampled and the new species candidates clustered in six OTUs, and their closest matches in the database were species of Clostridium (five out of six OTUs) and Bacteroides. Five isolated (ISOL_5A, ISOL_45, ISOL_106, ISOL_32, and ISOL_110) were unique to this study; consequently, these are considered endemic and restricted to Antarctic continent. On the other hand, OTU related to Clostridium perfringens cluster has a worldwide distribution and was found in various geographical locations with many representatives. Further biogeography studies based on microorganism distribution could be fundamental to understand the evolution of the Antarctic continent and, consequently, understand of why anaerobic bacteria are so prevalent at D. antarctica rhizosphere.
For phenotype biotechnological enzyme prospection, the study focused on two important industrial enzymes, the proteases and lipases. All isolates were tested for both enzymes activity. Overall, 38.46 % of isolates (35) exhibited some enzyme activity, but 26 isolates were considered positive (I 2 > 2) for proteolytic activity, and 12 isolates were positive for lipolytic activity (Supplementary Figure S4S). It is important to highlight that the treatment by HS enhanced the isolation of strains with positive enzymatic activity. The majority of strains with protease activity (80.77 %) and lipase activity (66.3 %) were isolated after HS treatment. The highest activity for proteolysis was found is Clostridium genus and the lipolytic activity was mostly seen in Bacillus, Clostridium, and Propionibacterium genus. It is important to highlight that six of the new candidate species were positive for these enzyme activities (ISOL25A, ISOL34A, ISOL68, ISOL8A, ISOL9, and ISOL45A) (Supplementary Figure S4S). This study isolates and cultivates anaerobe bacteria from the Antarctic rhizosphere and suggests the potential industry applications of these species.
Discussion
The biggest challenge of investigating a microbial community in all its complexity and interactions is not only the technique used, but also the paradigm of thought used. The study focused on microbial communities with low tolerance for atmospheric oxygen inhabiting an environment with harsh temperature conditions. Since the origin of life, microorganisms evolve from a low redox potential context to an environment with approximately 21 % of molecular oxygen atmosphere. Anaerobic bacteria harbor, in their genome, the accumulation of billions years of evolution based on different selective pressures. The anaerobic bacteria biodiversity found in this region is poorly characterized. Our results demonstrate that some anaerobic bacteria are exclusively found on the Antarctic continent with prominent members of the rhizosphere environment in vascular plants and could also contain biotechnological importance.
Although limited available cultivation techniques do not allow a deep analysis of all microbial community, our results show that simple changes in the culture medium physicochemical composition were enough to improve the isolation of microbial species from the majority of the anaerobe taxon present on the environment (revealed by molecular approaches in Teixeira et al. 2010). The only exception is the taxa Bifidobacteriales.
The most commonly isolated taxon in our data set was the genus Clostridium, together with a notable presence of taxon candidate to new species. The use of sample treatment before isolation favors some microbial groups over others. The use of HS treatment for example was directly correlated to the isolation of high numbers of representatives of new species candidates and also to selecting taxon with high enzymatic activity features. Our results represent strong evidence that Antarctic microbial soil harbors great enzymatic potential for industrial application with cold-adapted enzymes that are essential to various industrial processes that require high financial costs to fit into the new reality of green industry. Further experimental research will be required to elucidate the molecular proteins characterization and other enzymes.
In an ecological aspect, our results suggest that microbial anaerobic community in rhizosphere contributes to the maintenance of the D. antarctica vascular plants mainly through the biogeochemical cycle of nitrogen and phosphate. Plant-associated bacteria have a strong effect on plant development and health, including germination, growth, disease protection, and productivity. In the survey area, a majority of new species should be well studied to really understand the ecology and metabolism in these ecosystems. Further analyses are needed for a complete description of this new bacterial anaerobic diversity.
In this study, we used PICRUSt algorithms to understand the potential contribution of the isolated strains for five of those principal PGPB function. Using Bacillaceae family for comparison, which has several species commercialized as PGPB, our results revealed that some representatives presented higher metabolisms potential than Bacillaceae, particularly the Bacteroidaceae family in nitrogen metabolism. The highest numbers of candidates for new species were found within the Clostridiaceae, which has also shown similar features to Bacillaceae group. Those strains reveal an interesting applicability by their intrinsic adaptation to cold, so countries with a temperate climate could be potential beneficiaries of those varieties. Nevertheless, reassessment of the data from the previous molecular study validates our observations not only in terms of phylogeny, but also for metabolic functions profile (Teixeira et al. 2010).
In summary, the isolation of anaerobic bacteria species from Deschampsia antarctica rhizosphere allows us to further understand the nature of microbial communities in the Antarctic continent and to explore the biological potential of these microorganisms. This study solidifies the notion that anaerobic microbiota can support life of a higher organism even in extreme temperature conditions.
