
Abstract
A moderately thermophilic Gram-negative bacterium isolated from the Polok hot spring, Sikkim, India, was identified as a strain (PL17) of Tepidimonas fonticaldi by 16S rDNA sequencing. T. fonticaldi PL17 produces a Type IIP restriction endonuclease; named TfoI. Restriction mapping, run-off sequencing of TfoI-digests of dsDNA fragments, and end compatibility of TfoI with NdeI confirmed that the enzyme recognizes and cleaves the sequence 5′–T^TAA–3′, and is thus an isoschizomer of MseI. The TfoI restriction–modification genes in the T. fonticaldi PL17 genome were identified, and the annotated TfoI protein encodes a protein of 181 amino acid residues that shares 47.2% sequence identity with MseI. The native enzyme was purified using a four-column chromatography protocol, and its functional homogeneity was confirmed by standard quality control tests. The ESI-MS measured molecular weight of purified TfoI (20.696 kDa) is in agreement with that of the calculated monomeric molecular weight of the predicted TfoI protein sequence (20.694 kDa). TfoI exhibits optimal activity in the temperature range of 55–70 °C with Mg+2 or Co+2 as cofactor. Similar to its isoschizomers, TfoI can be used as the frequent cutter for genome analysis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Restriction–modification (RM) systems are largely accepted to be the cellular defense machinery of prokaryotes against invading bacteriophages, plasmids, or other foreign DNAs (Raleigh and Brooks 1998; Vasu and Nagaraja 2013). The RM system consists of a restriction endonuclease (REase, R) that recognizes and cleaves specific DNA sequences, and a cognate mehtyltransferase (MTase, M) which protects the self DNA from the REase by selectively methylating the adenine and/or cytosine bases of the same recognition sequence (Bickle and Kruger 1993). In addition to the role of RM systems in protecting cell against foreign DNA, there is emerging information on these as selfish genes to their involvement in stabilizing genomes, controlling speciation etc (Vasu and Nagaraja 2013). RM systems are observed to be widely distributed in eubacteria and archaebacteria (Oliveira et al. 2014). Even certain phages are known to carry the RM systems (Chaturvedi and Chakravorty 2003; Dempsey et al. 2005; Hadi et al. 1983; Rao et al. 2014; Xia et al. 1986; Xia and Etten 1986). The function of the phage-encoded RM enzymes are proposed to provide selective survival advantage by preventing infection of the host by other viruses (Agarkova et al. 2006; Joshi et al. 1982; Dempsey et al. 2005) and in degradation of the host DNA (Szekeres et al. 1983). REBASE (Roberts et al. 2015), a comprehensive database of proteins of RM systems, reports about 4290 different restriction enzymes genetically or biochemically characterized until date, and more than 48,175 putative REases are predicted from genome sequences (http://rebase.neb.com/rebase/statlist.html).
The REases are classified primarily into four categories, Type I–IV, based on the DNA recognition sequence, cleavage position, cofactor requirements, and the protein subunit composition (Loenen et al. 2014). Of the four types of REases, Type II enzymes are considered as the “work horses” of modern molecular biology (Roberts 2005; Loenen et al. 2014). Type II REases exhibit high sequence specificity with respect to both recognition and cleavage, and usually require Mg+2 ions as the only cofactor (Pingoud et al. 2014). Type II enzymes are further grouped into 11 subtypes, with the most commonly used enzymes that recognize Palindromic sequences belonging to subtype IIP. About 43% of the putative restriction enzymes are classified as Type II REases and greater than 96% of the characterized REases belong to the Type II category with 412 distinct specificities (http://rebase.neb.com/rebase/statlist.html).
Most restriction enzymes have been isolated from mesophilic bacteria (Roberts et al. 2015), but thermostable REases with both novel and known specificities (isochizomers) are desirable from enzyme properties and protein processing perspectives (Sharma et al. 2013). Thermophilic bacteria found in a broad range of geothermally active habitats, such as geysers, hot springs, and deep sea hydrothermal vents, are source of thermostable enzymes. In this paper, we describe isolation and purification of a moderately thermophilic REase, TfoI, from a thermophile (Tepidimonas fonticaldi PL17) found in the water of the Polok hot spring in Northeast India. Sequence analysis and biochemical characterization of purified TfoI reveal it to be a Type IIP REase, and as a new isoschizomer of MseI enzyme.
