
Abstract
A putative Type II restriction–modification system of Thermotoga neapolitana, TneDI, was cloned into Escherichia coli XL1-Blue MRF′ and characterized. Gene CTN_0339 specifies the endonuclease R.TneDI, while CTN_0340 encodes the cognate DNA methyltransferase M.TneDI. Both enzymes were purified simply by heating the cell lysates of E. coli followed by centrifugation. The enzymes were active over a broad range of temperatures, from 42°C to at least 77°C, with the highest activities observed at 77°C. R.TneDI cleaved at the center of the recognition sequence (CG↓CG) and generated blunt-end cuts. Overexpression of R.TneDI in BL21(DE3) was confirmed by both SDS-PAGE and Western blotting.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Thermotoga spp. are a group of hyperthermophilic bacteria with optimal growth around 80°C. These strict anaerobes hydrolyze a number of polysaccharides through fermentative catabolism and produce hydrogen gas as one of the final products (Schroder et al. 1994), which may be directly used as a clean fuel. In addition, many Thermotoga enzymes have been expressed in E. coli and display extraordinary stability and extended shelf life. There is an increasing demand for tractable tools to enable genetic analyses and manipulations of Thermotoga. A technical obstacle to any genetic engineering effort is the restriction–modification (R–M) systems of the host. If not properly modified, foreign DNA molecules will likely be restricted by host endonucleases as soon as they enter the new cell. Understanding the R–M systems of Thermotoga is a necessary step toward genetically modifying these organisms.
R–M systems represent a common mechanism that bacteria use to protect the integrity of their genetic material, DNA. Most R–M systems comprise pairs of intracellular enzymes with opposing catalytic activities: a restriction endonuclease (REase) and a modification methyltransferase (MTase) (Wilson and Murray 1991; Pingoud et al. 2005). The modification function adds a methyl group to an adenine or cytosine in a specific sequence context, i.e., a recognition site. In most cases, REases break DNA chains containing the recognition sites, but not if the site has been modified by the cognate MTases. Genes specifying REases are generally linked to the gene encoding the cognate MTases. R–M systems are often strain specific, allowing bacteria to differentially destroy invading DNA.
Thousands of R–M systems have been identified through massive screening of a large number of strains (Whitehead and Brown 1985; Hjorleifsdottir et al. 1996) or more recently by bioinformatic analysis of genome sequences (Matveyev et al. 2001; Ishikawa et al. 2005). These systems have been grouped into 4 classes (Types I, II, III, and IV) (Wilson and Murray 1991; Roberts et al. 2003). In a Type I system, three subunits, R (restriction), M (modification), and S (specificity) form a holoenzyme acting as both a REase and an MTase. Their recognition sequences are asymmetric and bipartite. Cleavage happens at random positions far away from the recognition site (~100 nt), and ATP is required for this process. Type II systems are composed of just two proteins, R and M, and they usually act independently. They break DNA at or near the recognition sites at specific positions. The restriction activity requires Mg2+ but not ATP. Type III systems also have just two polypeptides: R and M. The M subunit can function independently as a modification MTase, but the restriction activity requires a complex containing both R and M subunits. DNA cleavage occurs at a site ~25 bp away from the asymmetrical recognition sequence in the presence of Mg2+ and ATP (Sears et al. 2005; Adamczyk-Poplawska et al. 2009). Type IV systems cleave only modified DNA. DNA fragments cut by the same REase or its isoschizomers can be rejoined by DNA ligase, allowing rearrangement of DNA sequences. R–M systems serve as important biotechnology tools in modern molecular biological research and genetic engineering practices, particularly Type II R–M systems, hundreds of which have been commercialized.
Based on sequence comparison to related genes, the Restriction Enzyme Database (REBASE) (Roberts et al. 2010) predicts that there are three methyltransferase genes in the genome of T. neapolitana: CTN_0340, CTN_1203, and CTN_1590. It further suggests that CTN_0339 and CTN_0340 constitute a Type II R–M system recognizing CGCG sequences with an unclear cleavage site. In the NCBI database, CTN_0339 is annotated as a hypothetical gene, and CTN_0340 as an m4C-MTase gene. These two genes are clustered on the chromosome with a convergent orientation. The purpose of this study is to validate the functional assignments of the two genes made by REBASE and to facilitate the construction of genetic tools for Thermotoga.
