
Abstract
Lipases expressed in microbial hosts have great commercial value, but their applications are restricted by the high costs of production and harsh conditions used in industrial processes, such as high temperature and alkaline environment. In this study, an Escherichia coli–Bacillus subtilis shuttle vector (pHS-cotB-Tm1350) was constructed for the spore surface display of the lipase Tm1350 from hyperthermophilic bacterium Thermotoga maritima MSB8. Successful display of the CotB-Tm1350 fusion protein on spore surface was confirmed by Western blot analysis and activity measurements. The optimal catalytic temperature and pH of the spore surface-displayed Tm1350 were 80 °C and 9, respectively, which were higher than non-immobilized Tm1350 (70 °C and pH 7.5). Analysis of thermal and pH stability showed that spore surface-displayed Tm1350 retained 81 or 70 % of its original activity after 8 h of incubation at pH 8 or pH 9 (70 °C), which were 18 % higher than the retained activity of the non-immobilized Tm1350 under the same conditions. Meanwhile, recycling experiments showed that the recombinant spores could be used for up to three reaction cycles without a significant decrease in the catalytic rate (84 %). These results suggested that enzyme display on the surface of the B. subtilis spore could serve as an effective approach for enzyme immobilization, which has potential applications in the harsh biochemical industry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipases are a complex and crucially important group of enzymes that have been found in various organisms, including animals, plants, and microorganisms. However, only lipases expressed in microbial hosts by genetic engineering technology have commercial value, e.g., in the production of foods, oils, fats, chiral compounds, leather, cosmetics, and biodiesel (Miroliaei and Nemat-Gorgani 2002). Despite the great interest in the applications of lipases in various industries, the high costs and harsh industrial conditions, such as time-consuming purification processes, and elevated catalysis temperatures, often restrict their use as biocatalysts (Snellman et al. 2002). Enzymes from hyperthermophiles are promising candidates, suitable for harsh industrial processes, because of their high intrinsic thermal and chemical stability (Gomes and Steiner 2004); indeed, these enzymes have been proven to be stable at high temperatures and in the presence of organic solvents (Chandrayan et al. 2008; Masomian et al. 2013).
Enzyme immobilization is an important method for increasing enzyme enantioselectivity, stability, and recyclability (Chandrayan et al. 2008). Common methods for achieving enzyme immobilization include adsorption or covalent attachment to solid materials, and entrapment in organic or inorganic polymers or microemulsions (Costas et al. 2008). However, during the time-consuming immobilization procedures, recovery of enzyme activity is reduced, and the associated costs are substantial (Roessl et al. 2010). Moreover, enzymes displayed on the surface of living cells also exhibit various problems associated with membrane permeability or crossing, which prevents the display of multimeric large enzymes due to the general transport mechanism of target proteins across the cell membrane and cell wall. Furthermore, industrial applications of the enzyme are still limited by harsh industrial processes, which result in the rupture of living cells (Kim et al. 2005).
Recently, enzymes displayed on the surface of Bacillus subtilis spore have drawn widely attention. This is due to several factors: firstly, the spore displays high stability and robust resistance to heat, radiation, and chemicals in a harsh environment (Nicholson et al. 2000). Secondly, the B. subtilis spore is assembled by various coat proteins expressed in the mother cell, thereby eliminating transport steps that block the display of multimeric large enzyme, for example, Escherichia coli β-galactosidase, a multimeric protein, has been displayed on the surface of the B. subtilis spore (Kwon et al. 2007). Thirdly, the spore can be easily separated from the vegetative cell by centrifugation. Since B. subtilis is generally recognized as safe bioresources and are used as food additives and in oral bacteriotherapy and bacterioprophylaxis (Green et al. 1999). Display systems using CotB, CotC, and CotG as anchoring motifs have been widely used for developing an oral vaccine (Isticato et al. 2001; Mauriello et al. 2004; Duc et al. 2007), but less for studying the enzymology (Wang et al. 2011; Qu et al. 2014).
Our previous unpublished studies demonstrate that the lipase Tm1350 from Thermotoga maritima MSB8 is a thermophilic, alkali tolerant, and methanol-activated lipase. In the current work, the lipase Tm1350 displayed on the B. subtilis spore coat with CotB as an anchoring motif was reported, the results showed that the thermal stability, pH stability, and reusability ratio of the Tm1350 displayed on the spore surface were all higher than those of its non-immobilized Tm1350. These data indicated that display of the enzyme on the B. subtilis spore surface may be an effective approach for enzyme immobilization and has significant potential application in the harsh biochemical industry.