References
Bach Q, Kim S, Choi S, Oh Y (2005) Enhancing the intrinsic bioremediation of PAH-contaminated anoxic estuarine sediments with biostimulating agents. J Microbiol 43:319–324
Clarke A, Crame JA (1992) The Southern Ocean benthic fauna and climate change: a historical perspective. Phil Trans R Soc B Biol Sci 338:299–309
Connon SA, Giovannoni SJ (2002) High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl Environ Microb 68:3878–3885
Fernandez-Carazo R, Hodgson DA, Convey P, Wilmotte A (2011) Low cyanobacterial diversity in biotopes of the Transantarctic Mountains and Shackleton Range (80–82°S), Antarctica. FEMS Microbial Ecol 77:503–517
Hankin JM, Anagnostakis S (1975) The use of solid media for detection of enzyme production by fungi. Mycologia 67:597–607
Hankin I, Zucker M, Sands DC (1971) Improved solid medium for the detection and enumeration of pectolytic bacteria. Appl Microbiol 22:205–209
Harris JM, Tibbles BJ (1997) Factors affecting bacterial productivity in soils on isolated inland nunataks in continental Antarctica. Microb Ecol 33:106–123
Hasan F, Shah AA, Hameed A (2009) Methods for detection and characterization of lipases: a comprehensive review. Biotech Adv 27:782–798
Hugenholtz P (2002) Exploring prokaryotic diversity in the genomic era. Genome Biol 3:1–8
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Janssen PH (2006) Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72:1719–1728
Jeltsch A (2013) Oxygen, epigenetic signaling, and the evolution of early life. Trends Biochem Sci 38:172–176
Kowalchuk GA, Buma DS, de Boer W, Klinkhamer PGL, van Veen JA (2002) Effects of above-ground plant species composition and diversity on the diversity of soilborne microorganisms. Antonie Van Leeuwenhoek 81:509–520
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA et al (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821
Lealem F, Gashe BA (1994) Amylase production by gram-positive bacterium isolated for fermeting tef (Eraglostis tef.). J Appl Microbiol 77:348–352
Pointing SB, Chan Y, Lacap DC, Lau MC, Jurgens JA, Farrell RL (2009) Highly specialized microbial diversity in hyper-arid polar desert. Proc Natl Acad Sci USA 106:19964–19969
Rogers AD (2007) Evolution and biodiversity of Antarctic organisms: a molecular perspective. Philos Trans R Soc Lond B Biol Sci 362:2191–2214
Rondon MR, Goodman RM, Handelsman J (1999) The Earth’s bounty: assessing and accessing soil microbial diversity. Trends Biotechnol 17:403–409
Simas FNB, Schaefer CEGR, Albuquerque Filho MR, Francelino MR, Fernandes Filho EI, Costa LM (2008) Genesis, properties and classification of Cryosols from Admiralty Bay, maritime Antarctic. Geoderma 144:116–122
Smith JJ, Tow LA, Stafford W, Cary C, Cowan A (2006) Bacterial diversity in three different Antarctic cold desert mineral soils. Microb Ecol 51:413–421
Song J, Oh H-M, Cho J-C (2009) Improved culturability of SAR11 strains in dilution-to-extinction culturing from the East Sea, West Pacific Ocean. FEMS Microbiol Lett 295:141–147
Teixeira LCRS, Peixoto RS, Cury JC, Sul WJ, Pellizari VH, Tiedje J, Rosado AS (2010) Bacterial diversity in rhizosphere soil from Antarctic vascular plants of Admiralty Bay, maritime Antarctic. ISME J 4:989–1001
Tindall BJ (2004) Prokaryotic diversity in the Antarctic: the tip of the iceberg. Microb Ecol 47:271–283
Torsvik V, Ovreas L, Thingstad TF (2002) Prokaryotic diversity magnitude, dynamics, and controlling factors. Science 296:1064–1066
Versalovic J, Schneider M, De Bruijn F, Lupski J (1994) Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Methods Mol Cell Biol 5:25–402
Wagner ID, Wiegel J (2008) Diversity of thermophilic anaerobes. Ann N Y Acad Sci 1125:1–43
Wynn-Williams DD (1996) Antarctic microbial diversity: the basis of polar ecosystem processes. Biodivers Conserv 5:1271–1293
Yergeau E, Schoondermark-Stolk SA, Brodie EL, Déjean S, DeSantis TZ, Gonçalves O, Piceno YM, Andersen GL, Kowalchuk GA (2009) Environmental microarray analyses of Antarctic soil microbial communities. ISME J 3:340–351
Zengler K, Toledo G, Rappe M, Elkins J, Mathur EJ, Short JM, Keller M (2002) Cultivating the uncultured. Proc Natl Acad Sci USA 99:15681–15866
Acknowledgments
The authors also thank Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) for fellowships and financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by H. Atomi.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Peixoto, R.J.M., Miranda, K.R., Lobo, L.A. et al. Antarctic strict anaerobic microbiota from Deschampsia antarctica vascular plants rhizosphere reveals high ecology and biotechnology relevance. Extremophiles 20, 875–884 (2016). https://doi.org/10.1007/s00792-016-0878-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-016-0878-y