Materials and methods
Strains and cultivation conditions
Bacterial strains and vectors involved in this study are listed in Table 1. T. neapolitana ATCC 49049 (same as DSM 4359) was obtained from the American Type Culture Collection (http://www.atcc.org/) and was cultivated at 77°C in liquid SVO medium (Van Ooteghem et al. 2002). All DNA manipulations were carried out in E. coli XL1-Blue MRF′, and the expression of CTN_0339 and CTN_0340 was studied in both XL1-Blue MRF′ and E. coli BL21(DE3). E. coli strains were cultivated at 30, 37, or 42°C in Luria–Bertani (LB) medium (1% tryptone, 0.5% NaCl, 0.5% yeast extract) with 1.5% (w/v) agar for plates. Kanamycin and chloramphenicol were added when needed at 50 and 30 μg ml−1, respectively. Growth of E. coli strains was measured as optical density at 600 nm (OD600). For BL21(DE3) recombinant strains, 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) was added to induce the expression of CTN_0339 when culture density reached about 0.4–0.6. Induced cultures were incubated for another 4 h prior to further analyses.
Cloning of CTN_0339 into pET-24a(+)
The complete genome sequence of T. neapolitana is available at http://www.ncbi.nlm.nih.gov/genomeprj/21023. A DNA fragment corresponding to the coding region of CTN_0339 (672 bp, excluding the stop codon) was amplified from the genomic DNA of T. neapolitana with primers 5′-GGAATTCCATATGAGAAAAACGGATCCTCTCAT-3′, which allows the start codon to be embedded in an NdeI recognition site, and 5′-CACAAGCTTCTGTTGATATTTTTCTATCA-3′, which introduces a HindIII site at the end of sequence. The PCR product (691 bp, including NdeI and HindIII linkers) was purified by ethanol precipitation, digested with the two restriction enzymes, and inserted to the same sites of the E. coli expression vector pET-24a(+) to generate pJC339. The expressed protein has an appended His tag for immunodetection.
Cloning of CTN_0340 into pACYC184
CTN_0340 was cloned to pACYC184 under the control of the promoter for the tetracycline resistance gene (tet). First, the coding region of the tet gene as well as its downstream region (1581–3603) was removed from plasmid pACYC184 by inverse PCR using primers 5′-GAATTCCATATGCGGTGCCTGACTGCGTTAGC-3′ and 5′-GAATTCCATATGGAATTCCCGCGGATCCTGAGAAGCACACGGTCACACTGCTTCC-3′. The PCR product carries an NdeI site at its 5′ end and a BamHI–NdeI linker at its 3′ end. After digestion with NdeI, the fragment was allowed to recircularize to form pJC184. Meanwhile, primers 5′-GAATTCCATATGAAGAGAAGAAAATCAACAAG-3′ and 5′-CGCGGATCCTTATCAACAGTGATCTTCAA-3′ were used to amplify the 927-bp coding region of CTN_0340 from the chromosome of T. neapolitana. The primers were designed to have the start codon embedded in an NdeI site and the stop codon followed by a BamHI site. PCR fragment of CTN_0340 was then digested with NdeI and BamHI and was inserted to the corresponding sites of pJC184 to generate pJC340.