Materials and methods
Chemicals and antiserum
The genomic DNA of T. maritima was obtained from Professor Weilan Shao at Jiangsu University. The high-copy-number shuttle vector pLJ was provided by Mingming Yan from Northwest A&F University. Restriction enzymes, PrimeSTAR HS DNA polymerase, and DNA ligase were all purchased from TaKaRa Biotechnology (Dalian, China). The p-nitrophenyl esters were obtained from Sigma (St. Louis, USA). All chemicals were of analytical grade.
Expression and purification of the recombinant Tm1350 protein were performed as follows: The gene tm1350 was cloned into the prokaryotic expression vector pET28a (+), and transformed into BL21 (DE3). The recombinant protein was then expressed by induction with isopropyl β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 0.1 mM. After induction for 5 h at 37 °C, cells were harvested by centrifugation. The harvested cells were resuspended in buffer (50 mM Tris–HCl, pH 8.0, 50 mM NaCl), and disrupted by sonication. The disrupted cells were centrifuged at 8000×g for 10 min at 4 °C, and the supernatant was then loaded on a nickel column (GenScript, Nanjing, China). Three bed volumes of binding buffer (100 mM NaH2PO4, 10 mM Tris–HCl, pH 8.0) were added to the nickel column, followed by the addition of three bed volumes of wash buffer (100 mM NaH2PO4, 10 mM Tris–HCl, 10 mM imidazole, pH 8.0) to the nickel column. The recombinant enzymes were eluted with the elution buffer (100 mM NaH2PO4, 10 mM Tris–HCl, 100 mM imidazole, pH 8.0), and then dialyzed in buffer A (50 mM PBS, pH 7.5) overnight to remove imidazole before concentration using a microfilter (Micro-con YM-10, Millipore Corp; Bedford, MA, USA). To confirm that Tm1350 was expressed on the B. subtilis spore surface, rat antiserum against recombinant Tm1350 protein was prepared by immunization of rats with purified recombinant Tm1350 protein following emulsification in Freund’s adjuvant in accordance with the Handbook of Animal Experiments (Sambrook et al. 1989). Rat antiserum was prepared in the Laboratory Animal Centre of Jiangsu University, China.
Plasmids and strain construction
To display Tm1350 on the surface of B. subtilis DB403 spores, an E. coli–B. subtilis shuttle vector (pHS-cotB-Tm1350) was constructed, based on the plasmid pLJ (Yang et al. 2006). The recombinant plasmid backbone pHS was amplified to remove the promoter of the maltose utilization operon (Pglv-inframe) using pLJ as a template, with Phs-F and Phs-R as primers. The amplified sequence was digested with EcoRI and then self-looped with T4 DNA ligase. A 1196-bp fragment, which contained the promoter region (PcotB) and the 927-bp DNA fragment encoding the first 304 amino acids of CotB (GenBank: CAB07789.1) and a flexible linker (Gly–Gly-Gly–Gly-Ser), was amplified from the chromosomal DNA of B. subtilis with Pcotb-F and Pcotb-R, and ligated into pHS cloning vector to generate the plasmid pHS-cotB. The flexible linker (Gly–Gly-Gly–Gly-Ser) was inserted between the C terminus of CotB, and the N terminus of the displayed enzyme, which is beneficial to promote the formation of the correct conformation of enzyme. The gene tm1350 was PCR amplified using the T. maritima genome as a template with two primers: Ptm-F2 and Ptm-R2. The gene was then digested with SpeI and XbaI and inserted into SpeI–XbaI-digested pHS-cotB plasmid. The recombinant plasmid was called pHS-cotB-Tm1350 (Table 1).
Preparation of spores
The transformation of pHS-cotB and pHS-cotB-Tm1350 into B. subtilis DB403 was operated by the two-step (GM I, GM II) method (Nicholson and Setlow 1990). B. subtilis DB403 was cultivated in Difco sporulation medium (DSM). After cultivation in DSM at 37 °C for 36 h, the spores of B. subtilis DB403, which harbored recombinant plasmids, were harvested by centrifugation and resuspended in 0.1 M phosphate sodium buffer (pH 7.0). The purification of spore was carried out as described without phenylmethylsulfonyl fluoride (PMSF) (Xu et al. 2011).
