
Abstract
Phospholipases can catalyze the hydrolysis of one or more ester and phosphodiester bonds and have a considerable interest in the food, oil leather and pharmaceutical industries. In this report, a lysophospholipase gene from the hyperthermophilic archaeon Thermococcus kodakarensis KOD1 (LysoPL-tk) was cloned. The gene of 783 bp encodes a 260-amino acid protein with a molecular mass of 29 kDa. LysoPL-tk has a consensus motif (GxSxG) and a catalytic triad (S, D, H) of esterases in the deduced amino acid sequence. LysoPL-tk was expressed in Escherichia coli and purified to homogeneity. The enzyme can degrade substrates with both short and long acyl chain lengths. The apparent K m value for p-nitrophenyl butyrate was 607.1 μM with V max values of 95.5 U/mg. The enzyme was active at a broad range of pH (5–8) and temperatures (70–95 °C) with the optimum pH and temperature being 8.0 and 85 °C, respectively. The high yield, broad substrate range along with its thermo-stability indicates that LysoPL-tk is a potential enzyme in industrial application.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The lipolytic enzymes, including lipases, esterases/carboxylesterases, and phospholipases, have important function in lipid metabolism, lipoprotein metabolism, membrane homeostasis and signal transduction, etc. (Tirawongsaroj et al. 2008). Phospholipases are wide spread in archaea, eubacteria and eukaryotes, which can be further classified into phospholipase A, B, C and D depending on the site of hydrolyzed ester bond (Ramrakhiani and Chand 2011). Phospholipase A hydrolyzes the sn-1 and sn-2 fatty acid ester bond of glycerophospholipids. Phospholipase B catalyzes three distinct activities: a sn-1 and sn-2 fatty acid ester hydrolase, a lysophospholipase and a transacylase activity. C-type phospholipases catalyze the cleavage of membrane phospholipids to 1,2-diacyl glycerol and the organic phosphate. Phospholipase D hydrolyzes the phosphodiester bond of glycerophospholipids to generate phosphatidic acid and a free head group (Jiang et al. 2011; McDermott et al. 2004; Ramrakhiani and Chand 2011). All classes of phospholipases together can cleave glycerophospholipids into fatty acid and glycerol moiety completely. Among the phospholipases, lysophospholipases (LysoPLAs) (EC 3.1.1.5) can cleave the sn-1 or sn-2 ester bond of lysophospholipids (LysoPLs) producing a free fatty acid and a glycerolphosphate derivative (Farooqui et al. 1987). Lysophospholipids are glycerophospholipids in which one acyl chain is lacking and then only one hydroxyl group of the glycerol backbone is acylated (D’Arrigo and Servi 2010). LysoPLs are found only in small amounts in biological cell membranes but exhibit a wide range of diverse biological activities including reproduction, vascular development, cancer and nervous system function (Birgbauer and Chun 2006). As LysoPLs exhibit such diverse biological functions and LysoPLAs can control its level, LysoPLAs can also play a pivotal role in the regulation of cell functions.
Phospholipases have a great potential in industrial applications such as starch, oil, baking and poly-unsaturated fatty acid production (De Maria et al. 2007). Phospholipases also provides enantioselectivity, which makes them suitable for the reactions and is desired in pharmaceutical industry (Jaeger and Eggert 2002). However, the enzymes from normal environments are not suitable for industrial usage because of the stability problem. One approach to solve the problem is to digger the enzymes from thermophilic or hyperthermophilic microorganisms. Several thermophilic easterase, carboxylesterase and lysophospholipase have been purified and characterized from thermophilic archaea or bacteria. The putative lysophospholipase from Pyrococcus furiosus displayed both lipase and esterase activity at 70 °C (Chandrayan et al. 2008). In Pyrobaculum calidifontis VA1, the carboxylesterase can hydrolyze ester bond with short to medium chains at 90 °C (Hotta et al. 2002). Some thermophilic enzymes were isolated from thermal environmental metagenomic libraries, which included the lipase and esterase from Indonesia and Thailand (Rhee et al. 2005; Tirawongsaroj et al. 2008).