Purification and analyses of CTN_0339 and CTN_0340 gene products
One gram of E. coli wet cells grown at 37°C, containing pJC339 or pJC340 or both, were resuspended in 3 ml of lysis buffer (10 mM Tris–HCl, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT, pH 8.0) and were lysed with two passages through a French press at 14,000 pounds per square inch. After centrifugation at 27 k×g for 25 min, the supernatant was transferred to a new tube and heated at 77°C for 1 h to denature most of the host proteins, followed by another round of centrifugation at 17 k×g for 15 min. The supernatant containing purified Thermotoga proteins was stored at 4°C. Such extracts retained 100% of the restriction activity for up to 5 weeks, and ≥50% of the activity for at least 12 weeks. For expression analysis, heat-purified cell extracts were mixed with loading buffer and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) in 15% (w/v) gels. HisDetector Western Blot Kit (Kirkegaard & Perry Laboratories, Inc., Maryland, USA) was used for colorimetric immunodetection of histidine-tagged CTN_0339 protein. For restriction assays, heat-purified cell extracts containing REase were serially diluted in 2-fold steps with the lysis buffer and incubated with 5 μg of substrate DNA in a reaction volume of 50 μl. After 1 h of incubation at 50, 65, or 77°C, the reaction was terminated with 5 μl of stopping buffer (50 mM EDTA, 50% (w/v) glycerol, 0.02% (w/v) bromophenol blue, pH 8.0). Digested DNA fragments were resolved on 1% (w/v) agarose gels. One unit of REase activity is defined as the quantity required to completely digest 1 μg of pUC19 DNA in 1 h at 77°C. For modification assays, heat-purified cell extracts containing the MTase were serially diluted in 2-fold steps with the lysis buffer and incubated with 5 μg of substrate DNA in a total volume of 50 μl. After 1 h of incubation at 77°C, the reaction mix was then incubated with 0.5 U of REase at 77°C overnight. One unit of the MTase activity is defined as the quantity required to completely protect 1 μg of pUC19 DNA from the cleavage of REase for 1 h at 77°C.
Determination of the cleavage site of REase
The PCR product of CTN_0339 was completely digested by heat-purified REase. Two fragments were generated and retrieved from agarose gels. The smaller piece was directly analyzed by Sanger sequencing; the larger piece was further digested by NdeI and inserted into pUC19 at the NdeI–SmaI sites to form pDH22. The insertion region of the constructed vector was sequenced.
Results
Growth of E. coli recombinant strains
Plasmids pJC339 and pJC340 were separately constructed in E. coli strain XL1-Blue MRF′ by screening the transformants at 30°C. The cloned sequence of CTN_0339 (putative REase) and CTN_0340 (putative MTase) were confirmed by both restriction digestions and DNA sequencing. Plasmid pJC339 was introduced into XL1-Blue MRF′/pJC340 to obtain strain XL1-Blue MRF′/pJC339+pJC340. On these plasmids, CTN_0339 and CTN_0340 are under the control of T7 and tet promoters, respectively. Unlike BL21(DE3), XL1-Blue MRF′ has no chromosomal T7 RNA polymerase gene, thus CTN_0339 is transcribed merely at a basal level in an XL1-Blue MRF′ background. The tet promoter is constitutively active in most E. coli laboratory strains, including XL1-Blue MRF′ and BL21(DE3).
XL1-Blue MRF′ harboring pJC339 or along with pJC340 was cultivated at 30, 37, or 42°C, and the growth was monitored hourly. All strains grew equally well at 30 and 37°C (Fig. 1) and displayed no signs of stress from expressing the Thermotoga proteins, which are not expected to be much active at these temperatures. At 42°C, cells carrying pJC339 and pJC340 propagated as well as the control strain. With pJC339, however, the growth ceased at a low culture density. Therefore, at elevated temperatures, CTN_0339 inhibits the physiological well-being of host cells, but this inhibition is countered by CTN_0340. These observations are in agreement with the speculated restriction and modification activities of the two proteins.

XL1-Blue MRF′ recombinant strains grown at 30°C (a), 37°C (b), or 42°C (c)
Restriction and modification assays of the Thermotoga proteins
To provide further evidence to the functions of the two Thermotoga proteins, we first tested the restriction activity of CTN_0339 at 50, 65, and 77°C using pUC19 plasmid DNA prepared from strain XL1-Blue MRF′. Apparent REase activity was observed in cells expressing pJC339 at all tested temperatures, and the highest cleavage efficiency occurred at 77°C (Fig. 2). For example, 2.5 μl of CTN_0339 extract was required to completely digest 1 μg of pUC19 DNA within 1 h at 65 or 50°C, whereas only half of the amount was needed at 77°C (Fig. 2). The sizes of fully digested pUC19 fragments agree with the occurrence and locations of CGCG sites in this plasmid (Fig. 3).