Western blot analysis
The expression of CotB-Tm1350 was confirmed by Western blotting with specific rat antiserum obtained in immunized rats. The specificity of the antiserum was determined using prokaryotically expressed recombinant Tm1350 protein as the antigen. Spore coat protein was extracted from purified spore suspension by sodium dodecyl sulfate (SDS)-dithiothreitol (DTT) treatment at 65 °C for 10 min (Nicholson and Setlow 1990). Western blot analysis was performed using standard protocols (Isticato et al. 2001), and horseradish peroxidase (HRP)–goat anti-mouse IgG (TaKaRa, Dalian, China) was used for immunodetection of the fusion protein. An enhanced HRP-DAB chromogenic substrate color kit (Tiangen, Beijing, China) was used for the color reaction according to the manufacturer’s instructions.
Enzymatic activity assay
Analysis of optical density (OD) at 405 nm was used for determination of enzyme activity with p-nitrophenyl esters as substrate (Winkler and Stuckmann 1979). The standard reaction mixture, consisting of 50 mM sodium phosphate (pH 8.0) and the purified spore suspension, was pre-incubated at 70 °C for 10 min. The reaction was then initiated after addition of p-nitrophenyl butyrate (pNP-C4) at a final concentration of 2 mM. After incubation at 70 °C for 2 min, the enzyme reaction mixture was centrifuged at 10,000×g for 30 s at room temperature. Enzyme activity was measured by UV absorption at 405 nm. One unit of enzyme activity was defined as the amount of enzyme releasing 1 μmol of p-nitrophenyl per minute under the above conditions. The extinction coefficient (ε405) for pNP-C4 was 16,540 M−1 cm−1. The number of spores was calculated by counting directly on the LB, solid medium through the serial dilution. The concentration of the collected recombinant spores was 6 × 108 spore/ml. The volume of the reaction solution was 0.5 mL, and the concentration of the recombinant spores in reaction solution was 1 × 108 spore/ml.
The effect of temperature was determined by enzyme activity assays at various temperatures ranging from 30 to 90 °C in 50 mM sodium phosphate buffer (pH 8.0) with pNP-C4 as substrate. The thermostability of the spore surface-displayed Tm1350 was examined by incubation of the purified spore suspension in 50 mM sodium phosphate buffer (pH 8.0) at three different temperatures (60, 70, and 80 °C) for different time intervals ranging from 0 to 8 h. The effect of pH on enzyme activity was determined in various buffers ranging from pH 4 to pH 9 at 70 °C. Buffers included 50 mM sodium citrate (pH 4.0), sodium acetate (pH 4.0–6.0), sodium phosphate (pH 6.0–8.0), and Tris–HCl (pH 8.0–9.0). The temperature was constantly maintained at 70 °C. The pH stability of the spore surface-displayed Tm1350 was examined by pre-incubating the purified spore suspension in three different buffers (pH 8.0, 9.0, or 10.0) at 70 °C for different times ranging from 0 to 8 h. Samples were dispensed out at different time intervals during incubation, and centrifuged at 10,000×g for 30 s at room temperature. The relative activity was then quickly determined and calculated by defining the respective original activity as 100 %. Residual activities were measured by standard assay as described above. In order to eliminate the influence of the background hydrolysis of the substrate under different conditions, the following controls were applied: (1) the compounds of the corresponding reaction mixture and the reaction conditions were the same and (2) the DB403 spore without the surface-displayed Tm1350 was used instead of the recombinant DB403 spore.
The spores could be easily purified by centrifugation; therefore, the reusability of the surface-displayed protein was measured by determining the activity of centrifuged recombinant spores. All reactions were conducted under the optimal catalytic condition (70 °C, pH 8.0) with 2 mM pNP-C4 as the substrate. After one reaction cycle, the spores were isolated by centrifugation at 4000×g for 2 min, and reused for five cycles. The relative activity was calculated by defining the activity of the first reaction as 100 %.
To further confirm that Tm1350 was expressed on the B. subtilis spore surface, the activity of spore surface-displayed Tm1350 was determined following different treatments with pNP-C4 as the substrate. The untreated surface-displayed Tm1350 and DB403 (pHS-cotB) spores were used as controls. Purified recombinant spores were suspended in phosphate buffer (pH 8.0) containing 0.1 % trypsin, proteinase K, bromelain, and 1 mM PMSF for 1 h at 37 °C. To eliminate the influence of background hydrolysis of the substrate under different conditions, the following controls were applied: (1) the compounds of the corresponding reaction mixture and the reaction conditions were the same and (2) the isopyknic phosphate buffer (pH 8.0) was used without the spores. The activity of untreated surface-displayed Tm1350 was defined as 100 %. All tests were carried out in triplicate, and three independent experiments were preformed. The data are expressed as mean ± standard deviation.