Thermococcus kodakaraensis KOD1 is a thermophilic anaerobic archaeon belonging to the Thermococcaceae family, whose whole genome sequence has been reported (Atomi et al. 2004; Fukui et al. 2005). As a hyperthermophilic anaerobe living in deep-vent environments, T. kodakaraensis KOD1 is considered to be a model microorganism to study hyperthermophiles and a potential industrial enzymes source. There are two phospholipase (TK0999 and TK1563) annotated in the genome sequence, which may be applied industrially. In this study, we described the sequence characteristics, cloning and overexpression of TK0999 which was named lysophospholipase in T. kodakaraensis KOD1 (LysoPLA-tk). We further purified the soluble recombinant protein from E. coli and reported its biochemical properties.
Materials and methods
Cloning of LysoPLA-tk from T. kodakarensis KOD1
Polymerase chain reaction (PCR) with T. kodakarensis KOD1 genomic DNA as a template was performed to isolate LysoPLA-tk using the oligonucleotide primers: forward, 5′-GGAATTCATGGAAATCTACAAAGCCAA-3′; and reverse, 5′-CCCAAGCTTTCAAGCCTTCTCTGAATGCTTTC-3′, which included restriction enzyme sites for EcoRI and HindIII. The PCR products were ligated into the pET28(a) vector, transformed into Escherichia coli BL21 (DE3), and sequenced.
Expression and purification of LysoPLA-tk
E. coli BL21(DE3) cells containing the pET28a-LysoPLA-tk plasmid were cultured in 2 l of LB broth with 50 μg/ml kanamycin at 37 °C for 3 h. When the OD600 reached 0.7, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to induce protein expression. The cells were cultured in the presence of IPTG for 4 h with shaking, harvested by centrifugation at 6000 rpm for 10 min, and then resuspended in lysis buffer containing 50 mM Tris (pH 8.0), 300 mM NaCl, 20 mM 2-mercaptoethanol, and 20 mM imidazole. The cell suspension was sonicated and heated at 65 °C for 1 h. The thermo-stable components in the supernatant were collected following centrifugation and loaded on a Ni-NTA column. After washing the column with lysis buffer, LysoPLA-tk was eluted using an imidazole gradient (40–300 mM). The purified LysoPLA-tk was visualized after separation by 12 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The eluted proteins were dialyzed against 50 mM Tris buffer (pH 8.0) containing 300 mM NaCl and 20 mM 2-mercaptoethanol. Protein concentrations were estimated by the method of Bradford using bovine serum albumin (BSA) as a standard.
Enzyme assays
LysoPLA-tk enzyme activity was measured by following the increase in absorbance at 410 nm at 85 °C when the enzyme was incubated in a standard assay mixture (total volume 1.0 ml) that contained 50 mM HEPES (pH 8.0), 1 % acetone, and 1 mM substrates, paranitrophenyl butyrate (p-NP-butyrate), 0.1 mM paranitrophenyl caprylate (p-NP-caprylate), or 0.1 mM paranitrophenyl palmitate (p-NP-palmitate), respectively. The enzyme activity was determined from the initial velocity of the reaction. One unit LysoPLA-tk activity was defined as the amount of enzyme that catalyzes the formation of 1 μmol paranitrophenol/min under the conditions of the assay. The specific activity of the enzyme is determined by the ratio of enzyme activity to the amount of protein in the assay.
Kinetic study
For kinetic studies, the initial velocities of the enzymatic reaction were examined by varying the concentration of p-NP-butyrate (from 0.1 to 1 mM). Values of the Michaelis constants (K m) and maximal velocity (V max) were obtained by mathematical calculations according to Sigma Plot software. The parameters were determined by three separate experiments.
Effect of pH, organic solvents and metal ions on enzyme activity
50 mM sodium acetate (pH 3.0–5.0), 50 mM MES (pH 6.0–7.0), 50 mM HEPES (pH 7.0–8.0), and 50 mM glycine (pH 9.0–10.0) were used to determine the optimum pH of the enzyme under standard assay conditions. Absorbance at 348 nm was measured to avoid the effects of pH-dependent changes in the molar extinction coefficient.
Stability against organic solvents was measured by incubating the enzyme (100 μg/ml) in 20 mM HEPES (pH 8.0) containing 20 and 50 % organic solvent (v/v) at 30 °C for 60 min. To measure the residual activities, aliquots were taken from these mixtures and added as the enzyme sample in the standard assay.