Digestion of pUC19 DNA with extracts of XL1-Blue MRF′ carrying pJC339 at 50°C (a), 65°C (b), or 77°C (c). The amount of cell extract per μg DNA is labeled on top of each lane (in μl). M λ/HindIII. Analyzed with 1% (w/v) agarose gel

R.TneDI-digested pUC19 DNA analyzed with 2% (w/v) agarose gel. Sizes of the fully digested fragments match the occurrence and locations of CGCG sites in the plasmid
We next tested whether CTN_0340 was able to protect pUC19 DNA from the digestion of REase. The substrate DNA was incubated with CTN_0340 extract at 77°C for 1 h and then digested by REase. DNA treated with 10 μl of CTN_0340 extract was only partially digested by REase, even after overnight incubation at 77°C (Fig. 4a). If the CTN_0340 extract was diluted in 2-fold steps, the substrate DNA became more and more susceptible to the same amount of R.TneDI, and full digestion eventually occurred (Fig. 4a). In another experiment, cell extract containing both CTN_0339 and CTN_0340 was used to treat pUC19 DNA (Fig. 4b). After 1 h of incubation at 55°C, the majority of the DNA was still intact (Fig. 4b), whereas under the same conditions, cell extract containing just CTN_0339 would have substantially degraded the DNA (Fig. 2a). Low levels of partial digestion were noticed in some samples in Fig. 4b, reflecting a competition between the antagonistic activities of CTN_0339 and CTN_0340. Taking together the above findings (Figs. 1, 2, 3, 4), it is conclusive that CTN_0339 encodes REase R.TneDI, and CTN_0340 the MTase M.TneDI.

Protection of pUC19 DNA by M.TneDI. a Plasmid DNA was treated with various amounts of cell extract of XL1-Blue MRF′ expressing CTN_0340, as labeled on top of each lane. The DNA was then subjected to the digestion of R.TneDI (0.1 U per μg DNA). b Plasmid DNA was treated with various amounts of cell extract of XL1-Blue MRF′ expressing both CTN_0340 and CTN_0339. The amount of cell extract per μg DNA is labeled on top of each lane (in μl)
Overexpression of R.TneDI
Although REase activity was detected in XL1-Blue MRF′/pJC339, we were unable to observe the R.TneDI protein with either SDS-PAGE or immunoblotting, probably due to its extremely low expression level. To prepare a large quantity of R.TneDI for further applications, pJC340 and pJC339 were co-transformed to BL21(DE3), and the cell extract of the recombinant strain was prepared and analyzed by SDS-PAGE. A distinctive band with an apparent size of ~29 kDa was revealed (Fig. 5, top). Western blotting analysis confirmed that the specific band was the histidine-tagged R.TneDI (Fig. 5, bottom), which has a theoretical molecular weight of 28.3 kDa. Thus, it is feasible to overproduce R.TneDI in an E. coli host and subsequently purify it with an affinity column. The synthesis of M.TneDI, which has a theoretical molecular weight of 35.3 kDa, was below the detection limit of SDS-PAGE in both hosts. This is not surprising though, since pACYC184 is a low copy number vector and the tet promoter is not typically used for overexpression. Immunoblotting for this MTase was not attempted.

SDS-PAGE (top) and Western blotting (bottom) analyses of extracts of BL21(DE3) carrying both pJC339 and pJC340. Arrows point to the position of R.TneDI. Cells carrying the parent plasmids pET-24a(+) and pJC184 were used as the control
Determination of the cleavage site of R.TneDI
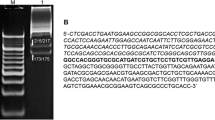
Sequence analysis uncovered a recognition site (CGCG) in the coding region of R.TneDI. The PCR product of this region was digested by R.TneDI, generating an upstream fragment of 502 bp and a downstream fragment of 189 bp. Sequencing of the smaller piece revealed that R.TneDI cut immediately after the first G in its recognition sequence (CG↓CG) (Fig. 6a). To further confirm that R.TneDI is a blunt-end cutter, the larger fragment was digested with NdeI and inserted into the NdeI–SmaI sites of pUC19. Sequencing of the new vector in the insertion region showed that the two DNA pieces, cut by R.TneDI and SmaI, respectively, were joined together seamlessly (Fig. 6b).