Materials and methods
Strain and growth conditions
T. fonticaldi PL17 used in this study was isolated from one of the two hot springs in Polok (Latitude: 27°21.00°, Longitude: 88°19.353°) in Sikkim, Northeast India (Thakur et al. 2013). The temperature and pH of water in this hot spring was measured to be at 58 °C and 8.0, respectively. Bacteria were typically cultured in R2A broth (Reasoner and Geldreich 1985) (HiMedia) at 55 °C, which was determined to be optimal based on the temperature dependence of growth of the isolate. To determine the optimum time of growth for maximum production of the TfoI, 100 ml of R2A broth inoculated with 2% overnight seed culture was grown at 55 °C for 20 h. Cells were harvested from 1.5 ml of the culture broth at different hours of growth and stored at −80 °C until further use.
16S rDNA amplification, sequencing, and phylogenetic analysis
The 16S rDNA gene sequence was PCR amplified using the genomic DNA of the isolate (ZR Fungal/Bacterial DNA MiniPrep kit) as a template and E. coli 16S rDNA specific primers, 27F and 1492R (Weisburg et al. 1991). The PCR product was directly subjected to ExoSap (Affymetrix, China) treatment before sequencing. DNA sequencing was performed using primers, 27F, 536R, and 1492R; the numbers correspond to the E. coli 16S rDNA positions (Weisburg et al. 1991; Wang and Qian 2009). The individual sequences were aligned and the complete 16S rDNA sequence was assembled using SeqManPro in DNAStar (v7.0). The ez-taxon module in EzBioCloud (Kim et al. 2012) was used to determine the phylogenetic neighbors and the phylogenetic tree was constructed using the Neighbor-Joining method (Saitou and Nei 1987) with 1000 bootstrap replications to assess the nodal support in the tree. The dendrogram was created from the Molecular Evolutionary Genetics Analysis software (MEGA, version 6).
Restriction endonuclease (REase) activity assay in cell lysate
Frozen cells from 1 to 1.5 ml of the isolate grown in R2A broth were thawed, and re-suspended in 250 µl of 50 mM Tris (pH 7.4). Re-suspended cells were lysed by sonication (30% amplitude; 10 s on-15 s off per cycle; total time of 2 min) and the supernatant (soluble cell lysate) was separated by centrifugation at about 18,000×g for 15 min. The supernatant (2 µl) was used to digest about 200 ng of plasmid DNA (pBR322, Fermentas; pUC18, Merck) and phage DNA (Lambda DNA, Takara or NEB; M13mp18, Merck and φX174, Merck) in a reaction volume of 10 µl for 1 h at 55 °C and the digestion patterns were obtained on a 1% agarose gel. The fragmentation pattern obtained for various DNA substrates cleaved with the enzyme present in the cell lysate were compared with that obtained for the same substrates digested with the purified TfoI (data not shown).
In the experiment to determine the TfoI production as a function of the cell growth, the total protein content in the cell lysate at different timepoints of the growth curve was estimated using the BCA protein estimation kit (Thermo Scientific). The pBR322 plasmid DNA (100 ng) was then digested with around 1–4 µl of cell lysate containing 500 ng of total protein in a reaction volume of 15 µl for 10 min at 55 °C in buffer B (Thermo Scientific). The digestion products were visualized on a 1% agarose gel.
Purification of TfoI
Cell pellet from two liters growth of T. fonticaldi PL17 (∼3 g of cell wet-weight per 2 L of culture) at 55 °C for 12 h at 200 rpm in R2A broth (HiMedia) was obtained after centrifugation of culture at 2430×g for 20 min at 4 °C. The cell pellet was re-suspended in 40 ml of 50 mM Tris, pH 7.4 (USB) and stored at −80 °C until further use. The frozen cells were thawed, sonicated (amplitude 30%, 10 s on-15 s off per cycle) for a total time of 30 min. The cell lysate was clarified by centrifugation at 18,500×g for one hour at 4 °C.