In order to determine the effects of immobilization, the thermostability and alkaline tolerance of free Tm1350 were determined as described above for detection of the activity of surface-displayed Tm1350.
Results
Construction of the shuttle vector pHS-cotB-Tm1350
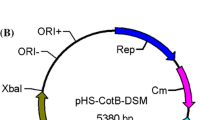
The expression vector (pHS-cotB-Tm1350) was constructed on the foundation of the plasmid pLJ (E. coli–B. subtilis shuttle vector) (Fig. 1). A flexible linker (Gly–Gly-Gly–Gly-Ser) was inserted between the C terminus of the anchoring motif of CotB with the termination codon eliminated and the N terminus of the displayed enzyme Tm1350. The relative flexibility of the short peptide allows substantial movement of structural domains, which is important for preventing the disturbance of domain functions. The short peptide is beneficial to promote the formation of the correct conformation of enzyme (Wriggers et al. 2005). Each target gene was amplified by PCR with specifically designed primers to ensure precise construction of recombinant plasmids, which were sequenced to ensure that no unintended mutation had occurred.

Map of the expression vector pHS-cotB-Tm1350. ORI+, ORI− and Rep represent the single-strand replication origin, the double strand origin and replication protein in B. subtilis, respectively. CoEI and Cm represent E. coli CoEI replicon and chloramphenicol-resistance marker, respectively. CotB a B. subtilis spore coat protein encoding gene containing the promoter of cotB. Tm1350, lipases protein encoding gene
Determination of CotB-Tm1350 expression on the B. subtilis spore surface
The specificity of the rat antiserum against Tm1350 protein was determined by Western blot analysis using total protein from E. coli (pET-28a-Tm1350) expressing recombinant Tm1350 protein antigens (Fig. 2a). Because the molecular mass calculated from the predicted amino acid sequence was 32.3 kDa, the results of Western blot analysis suggested that the rat antiserum had high specificity against the Tm1350 protein.

Western blot analysis: a The specificity of the rat antiserum against Tm1350 protein. M protein marks; lane 1 protein expressed with the empty pET-28a plasmid vector; lane 2 crude cell extract E. coli expressing recombinant Tm1350 protein; b The recombinant spores expressing CotB-Tm1350 fusion proteins. M molecular mass standards; Lane 1 proteins extracted from spores of B. subtilis DB403 (pHS-cotB); lane 2 proteins extracted from purified recombinant spores (pHS-CotB-Tm1350)
The expression of CotB-Tm1350 was confirmed by Western blotting with the previously prepared rat antiserum (Fig. 2b). A band around 70 kDa was detected in the extracts from recombinant spores, while no similar band was detected in the control lane; this 70 kDa was consistent with the expected molecular weight of CotB-Tm1350 (66 kDa). These data indicated that the fusion protein CotB-Tm1350 was expressed on the surface of recombinant spores. The activity of spore surface-displayed Tm1350 under different treatments was determined using pNP-C4 as the substrate (Fig. 3). The activities of purified recombinant spores treated with 0.1 % trypsin, proteinase K, bromelain, and 1 mM PMSF were 23, 31, 52, and 46 %, respectively. The DB403 (pHS-CotB) spores demonstrated significantly lower activity (11 %) compared to that of untreated DB403 (pHS-CotB-Tm1350) spores (100 %). Recombinant spores subjected to different treatments had lower activity than did intact spores, which were close to the activity of the untreated DB403 (pHS-CotB) spores. This result indicated that Tm1350 was located on the surface of spores.