The effects of EDTA and various metal ions on the hydrolytic activity were determined by detecting the activity against p-NP-butyrate in 20 mM HEPES buffer (pH 8.0) at 80 °C in the presence of 5 mM EDTA, CaCl2, MgCl2, NiCl2 BaCl2, CuCl2, and ZnCl2, respectively.
Result
Sequence homolog of LysoPLA-tk
The DNA sequence encoding LysoPLA-tk has an open reading frame of 783 nucleotides, which was predicted to encode a protein of 260 amino acid residues with a theoretical molecular weight of 28990.40 and pI of 6.38. The homologs of LysoPLA-tk were searched using BLAST in the NCBI database. Sequences were aligned using Clustal W. LysoPLA-tk exhibited 83 % identity to previously characterized LysoPLA from archaea, such as the LysoPLA from P. furiosus (Pf0480) (Chandrayan et al. 2008). It also shared a significant level of identity with LysoPLA from Archaeoglobus fulgidus (35 %), monoglyceride lipase from Homo sapiens (30 %) (Labar et al. 2010), Yju3p from Saccharomyces cerevisiae (28 %) (Heier et al. 2010) (Fig. 1). Based on the alignment, LysoPLA-tk was predicted to be a serine hydrolase harboring a G-X-S-X-G motif and an α/β hydrolase fold, which are both characteristics for lipolytic enzymes and the catalytic triad of LysoPLA-tk can be assigned to Ser87, Asp203, and His233 (Fig. 1). Phylogenetic analysis of LysoPLA-tk and other lipases reveals that LysoPLA-tk belongs to family VI (Supplementary Fig. S1) (Bornscheuer 2002; Arpigny and Jaeger 1999).

Multiple amino acid alignment of LysoPLA homologs. The proteins are LysoPLA from T. kodakarensis KOD1 (LPLA-tk, YP_183412), P. furiosus (LPLA-pf, NP_578209) and A. Fulgidus (LPLA-af, NP_070581); monoglyceride lipase from Homo sapiens (ML-hs, NP_001003794); Yju3p from S. cerevisiae (Yju3p-sc, EGA61615) and carboxylesterase from E. coli (CE-ec, P_002414522) and B. subtilis (CE-bs, CAA58063). The regions with black shading and white lettering are conserved residues. Asterisk symbols indicate conserved catalytic triad. The numbers at the ends of each line on the right-hand side refer to the numbers of amino acid residues. The conserved GXSXG in the serine hydrolase is shown by the solid line
Expression and purification of LysoPLA-tk
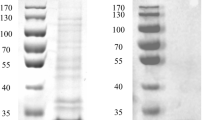
The gene of LysoPLA-tk was cloned in pET expression vectors to purify the protein and understand the catalytic mechanism. LysoPLA-tk could be expressed as soluble form in E. coli after IPTG induction. The purification of LysoPLA-tk was performed by heating a total protein extract of the induced cells and removing the denatured proteins by centrifugation firstly. The thermo-stable LysoPLA-tk in the supernatant was then purified using an Ni-NTA affinity chromatography methods as described in “Materials and methods”. SDS-PAGE analysis of recombinant LysoPLA-tk revealed a molecular weight of approximately 32 kDa (Fig. 2). The yield of purified protein was about 4.12 mg from 1 l E. coli.

Purification of LysoPLA-tk. Lane 1 crude protein extract from non-induced cells, lane 2 soluble extract after heating at 65 °C for 1 h from IPTG-induced cells, lane 3 unbound proteins eluted from the Ni-NTA column, lane 4 proteins eluted by lysis buffer, lane 5–7 proteins eluted by 50, 100, and 250 mM imidazole. Lane 8 protein markers. The molecular mass standards are indicated at the right
Activity of LysoPLA-tk
p-NP-butyrate, p-NP-caprylate, p-NP-palmitate were used as substrates to study the specificity of LysoPLA-tk at 85 °C and pH 8.0. The result showed that LysoPLA-tk degraded all of them, demonstrating it is not only an esterase (substrates with acyl chain lengths of <10 carbon atoms) but also a lipase (substrates with acyl chain lengths of >10 carbon atoms) (Rhee et al. 2005). However, the esterase activity of LysoPLA-tk to p-NP- butyrate is 8 times greater than lipase activity to p-NP-palmitate, indicating a preference for short chain fatty acid ester (Fig. 3a).