Four different chromatography matrices, Q-Sepharose fast flow (GE Healthcare), SP-Sepharose fast flow (GE Healthcare), Affi-Gel Blue gel (BioRad), and phenyl-Sepharose fast flow (GE Healthcare), were used in sequence for purification of the active protein. The buffer used for purification with all columns was 50 mM Tris, pH 7.4 and protein elution was carried out in a step gradient mode with 50 mM Tris (pH 7.4) containing NaCl at varying concentrations. In brief, the supernatant after cell lysis was loaded on the Q-Sepharose resin (30 ml); active protein primarily present as unbound and in low salt (0.1 M NaCl) wash fractions was pooled and loaded on the SP-Sepharose resin (20 ml). Active TfoI maximally eluted in buffer containing 0.2 M NaCl and its NaCl concentration was adjusted to a final concentration of 0.4 M before loading on to the Affi-Gel Blue gel matrix (12 ml). Active protein which eluted between 0.5 M and 1.0 M NaCl containing buffer was pooled, and the NaCl concentration was increased to a final concentration of 2 M before applying it on the phenyl-Sepharose fast flow resin (2 ml). The final active protein was eluted in water from this column and further transferred into 50 mM Tris (pH 7.4) using 1 M Tris stock solution. Fractions containing active TfoI in each step of the purification protocol were determined by REase activity assay using pBR322 as substrate. The total protein content in active fractions for each of the purification steps was estimated using the BCA protein estimation kit (Thermo Scientific), and the pooled fractions with active TfoI were analyzed on a silver-stained 15% SDS–PAGE.
Unit activity determination
To determine the total TfoI enzyme units at different stages in the purification process, one µg of Lambda DNA in buffer B (Thermo Scientific) was digested with different volumes of pooled active fractions in a reaction volume of 50 µl and the reaction mixture was incubated at 55 °C for 1 h. The digested products were analyzed on a 1% agarose gel. One unit of TfoI activity was estimated to be present in the minimum volume of the active fraction that resulted in complete digestion of one µl of Lambda DNA under the above mentioned reaction condition.
Assay for ATP-dependent nucleases
Lambda DNA (200 ng) was digested with two units of TfoI for 1 h at 55 °C in buffer B (Thermo Scientific) in the presence and absence of 1 mM ATP (Thermo Scientific) from different stages of purification. The reactions were analyzed on a 1% agarose gel.
TfoI recognition and restriction site determination
REase activity assay was performed using two units of purified TfoI with 500 ng of plasmid DNA (pBR322, Fermentas; pUC18, Merck) and 300 ng of phage DNA substrates (EcoKI methylated-Lambda DNA, Takara; M13mp18, Merck and φX174, Merck) as described earlier. The fragmentation pattern from the digestion of these DNA substrates was input in the REBpredictor (http://tools.neb.com/maint.php/REBpredictor/) to predict the likely recognition sequence. The largest DNA fragment (of about 1.6 kb) from digesting pBR322 DNA with TfoI was purified from gel, and sequenced using a primer designed as an internal primer, 1500F, based on the prediction by REBpredictor. A 1045 bp pBR322 fragment (base position 871–1915) containing a single predicted recognition site by REBpredictor was PCR amplified using primers 871F and 1915R. About 500 ng of the PCR product was digested with ten units of purified TfoI at 55 °C for 1 h in buffer B (Thermo Scientific). The two fragments thus obtained were gel-purified and sequenced using the 1500F and 1915R primers, respectively. The number in all primer names is with respect to the start base position in the pBR322 plasmid that these are derived from; and forward and reverse orientations of the primers are referred to as ‘F’ and ‘R’, respectively.
TfoI cleavage site was confirmed by testing for TfoI end compatibility with the NdeI restriction site using a directional cloning approach. The pET28a(+) plasmid was double digested with NdeI and XhoI restriction enzymes to generate the vector for cloning. A fragment of pET29b(+) plasmid encompassing its multiple cloning site was PCR amplified using primers corresponding to T7 promoter and T7 terminator sequences, and was subjected to TfoI-XhoI restriction digestion to generate the insert for cloning. The vector and insert fragments purified from gel were ligated using T4 DNA ligase (NEB), and transformed into E. coli DH5α competent cells. Plasmid was isolated from the transformant cells, and DNA sequenced to determine the ligation sites. The NdeI-XhoI and MseI-XhoI digests of pET29b(+) PCR fragment were used as control inserts in this experiment.