The activity of spore surface-displayed Tm1350 with different treatments. Control the spores of DB403(pHS-CotB) without treated, untreated the recombinant spores without treated, PMSF, blomelain, proteinase K, trypsin purified recombinant spores were treated with 0.1 % trypsin, proteinase K, blomelain and 1 mM PMSF. The treatments were all treated at 37 °C for 1 h, then the reaction was started after p-nitrophenyl butyrate (pNP-C4) was added to the final concentration of 2 mM. After incubated at 70 °C for 2 min, the enzyme reaction mixture was centrifuged at 10,000×g for 30 s at room temperature, then quickly determined. Residual activities were measured by the standard assay. The activity of untreated was defined as 100 %. All tests were implemented in triplicates from 3 independent experiments, and the data were expressed as mean ± deviations
Effect of temperature on enzyme activity and stability
The optimum temperature of the spore surface-displayed Tm1350 was tested at various temperatures, ranging from 30 to 90 °C, in 50 mM sodium phosphate buffer (pH 8.0) (Fig. 4a). The optimum temperature of Tm1350 was 80 °C, at which Tm1350 exhibited specific activity (2.53 × 10−8 U/spore) similar to that at 70 °C (2.42 × 10−8 U/spore, 95.5 % of the maximum specific activity), consistent with the optimal growth temperature of T. Maritima (approximately 80 °C).The thermostability of the spore surface-displayed Tm1350 was detected at three different temperatures (60, 70, and 80 °C) by measuring the residual activity with increasing incubation times of up to 8 h (Fig. 4b). The spore surface-displayed Tm1350 retained 81, 84, and 30 % of its original activity after 8 h of incubation at pH 8.0 and at 60, 70, and 80 °C, respectively, while non-immobilized Tm1350 retained only 66, 65, and 22 %, respectively, of its original activity under the same conditions (Fig. 6a). The thermostability data showed that Tm1350 displayed on the spore surface was more stable at 70 °C, even after incubation for 8 h and retained 55 % of its original activity at 80 °C, after 1 h incubation. These data indicated that the enzyme may be denatured following prolonged incubation under high temperatures. To further characterize the spore surface-displayed enzyme, we chose 70 °C as the experimental temperature in subsequent analyses.

Temperature optima (a) and thermostability (b) of the spore-surfaced Tm1350. a Temperature optima was determined with pNP-C4 as substrates in 50 mM sodium phosphate buffer (pH 8.0) at temperatures ranging from 30 to 90 °C. Activity at 80 °C (optimum temperature) was taken as 100 %. b Thermostability. The residual enzyme activity was measured after incubation at 60 °C (black boxes), 70 °C (red circle), 80 °C (blue triangle), respectively. Relative activity was calculated by defining respective original activity as 100 %. The values are means of three independent experiments
Effect of pH on enzyme activity and stability
The optimum pH of the spore surface-displayed Tm1350 was determined in various buffers with pH values ranging from pH 4 to pH 9 at 70 °C (Fig. 5a). The surface-displayed Tm1350 had maximal activity in sodium phosphate buffer (pH 9.0, 50 mM), and the specific activity reached 2.54 × 10−8 U/spore which could be defined as 100 %. The specific activity reached 2.42 × 10−8 U/spore at pH 8.0 and 70 °C. The alkali resistance of the spore surface-displayed Tm1350 was detected at three different pH values (8.0, 9.0, and 10.0) with increase in incubation times up to 8 h by measuring the residual activity (Fig. 5b). Most of the activity of Tm1350 was maintained at 70 °C within 2 h in pH 8.0 buffer. The spore surface-displayed Tm1350 retained 81, 70, and 24 % of its original activity after 8 h of incubation at pH 8, 9, and 10, while non-immobilized Tm1350 retained only 62 and 52 % of its original activity at pH 8 and pH 9, respectively. No activity was observed after 8 h of incubation at pH 10 (Fig. 6b).

pH optima (a) and alkaline tolerance (b) of the spore-surfaced Tm1350. a pH optima at pHs from 3 to 9 was measured by standard assay. The buffers were 50 mM of sodium citrate (pH 3.0), sodium acetate (pH 4.0–6.0), sodium phosphate (pH 7.0–8.0), and Tris–HCl (pH 8.5–9.0). Activity at pH 9.0 was taken to be 100 %. b Alkaline tolerance. The residual activity was measured with pNP-C4 as substrate after incubation of the enzyme at pH 8.0 (black boxes), 9.0 (red circle), 10.0 (blue triangle). Relative activity was calculated by defining respective original activity as 100 %. The values are means of three independent experiments

a Thermostability of the free Tm1350. The residual enzyme activity was measured in pH 7.5 after incubation at 60 °C (black boxes), 70 °C (red circle), 80 °C (blue triangle). b Alkaline tolerance of the free Tm1350. The residual activity was measured with pNP-C4 as substrate at 70 °C after incubation of the enzyme at pH 7.0 (black boxes), 8.0 (red circle), 9.0 (blue triangle) and 10.0 (green inverted triangle). Relative activity was calculated by defining respective original activity as 100 %
Specific activity and reusability of spore surface-displayed Tm1350
Under optimal temperature and pH conditions (70 °C, pH 8.0), the activity of the spore surface-displayed Tm1350 was determined using pNP-C4 as the substrate. The concentration of the recombinant spores was 108 spore/mL. The specific activity reached 2.42 × 10−8 U/spore. Our unpublished data showed that the activity of the purified recombinant lipase expressed in E. coli (pET-28a-Tm1350) was 64.26 U/mg. This suggested that 26.6 mL of spore liquid would be necessary to yield the activity of the purified recombinant lipase (1 mg). The reusability of the surface-displayed Tm1350 was measured by determining the activity of centrifuged recombinant spores with pNP-C4 as the substrate (Fig. 7). The recombinant spores could be used for up to three reaction cycles without any significant decrease in activity. After five reaction cycles, centrifuged recombinant spores retained 62 % of their original activity.