Activity assay of LysoPLA-tk. A The effect of substrate length on LysoPLA-tk activity. Ba Optimal temperature of LysoPLA-tk activity. b Arrhenius plot for the reaction. Solid line is linear fit (R 2 = 0.98), with intercept of ln(A) = 4.86, and slope of 3.90 corresponding to an Arrhenius activation energy of E a = 32.28 KJ mol−1. C Optimal pH of LysoPLA activity. Different buffers were used for the different pH solutions used in this assay. Sodium acetate was used for pHs 3.1 and 4.1; MES buffer was used for pHs 5.0 and 6.10; HEPES buffer was used for pHs 7.0 and 8.0; glycine buffer was used for pHs 9.0 and 10.1. The concentrations of the buffers were 50 mM. D Semi-logarithmic plots for the inactivation of LysoPLA-tk at 85 °C (filled diamonds) and at 95 °C (filled triangles). E Effect of substrate concentration on velocity of LysoPLA-tk. The assays were carried out a as described under “Materials and methods”
To study the optimum catalytic conditions, the influence of temperature and pH on purified LysoPLA-tk activity to p-NP-butyrate was examined. Hydrolysis of p-NP-butyrate by LysoPLA-tk was measured at temperatures from 30 to 95 °C at pH 8.0 (Fig. 3Ba). The enzyme exhibited highest activity at 85 °C with the activation energy 32.38 KJ mol−1 calculated using the corresponding linear Arrhenius plot (Fig. 3Bb). The activation energy of LysoPLA-tk is much higher than that of esterase from P. calidifontis VA1 and a metagenomic library with 26.4 and 21.1 KJ mol−1, respectively (Hotta et al. 2002; Rhee et al. 2005). The specific activity of purified LysoPLA-tk was also examined in the pH range 3.0–10.0 using a mixture of different buffers including sodium acetate, MES, HEPES, and glycine (Fig. 3c). LysoPLA-tk showed high activity among 5.0–8.0 and the pH optimum for enzyme activity was approximately 8.0.
The thermostability of LysoPLA-tk was examined at 85 and 95 °C (Fig. 3d). LysoPLA-tk was incubated for 20 min to 120 min at the indicated temperatures and the residue enzyme activity was then measured under optimum conditions. Half-lives of enzyme activity were calculated from semi-logarithmic plots of activity versus incubation time. The results demonstrated that LysoPLA-tk was a thermo-stable protein with half-life 5 h at 85 °C which is the suitable temperature for the growth of T. kodakarensis KOD1. At 95 °C, LysoPLA-tk is thermo-stable with half-life 1 h.
The K m value for p-NP-butyrate was determined by varying the concentration of the substrate. The kinetic parameters reported here are the mean of three determinations. The K m and V max values were 607.1 μM and 95.5 U/mg, respectively (Fig. 3e).
The effects of a chelating agent and divalent ions on the esterase activity are summarized in Table 1. The enzyme activity increased slightly in the presence of divalent cations, such as Ca2+ and Ni2+. However, the enzyme was inhibited in the presence of 5 mM Cu2+ and Zn2+. The enzyme could not be inhibited by 5 mM EDTA, suggesting that the protease is not a metalloenzyme.
As shown in Table 2, no obvious inactivation of LysoPLA-tk against p-NP-butyrate was observed in the presence of organic solvents tested. More than 90 % of the enzyme activity retained after incubation with acetonitrile. Interestingly, methanol, ethanol, and isopropanol even increased the activity to 111, 136, and 133 %, respectively.
Discussion
In this study, we reported a lysophospholipase from T. kodakarensis KOD1 (LysoPLA-tk) sharing structural and functional homology with eukaryotic monoglyceride lipase. Furthermore, LysoPLA-tk can hydrolyze both ester bonds with short acyl chain and long acyl chain, but the best substrate was short chain fatty acid ester. The optimum pH and temperature of the enzyme to short acyl chain were around 8.0 and 85 °C with K m = 607.1 μM and V max = 95.5 U/mg.