Cut-ligate-cut assay
One µg of Lambda DNA was digested with ten units of purified TfoI in a reaction mixture of 50 µl for 1 h followed by heat inactivation of TfoI at 80 °C for 20 min. About 200 ng DNA (cut sample) was removed from this reaction mixture and stored for later analysis. The remaining reaction mixture was subjected to overnight DNA ligation at 16 °C by the addition of ligation buffer and T4 DNA ligase (NEB). The T4 DNA ligase was inactivated at 65 °C for 20 min and 200 ng DNA (ligated sample) was removed for analysis. Ten units of purified TfoI was added to the remaining ligated reaction mixture and incubated for 1 h at 55 °C (re-cut sample). About 200 ng of re-cut DNA from the final reaction mixture was removed and all three (cut, ligated and re-cut samples) were analyzed on a 1% agarose gel.
Overdigestion assay
Increasing amount of purified TfoI (10, 20 and 30 units) was used to digest one µg of Lambda DNA in buffer B (Thermo Scientific) in a reaction volume of 50 µl for 16 h. About 200 ng DNA from the reaction mixtures was loaded on a 1% agarose gel for analysis.
Optimal temperature and buffer conditions for TfoI activity
The temperature dependence of TfoI activity was tested by incubating one µg of Lambda DNA and pBR322 plasmid DNA with one unit of the enzyme at different temperatures (25–75 °C) for 1 h. TfoI activity in buffers supplied with commercial REases from NEB (NEBuffers 1, 2, 3 and CutSmart) and Thermo Scientific (TANGO, B, G, O, R, and FastDigest) was assessed by digesting one µg of Lambda DNA with one unit of the purified enzyme for 1 h at 55 °C. About 200 ng DNA from the reaction mixture in both sets of the reaction were analyzed on a 1% agarose gel.
Temperature effect on TfoI activity
Two units of TfoI in buffer B (Thermo Scientific, without DNA substrate) was incubated at 55, 65, and 80 °C for 30 min. The reaction was brought to room temperature and 100 ng of pBR322 DNA was added as substrate followed by incubation at 55 °C for 30 min and the digestion products were analyzed on a 1% agarose gel. As controls, 100 ng of pBR322 DNA was digested with two units of TfoI at 55, 65, and 80 °C for 30 min. Heat inactivation of TfoI at 80 °C was also assessed by performing restriction digestion of a DNA substrate (plasmid DNA or genomic DNA) at 55 °C followed by heating the enzyme at 80 °C for 20 min and then assaying for residual activity of heat-inactivated TfoI in digesting a second DNA substrate (Fig. A9).
Dependence of the restriction enzyme activity on divalent metal ions and NaCl
About 100 ng of pBR322 DNA was digested with one unit of TfoI for 30 min at 55 °C in buffer B (Thermo Scientific) containing MgCl2 (Fisher Scientific), MnCl2 (Fisher Scientific), CoCl2 (Sigma) and CaCl2 (Sigma) at a final concentration of 10 mM. About 100 ng of pBR322 DNA was digested with one unit of TfoI for 30 min at 55 °C in buffer B containing increasing concentrations of Mg2+ (0–200 mM, supplied as MgCl2 salt). The effect of NaCl was measured by incubating 100 ng pBR322 DNA with one unit of TfoI for 30 min at 55 °C in buffer B in the presence of 0–500 mM NaCl. All digests were analyzed on a 1% agarose gel.
Effect of disulfide-reducing agents and organic solvents on enzyme activity
One unit of TfoI was incubated in buffer B (Thermo Scientific) containing 0–5 mM dithiothrietol (DTT, Sigma) and 0–5 mM beta-mercaptoethanol (β-ME, Sigma) for 12 h at 4 °C. The plasmid pBR322 (100 ng) was added as substrate to the mixture followed by incubation at 55 °C for 30 min. About 100 ng of pBR322 DNA was digested with one unit of TfoI in buffer B (Thermo Scientific) in the presence of 0 to 15% (v/v) of glycerol (Sigma) and 0 to 10% (v/v) dimethyl sulfoxide (DMSO, Bio Basic Inc.) for 30 min at 55 °C. All reactions were analyzed on a 1% agarose gel.