Reuse of the spore-displayed Tm1350. All reactions were conducted at 70 °C, pH 8.0 for 2 min with 2 mM pNP-C4 as the substrates. The standard reaction mixture was pre-incubated for 10 min, then the first reaction was started after p-nitrophenyl butyrate (pNP-C4) was added. After incubated at 70 °C for 2 min, the enzyme reaction mixture was centrifuged at 4000×g for 2 min at 4 °C, then quickly determined. The spores were cleaned using PBS (pH 8), then recovered to conduct the second reaction by centrifugation at 4000×g for 2 min at 4 °C. The relative activity was calculated by defining the activity of first reaction as 100 %. All tests were implemented in triplicates from 3 independent experiments, and the data were expressed as mean ± deviations
Discussion
Mesophilic enzymes are often not well suited for the harsh reaction conditions required in industrial processes due to lack of enzyme stability at high temperatures and in the presence of organic solvents (Demirjian et al. 2001). Enzymes from extremophilic microorganisms have a great impact on the field of biocatalysis, since they generally display high intrinsic thermal and chemical stability (Snellman et al. 2002). T. maritima is a hyperthermophilic bacterium that grows optimally at 80 °C, which makes it a potential source for thermostable enzymes. Some carboxylic ester hydrolases from T. maritima have been identified and characterized (Levisson et al. 2009). In unpublished studies, we characterized a new lipase (Tm1350) from T. Maritima. Our data showed that this enzyme was more stable at high temperatures, under alkaline conditions, and in some organic solvents such as methanol. These data suggested that the lipase Tm1350 has great potential for applications in the biochemical industry. In order to produce an enzyme that is useful for industrialization production, we displayed Tm1350 on the surface of B. subtilis spore using CotB as the anchoring motif and investigated the catalytic capability of the spore surface-displayed Tm1350 using p-NP-butyrate as a substrate.
The structure of the B. subtilis spore consists of three concentric compartments separated by two membranes (Kim and Schumann 2009). A spore coat encases the spore and helps protect it from noxious environmental agents; this spore coat is composed of about 70 different proteins, some of which have been studied in detail (Henriques and Moran 2000; Kim et al. 2006). CotB was shown to be abundantly expressed and externally exposed on the surface of the spore. Additionally, CotB was the first spore coat protein to be used for anchoring heterologous proteins on the spore surface as anchoring motifs (Isticato et al. 2001; Imamura et al. 2010). Moreover, CotC, CotG, CotX, CotE, and OxdD are used as anchoring motifs in the spore display system, but the majority of which were used for vaccination and immunogenicity, while a few were used for enzymology (Demirjian et al. 2001; Potot et al. 2010; Xu et al. 2011; Qu et al. 2014). Hinc et al., compared the ability of the anchoring motifs CotB, CotC, and CotG for displaying UreA, and showed that CotG yielded results similar to those of CotC, which was shown to allow higher efficiency of expression, whereas its passenger protein was not displayed outside the spore. However, the passenger protein of CotB was exposed on the spore surface; thus, CotB was considered as an appropriate carrier for the display of heterologous proteins (Hinc et al. 2010). To the best of our knowledge, the current study is the first to describe the display of lipase on the surface of B. subtilis spores using CotB as the anchoring motif.
The target gene was inserted into the chromosomal DNA of B. subtilis by double crossover using a recombinant integrative plasmid and an amylase-inactivated mutant that is generally used in the spore surface display system was produced (Potot et al. 2010; Wang et al. 2011). However, this method has some disadvantages, such as complex operation, low success rate of conversion, and low expression levels. While shuttle vectors have been shown to overcome these shortcomings, few have been used in spore surface display systems (Xu et al. 2011; Qu et al. 2014). The construction of the high-copy-number E. coli–B. subtilis shuttle vector pEB03 was based on the high-copy-number vector pGJ103 and used for displaying the enzyme on the spore surface, showing that the activity of the enzyme was much higher using pEB03 as the expression vector than that obtained using the low-copy-number pHP13 as the expression vector (Xu et al. 2011). In this study, we constructed an E. coli–B. subtilis shuttle vector (pHS-cotB-Tm1350), based on the high-copy-number plasmid pLJ, which was also based on the vector pGJ103 (Yang et al. 2006). A 66 kDa fusion protein, CotB-Tm1350, was successfully displayed on the spore surface of B. subtilis DB403 and was confirmed by Western blot analysis using a previously prepared rat antiserum and by detection of enzyme activities.