R.TneDI cleaved at the center of its recognition sequence (CG↓CG). a The sequence of the smaller fragment of CTN_0339/R.TneDI ended at CG. The extra A at the 3′-end, donated by an asterisk, was a template-independent addition by Taq polymerase (Clark 1988; Stier and Kiss 2010). b The larger fragment of CTN_0339/R.TneDI was ligated with pUC19/SmaI with their blunt ends. The half recognition sites of the enzymes are underlined. The shaded nucleotides represent CTN_0339 DNA sequences
Discussion
In this study, we demonstrated that genes CTN_0339 and CTN_0340 of T. neapolitana encode the TneDI Type II R–M system. R.TneDI may serve as a handy tool for specific cleavage of DNA molecules, and an E. coli strain containing M.TneDI should be a good host for preparing the DNA to be introduced to a Thermotoga host. Type II REases recognizing CGCG sequence have been discovered before, such as AccII, FnuDII, ThaI, BsuE, Bsh1236I, BepI, and MvnI (Lui et al. 1979; Strobl and Thompson 1984; Gaido and Strobl 1987; Gaido et al. 1988; Thomm et al. 1988; Venetianer and Orosz 1988), all of which cleave at the center of the recognition site and create blunt ends for their DNA substrates. A notable exception is SelI, which cleaves at the ends of the recognition site (↓CGCG) (Miyake et al. 1992).
To protect host chromosomal DNA, REases usually have to be cloned in the presence of the cognate MTases (Howard et al. 1986). Alternatively, in vitro transcription and translation approaches could be used (Ishikawa et al. 2005). Here, we were able to directly clone R.TneDI to XL1-Blue MRF′ by imposing its expression under the control of the T7 promoter. The fact that R.TneDI is less active at mesophilic temperatures probably also contributed to the survival of the host cells. Similar success has been achieved before with other thermophilic R–M systems, such as TaqI (Slatko et al. 1987).
In a modification reaction, AdoMet (S-adenosyl-l-methionine) serves as the methyl donor and is thus an essential cofactor for methylases. In our modification assays (Fig. 4), supplementation of AdoMet was not attempted, because the E. coli cell extracts were expected to carry enough of the cofactor, even after the heat treatment. Contrary to our common belief, AdoMet is reasonably stable in solutions. For instance, if crude yeast extract is incubated at 37°C, ~70% of the original AdoMet can be found after 6 days and ~25% for 15 days (Morana et al. 2002). Therefore, a fair amount of AdoMet is expected to survive after incubation at 77°C for 1 h in the cell extract of E. coli. Moreover, our cell extracts were prepared from 1 g of wet cells (equivalent to 1 L at OD600 ~1.0) in 3 ml of lysis buffer. The cell density was extremely high. In our assays, the cell extracts were diluted for no more than 64 times. Consequently, even the most diluted sample should have enough AdoMet.
We have not determined which cytosine at the recognition site is modified by M.TneDI. Also unknown is the exact modification position of the cytosine, though motif analysis almost certainly identifies M.TneDI as an m4C MTase (Malone et al. 1995). Comparison of M.TneDI and M.PvuII, a m4C MTase with a known structure (Gong et al. 1997), revealed the conserved catalytic center (TSPPY/F) (in motif IV) and the Rossman fold serving as the AdoMet binding site (FxGxG/N) (in motif I) (Fig. 7). The alignment is more gapped in the target recognition domains, which is not surprising as PvuII system recognizes CAGCTG instead of CGCG. It has been suggested that m4C is more common in thermophiles than m5C, because an m5C residue has a higher risk of deamination at elevated temperatures (Ehrlich et al. 1985).