Lipids with great variety of structures in various archaea play a key role to overcome the destabilizing conditions encountered in extreme environments as hot acidic springs and submarine volcanic fields. The biosynthesis of lipid in archaea was well studied (Koga and Morii 2007; Ulrih et al. 2009), but the knowledge of degradation is limited. β-oxidation system exists in hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus (Klenk et al. 1997), but the enzymes involved in fatty acids degradation were not clear in Thermococcus. LysoPL-tk was classified into the enzymes in central intermediary metabolism from genome sequence analysis (Fukui et al. 2005), and our result also proved that LysoPL-tk can degrade ester bonds with different length acyl chain. These results suggested that LysoPL-tk may have a role in lipid degradation. On the other hand, sequence analysis showed that LysoPLA-tk from thermophilic archaeon have high similarity with Yju3p from yeast and monoglyceride lipase from human (Heier et al. 2010; Labar et al. 2010). Monoglyceride lipase from mammalian was assumed to be responsible for breakdown of monoacylglycerol in the metabolism of triglycerides. Meanwhile, monoglyceride lipase has also been implicated in the degradation of the bioactive 2-arachidonoyl glycerol, which is known to be a potent endogenous agonist of cannabinoid receptors (Dinh et al. 2002; Lass et al. 2011). The similar sequence between LysoPLA-tk and monoglyceride lipase suggested the similar function. We can speculate that LysoPLA-tk function to hydrolyze ester compounds, providing short chain carboxylic acids to the cell, though the real function has not been revealed. In addition, LysoPLA-tk (TK0999), TK0997 and TK0998 are located in the same operon and under the same promoter elements (data not shown). The two proteins (TK0997 and TK0998) are transcription factor and nucleic binding protein, respectively. This information suggests that LysoPLA-tk may have alternative function to regulate gene expression.
Lipases are the most widely used group of biocatalysts for biotechnology, in fine chemical applications, mainly because they can be applied efficiently in the production of optically pure compounds (Hess et al. 2008). However, low thermodynamic stability and susceptibility to aggregation are the two very undesirable traits of most of the natural proteins that limit their usefulness in many biotechnological applications (Kamal et al. 2011). For example, Candida rugosa lipase and carboxylesterase NP from B. subtilis ThaiI-8 have been evaluated for their capacity to resolve ester derivatives of naproxen with high enantioselectivity, which is a popular nonsteroidal anti-inflammatory drugs used in the treatment of human connective tissue diseases (Sehgal and Kelly 2003). But the two mesophilic enzymes did not show notable thermostability. The LysoPLA from P. furiosus, which showed 83 % identity to LysoPLA-tk, was selected as substitute for the mesophilic enzymes in the pharmaceutical industry. The protein was found to deposit into inclusion bodies in recombinant E. coli and the yield is very low (0.23 mg from 1 l culture) though it displayed optimal activity at 70 °C. Compared with LysoPLA from P. furiosus (Chandrayan et al. 2008), LysoPLA-tk displayed both remarkable thermostability (optimal temperature at 85 °C) and high yield. Furthermore, this enzyme was also stable in the presence of certain organic solvents. Together with its potential biological function, we suggest LysoPLA-tk may have important roles in vivo and have useful applications in pharmaceutical or other industry.