In our previous studies, we achieved expression, purification, and identification of free Tm1350. The optimum reaction temperature of the spore surface-displayed Tm1350 was similar to that non-immobilized Tm1350. In contrast, the optimum pH differed that of the spore surface-displayed Tm1350 was 9, while that of the free Tm1350 was 7.5. This difference may have resulted from changes in the surface charge of the surface-displayed enzyme. After 8 h of incubation under different conditions, the retained activity of spore surface-displayed Tm1350 was about 18 % higher than the retained activity of non-immobilized Tm1350 under the same conditions. These results indicated that Tm1350 displayed on the surface of spores could enhance both thermal and pH stability, consistent with a previous study of hydrolases displayed on the surface of B. subtilis 168 spores (Xu et al. 2011).
The reusability of a biocatalyst is one of the most important factors to be considered in its industrial application, and spores have the characteristic of easy purification by centrifugation. In this study, the surface-displayed Tm1350 retained 84 % of its original activity after three uses, which was higher than that reported for lipase SMG1 immobilized on macroporous resin (Wang et al. 2014), and was similar to that reported for CALB immobilized on PPS and spore surface-displayed Neu5Ac aldolase (Xu et al. 2011; Abdallah et al. 2014). Therefore, display of enzymes on the surface of B. subtilis spores may be an effective approach for enzyme immobilization and has great potential for application in the harsh conditions required in the biochemical industry.
References
Abdallah NH, Schlumpberger M, Gaffney DA, Hanrahan JP, Tobin JM, Magner E (2014) Comparison of mesoporous silicate supports for the immobilisation and activity of cytochrome c and lipase. J Mol Catal B Enzym 108:82–88
Chandrayan SK, Dhaunta N, Guptasarma P (2008) Expression, purification, refolding and characterization of a putative lysophospholipase from Pyrococcus furiosus: retention of structure and lipase/esterase activity in the presence of water-miscible organic solvents at high temperatures. Protein Expres Purif 59:327–333
Costas L, Bosio VE, Pandey A, Castro GR (2008) Effects of organic solvents on immobilized lipase in pectin microspheres. Appl Biochem Biotechnol 151:578–586
Demirjian DC, Moris-Varas F, Cassidy CS (2001) Enzymes from extremophiles. Curr Opin Chem Biol 5:144–151
Duc LH, Hong HA, Atkins HS, Flick-Smith HC, Durrani Z, Rijpkema S, Titball RW, Cutting SM (2007) Immunization against anthrax using Bacillus subtilis spores expressing the anthrax protective antigen. Vaccine 25:346–355
Gomes J, Steiner W (2004) The biocatalytic potential of extremophiles and extremozymes. Food Technol Biotech 42:223–235
Green DH, Wakeley PR, Page A, Barnes A, Baccigalupi L, Ricca E, Cutting SM (1999) Characterization of two bacillus probiotics. Appl Environ Microbiol 65:4288–4291
Henriques AO, Moran CP Jr (2000) Structure and assembly of the bacterial endospore coat. Methods 20:95–110
Hinc K, Isticato R, Dembek M, Karczewska J, Iwanicki A, Peszynska-Sularz G, De Felice M, Obuchowski M, Ricca E (2010) Expression and display of urea of Helicobacter acinonychis on the surface of Bacillus subtilis spores. Microb Cell Fact 9:369–381
Imamura D, Kuwana R, Takamatsu H, Watabe K (2010) Localization of proteins to different layers and regions of Bacillus subtilis spore coats. J Bacteriol 192:518–524
Isticato R, Cangiano G, Tran HT, Ciabattini A, Medaglini D, Oggioni MR, De Felice M, Pozzi G, Ricca E (2001) Surface display of recombinant proteins on Bacillus subtilis spores. J Bacteriol 183:6294–6301
Kim J, Schumann W (2009) Display of proteins on Bacillus subtilis endospores. Cell Mol Life Sci CMLS 66:3127–3136
Kim JH, Lee CS, Kim BG (2005) Spore-displayed streptavidin: a live diagnostic tool in biotechnology. Biochem Biophys Res Commun 331:210–214