Sequence alignment of M.TneDI and M.PvuII generated by CLUSTAL W (1.81) (Thompson et al. 1994) and shaded by Boxshade 3.3.1. Conserved residues are highlighted in black and similar residues in gray. Nine possible structural motifs (IV–III), which are well conserved among group β MTase (Malone et al. 1995), are identified in M.TneDI and underlined
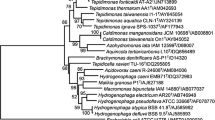
Orthologs of the TneDI system have been found in other members of the Thermotogaceae family. The locus IDs and identities of the orthologs pertaining to the TneDI system at the protein level are summarized in Table 2. The neighborhood genes in the six Thermotogaceae genomes share nearly perfect synteny. Of even greater interest, given the low level of sequence conservation among REases in general, is the presence of an orthologous R–M system in the archaeon Ferroglobus placidus. While F. placidus is phylogenetically distant from Thermotogaceae, they live in the same high-temperature anaerobic environment, suggesting a recent horizontal gene transfer event between the two groups.
References
Adamczyk-Poplawska M, Lower M, Piekarowicz A (2009) Characterization of the NgoAXP: phase-variable type III restriction–modification system in Neisseria gonorrhoeae. FEMS Microbiol Lett 300(1):25–35
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Belkin S, Wirsen CO, Jannasch HW (1986) A new sulfur-reducing, extremely thermophilic eubacterium from a submarine thermal vent. Appl Environ Microbiol 51(6):1180–1185
Clark JM (1988) Novel non-templated nucleotide addition reactions catalyzed by procaryotic and eucaryotic DNA polymerases. Nucleic Acids Res 16(20):9677–9686
Ehrlich M, Gama-Sosa MA, Carreira LH, Ljungdahl LG, Kuo KC, Gehrke CW (1985) DNA methylation in thermophilic bacteria: N4-methylcytosine, 5-methylcytosine, and N6-methyladenine. Nucleic Acids Res 13(4):1399–1412
Gaido ML, Strobl JS (1987) Methylation sensitivity of the restriction enzymes FnuDII and AccII. Arch Microbiol 146(4):338–340
Gaido ML, Prostko CR, Strobl JS (1988) Isolation and characterization of BsuE methyltransferase, a CGCG specific DNA methyltransferase from Bacillus subtilis. J Biol Chem 263(10):4832–4836
Gong W, O’Gara M, Blumenthal RM, Cheng X (1997) Structure of pvuII DNA-(cytosine N4) methyltransferase, an example of domain permutation and protein fold assignment. Nucleic Acids Res 25(14):2702–2715
Hjorleifsdottir S, Petursdottir SK, Korpela J, Torsti AM, Mattila P, Kristjansson JK (1996) Screening for restriction endonucleases in aerobic, thermophilic eubacteria. Biotechnol Tech 10(1):13–18
Howard KA, Card C, Benner JS, Callahan HL, Maunus R, Silber K, Wilson G, Brooks JE (1986) Cloning the DdeI restriction–modification system using a two-step method. Nucleic Acids Res 14(20):7939–7951
Ishikawa K, Watanabe M, Kuroita T, Uchiyama I, Bujnicki JM, Kawakami B, Tanokura M, Kobayashi I (2005) Discovery of a novel restriction endonuclease by genome comparison and application of a wheat-germ-based cell-free translation assay: PabI (5’-GTA/C) from the hyperthermophilic archaeon Pyrococcus abyssi. Nucleic Acids Res 33(13):e112
Lui AC, McBride BC, Vovis GF, Smith M (1979) Site specific endonuclease from Fusobacterium nucleatum. Nucleic Acids Res 6(1):1–15
Malone T, Blumenthal RM, Cheng X (1995) Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J Mol Biol 253(4):618–632
Matveyev AV, Young KT, Meng A, Elhai J (2001) DNA methyltransferases of the cyanobacterium Anabaena PCC 7120. Nucleic Acids Res 29(7):1491–1506