References
Arpigny JL, Jaeger KE (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343:177–183
Atomi H, Fukui T, Kanai T, Morikawa M, Imanaka T (2004) Description of Thermococcus kodakaraensis sp. nov., a well studied hyperthermophilic archaeon previously reported as Pyrococcus sp. KOD1. Archaea 1:263–267
Birgbauer E, Chun J (2006) New developments in the biological functions of lysophospholipids. Cell Mol Life Sci 63:2695–2701
Bornscheuer UT (2002) Microbial carboxyl esterases: classification, properties and application in biocatalysis. FEMS Microbiol Rev 26:73–81
Chandrayan SK, Dhaunta N, Guptasarma P (2008) Expression, purification, refolding and characterization of a putative lysophospholipase from Pyrococcus furiosus: retention of structure and lipase/esterase activity in the presence of water-miscible organic solvents at high temperatures. Protein Express Purif 59:327–333
D’Arrigo P, Servi S (2010) Synthesis of lysophospholipids. Molecules 15:1354–1377
De Maria L, Vind J, Oxenbøll K, Svendsen A, Patkar S (2007) Phospholipases and their industrial applications. Appl Microbiol Biotechnol 74:290–300
Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA 99:10819–10824
Farooqui A, Taylor W, Horrocks L (1987) Phospholipases, lysophospholipases, and lipases and their involvement in various diseases. Mol Chem Neuropathol 7:99–128
Fukui T, Atomi H, Kanai T, Matsumi R, Fujiwara S, Imanaka T (2005) Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res 15:352–363
Heier C, Taschler U, Rengachari S, Oberer M, Wolinski H, Natter K, Kohlwein SD, Leber R, Zimmermann R (2010) Identification of Yju3p as functional orthologue of mammalian monoglyceride lipase in the yeast Saccharomyces cerevisiae. Biochim Biophys ActaBBA-Mol Cell Biol L 1801:1063–1071
Hess M, Katzer M, Antranikian G (2008) Extremely thermostable esterases from the thermoacidophilic euryarchaeon Picrophilus torridus. Extremophiles 12:351–364
Hotta Y, Ezaki S, Atomi H, Imanaka T (2002) Extremely stable and versatile carboxylesterase from a hyperthermophilic archaeon. Appl Environ Microbiol 68:3925–3931
Jaeger K-E, Eggert T (2002) Lipases for biotechnology. Curr Opin Biotechnol 13:390–397
Jiang F, Huang S, Imadad K, Li C (2011) Cloning and expression of a gene with phospholipase B activity from Pseudomonas fluorescens in Escherichia coli. Bioresour Technol 104:518–522
Kamal MZ, Ahmad S, Molugu TR, Vijayalakshmi A, Deshmukh MV, Sankaranarayanan R, Rao NM (2011) In vitro evolved non-aggregating and thermostable lipase: structural and thermodynamic investigation. J Mol Biol 413:726–741
Klenk HP, Clayton RA, Tomb JF, White O, Nelson KE et al (1997) The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature 390:364–370
Koga Y, Morii H (2007) Biosynthesis of ether-type polar lipids in archaea and evolutionary considerations. Microbiol Mol Biol Rev 71:97–120
Labar G, Bauvois C, Borel F, Ferrer JL, Wouters J, Lambert DM (2010) Crystal structure of the human monoacylglycerol lipase, a key actor in endocannabinoid signaling. ChemBioChem 11:218–227
Lass A, Zimmermann R, Oberer M, Zechner R (2011) Lipolysis—a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res 50:14–27
McDermott M, Wakelam MJO, Morris AJ (2004) Phospholipase D. Biochem Cell Biol 82:225–253
Ramrakhiani L, Chand S (2011) Recent progress on phospholipases: different sources, assay methods, industrial potential and pathogenicity. Appl Biochem Biotechnol 164:991–1022
Rhee JK, Ahn DG, Kim YG, Oh JW (2005) New thermophilic and thermostable esterase with sequence similarity to the hormone-sensitive lipase family, cloned from a metagenomic library. Appl Environ Microbiol 71:817–825
Sehgal AC, Kelly RM (2003) Strategic selection of hyperthermophilic esterases for resolution of 2-arylpropionic esters. Biotechnol Prog 19:1410–1416
Tirawongsaroj P, Sriprang R, Harnpicharnchai P, Thongaram T, Champreda V, Tanapongpipat S, Pootanakit K, Eurwilaichitr L (2008) Novel thermophilic and thermostable lipolytic enzymes from a Thailand hot spring metagenomic library. J Biotechnol 133:42–49
Ulrih NP, Gmajner D, Raspor P (2009) Structural and physicochemical properties of polar lipids from thermophilic archaea. Appl Microbiol Biotechnol 84:249–260
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by H. Atomi.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Cui, Z., Wang, Y., Pham, B.P. et al. High level expression and characterization of a thermostable lysophospholipase from Thermococcus kodakarensis KOD1. Extremophiles 16, 619–625 (2012). https://doi.org/10.1007/s00792-012-0461-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-012-0461-0