Kim H, Hahn M, Grabowski P, McPherson DC, Otte MM, Wang R, Ferguson CC, Eichenberger P, Driks A (2006) The Bacillus subtilis spore coat protein interaction network. Mol Microbiol 59:487–502
Kwon SJ, Jung HC, Pan JG (2007) Transgalactosylation in a water-solvent biphasic reaction system with beta-galactosidase displayed on the surfaces of Bacillus subtilis spores. Appl Environ Microbiol 73:2251–2256
Levisson M, van der Oost J, Kengen SW (2009) Carboxylic ester hydrolases from hyperthermophiles. Extremophiles 13:567–581
Masomian M, Rahman RNZRA, Salleh AB, Basri M (2013) A new thermostable and organic solvent-tolerant lipase from Aneurinibacillus thermoaerophilus strain HZ. Process Biochem 48:169–175
Mauriello EMF, Duc LH, Isticato R, Cangiano G, Hong HYA, De Felice M, Ricca E, Cutting SM (2004) Display of heterologous antigens on the Bacillus subtilis spore coat using cotc as a fusion partner. Vaccine 22:1177–1187
Miroliaei M, Nemat-Gorgani M (2002) Effect of organic solvents on stability and activity of two related alcohol dehydrogenases: a comparative study. Int J Biochem Cell B 34:169–175
Nicholson W, Setlow P (1990) Sporulation, germination and outgrowth. In: Harwood C, Cutting S (eds) Molecular biological methods for Bacillus. Wiley, Chichester, pp 391–450
Nicholson WL, Munakata N, Horneck G, Melosh HJ, Setlow P (2000) Resistance of bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol Mol Biol Rev 64:548–572
Potot S, Serra CR, Henriques AO, Schyns G (2010) Display of recombinant proteins on Bacillus subtilis spores, using a coat-associated enzyme as the carrier. Appl Environ Microbiol 76:5926–5933
Qu YY, Wang JW, Zhang ZJ, Shi SN, Li DX, Shen WL, Shen E, Zhou JT (2014) Catalytic transformation of hodas using an efficient meta-cleavage product hydrolase-spore surface display system. J Mol Catal B Enzym 102:204–210
Roessl U, Nahalka J, Nidetzky B (2010) Carrier-free immobilized enzymes for biocatalysis. Biotechnol Lett 32:341–350
Sambrook J, Fritsch E, Maniatis T (1989) Molecular cloning: a laboratory manual. Corporation for Supportive Housing, New York
Snellman EA, Sullivan ER, Colwell RR (2002) Purification and properties of the extracellular lipase, lipa, of acinetobacter sp rag-1. Eur J Biochem 269:5771–5779
Wang N, Chang C, Yao Q, Li G, Qin L, Chen L, Chen K (2011) Display of Bombyx mori alcohol dehydrogenases on the Bacillus subtilis spore surface to enhance enzymatic activity under adverse conditions. PLoS One 6:e21454
Wang WF, Xu Y, Qin XL, Lan DM, Yang B, Wang YH (2014) Immobilization of lipase smg1 and its application in synthesis of partial glycerides. Eur J Lipid Sci Tech 116:1063–1069
Winkler UK, Stuckmann M (1979) Glycogen, hyaluronate, and some other polysaccharides greatly enhance the formation of exolipase by Serratia marcescens. J Bacteriol 138:663–670
Wriggers W, Chakravarty S, Jennings PA (2005) Control of protein functional dynamics by peptide linkers. Biopolymers 80:736–746
Xu XM, Gao C, Zhang XF, Che B, Ma CQ, Qiu JH, Tao F, Xu P (2011) Production of N-acetyl-d-neuraminic acid by use of an efficient spore surface display system. Appl Environ Microbiol 77:3197–3201
Yang MM, Zhang WW, Zhang XF, Cen PL (2006) Construction and characterization of a novel maltose inducible expression vector in Bacillus subtilis. Biotechnol Lett 28:1713–1718
Acknowledgments
This study was supported by the National Key Basic Research Program of China (973 Program, No. 2011CBA00800), the Open Funding Project of National Key Laboratory of Biochemical Engineering, and the Key Agriculture Support Project of Jiangsu Province, China (No. BE2013400).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Robb.
Rights and permissions
About this article

Cite this article
Chen, H., Tian, R., Ni, Z. et al. Surface display of the thermophilic lipase Tm1350 on the spore of Bacillus subtilis by the CotB anchor protein. Extremophiles 19, 799–808 (2015). https://doi.org/10.1007/s00792-015-0755-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-015-0755-0