Miyake M, Kotani H, Asada Y (1992) Isolation and identification of restriction endonuclease, SelI from a cyanobacterium, Synechococcus elongatus. Nucleic Acids Res 20(10):2605
Morana A, Stiuso P, Colonna G, Lamberti M, Carteni M, De Rosa M (2002) Stabilization of S-adenosyl-l-methionine promoted by trehalose. Biochim Biophys Acta 1573(2):105–108
Pingoud A, Fuxreiter M, Pingoud V, Wende W (2005) Type II restriction endonucleases: structure and mechanism. Cell Mol Life Sci 62(6):685–707
Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev S, Dryden DT, Dybvig K, Firman K, Gromova ES, Gumport RI, Halford SE, Hattman S, Heitman J, Hornby DP, Janulaitis A, Jeltsch A, Josephsen J, Kiss A, Klaenhammer TR, Kobayashi I, Kong H, Kruger DH, Lacks S, Marinus MG, Miyahara M, Morgan RD, Murray NE, Nagaraja V, Piekarowicz A, Pingoud A, Raleigh E, Rao DN, Reich N, Repin VE, Selker EU, Shaw PC, Stein DC, Stoddard BL, Szybalski W, Trautner TA, Van Etten JL, Vitor JM, Wilson GG, Xu SY (2003) A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31(7):1805–1812
Roberts RJ, Vincze T, Posfai J, Macelis D (2010) REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 38(Database issue):D234–D236
Schroder C, Selig M, Schonheit P (1994) Glucose fermentation to acetate, CO2 and H2 in the anaerobic hyperthermophilic eubacterium Thermotoga Maritima: involvement of the Embden–Meyerhof pathway. Arch Microbiol 161(6):460–470
Sears A, Peakman LJ, Wilson GG, Szczelkun MD (2005) Characterization of the Type III restriction endonuclease PstII from Providencia stuartii. Nucleic Acids Res 33(15):4775–4787
Slatko BE, Benner JS, Jager-Quinton T, Moran LS, Simcox TG, Van Cott EM, Wilson GG (1987) Cloning, sequencing and expression of the TaqI restriction–modification system. Nucleic Acids Res 15(23):9781–9796
Stier I, Kiss A (2010) The type II restriction endonuclease MvaI has dual specificity. Nucleic Acids Res 38(22):8231–8238
Strobl JS, Thompson EB (1984) Methylation of either cytosine in the recognition sequence CGCG inhibits ThaI cleavage of DNA. Nucleic Acids Res 12(21):8073–8083
Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Meth Enzymol 185:60–89
Thomm M, Frey G, Bolton BJ, Laue F, Kessler C, Stetter KO (1988) MvnI: a restriction enzyme in the archaebacterium Methanococcus vannielii. FEMS Microbiol Lett 52(3):229–233
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Van Ooteghem SA, Beer SK, Yue PC (2002) Hydrogen production by the thermophilic bacterium Thermotoga neapolitana. Appl Biochem Biotechnol 98:177–189
Venetianer P, Orosz A (1988) BepI restriction endonuclease, a new isoschisomer of FnuDII. Nucleic Acids Res 16(1):350
Whitehead PR, Brown NL (1985) A simple and rapid method for screening bacteria for type II restriction endonucleases: enzymes in Aphanothece halophytica. Arch Microbiol 141(1):70–74
Wilson GG, Murray NE (1991) Restriction and modification systems. Annu Rev Genet 25:585–627
Acknowledgments
We are grateful to Dr. Robert M. Blumenthal at the University of Toledo College of Medicine for insightful discussions and critical reading of the manuscript. This work was supported by the BGSU internal funds to ZX.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Huang.
Rights and permissions
About this article
Cite this article
Xu, Z., Han, D., Cao, J. et al. Cloning and characterization of the TneDI restriction–modification system of Thermotoga neapolitana . Extremophiles 15, 665 (2011). https://doi.org/10.1007/s00792-011-0397-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00792-011-0397-9