Identification of TfoI RM gene cluster
The T. fonticaldi PL17 whole genome shotgun sequencing was performed on MiSeq Illumina sequencing platform using MiSeq reagent kit, version 2 (Genotypic Technology Pvt. Ltd.). The de novo genome was assembled using CLC Genomics Workbench 7.5.1 to obtain minimum contig length of 500 bp and the 67 contigs obtained are submitted to the NCBI database (accession number: LZDH00000000). The protein coding sequences were annotated by NCBI Prokaryotic Genome Annotation Pipeline (PGAP) version 3.3 (Tatusova et al. 2016), Rapid Annotation using Subsystems Technology (RAST) version 2.0 (Aziz et al. 2008), and GLIMMER-3 (Delcher et al. 1999). The annotated genes belonging to restriction–modification systems were analyzed. The absence of a complete Type II RM system in the annotation led us to further analyze the only two MTase subunit genes one each as part of the Type II (YeeA type) and Type III RM systems that were annotated by RAST and GLIMMER. BLAST analysis of these two-gene products revealed that the protein corresponding to the MTase subunit annotated as Type III MTase shares 53.9% sequence identity with MseI MTase. The DNA sequence encompassing this MTase gene was analyzed next for the presence of an open reading frame (ORF) in its vicinity. An un-annotated ORF was found downstream of the MTase subunit of the Type III MTase and was subsequently used as a query sequence to search the Genbank sequences using protein BLAST. Gene identity search based on the sequence alignment of this RAST-annotated Type III MTase along with its immediate downstream un-annotated ORF using BLAST indicates this two-gene cluster to be a –TTAA– recognizing RM system (described in “Results”).
Determination of methylation status of genomic DNA of T. fonticaldi PL17
The genomic DNA (50 ng) of T. fonticaldi PL17 was digested with ten units of purified TfoI, Tru1I (Thermo Scientific, isoschizomer), and HaeIII (NEB, 5′GG^CC3′) for 1 h. The reaction conditions were used as appropriate for each enzyme. The genomic DNA of Pseudomonas aeruginosa MTCC1934 and E. coli BL21(DE3) was also subjected to digestion with all three REases and served as controls.
Results
One of the Gram-negative, moderately thermophilic bacterium isolated in our study of microbial biodiversity in water of a hot spring in Polok, Sikkim, Northeast India screened positive for REase activity (Fig. A1). Comparison of the 16S rRNA gene sequence of this isolate clustered it with the Tepidimonas genus (Fig. 1). Based on its 99.86% sequence identity with Tepidimonas fonticaldi AT-A2T, the isolate is assigned to be Tepidimonas fonticaldi PL17. The REase produced by T. fonticaldi PL17 was named TfoI according to REase nomenclature rules (Roberts et al. 2003).

Phylogenetic positioning of T. fonticaldi PL17. The phylogenetic tree of T. fonticaldi PL17 was derived from the 16S rDNA gene sequence comparison of 20 closely related bacterial species with sequence identity >94% using the Neighbor-Joining method. The numbers indicated at the branch nodes are percentage of 1000 bootstrap replications and only values >50% are shown here. E. coli ATCC11775 was used as an outgroup. The bar represents 0.02 substitutions per nucleotide position
TfoI Purification
REase activity assay in cell lysates performed as a function of growth of T. fonticaldi PL17 indicates that cells produce the enzyme from early log phase until the stationary phase of growth (Fig. A1). The enzyme produced by the isolate after 12 h of growth was purified to homogeneity using a four-step purification procedure (Fig. 2a). The final yield of the purified protein was about 2500 units from a liter of culture which was a recovery of around 15% of the total enzyme produced with specific activity of at least 150,000 U/mg of the protein. REase activity assayed in the presence of ATP at different stages of the purification indicates that ATP-dependent nuclease contamination is successfully removed after the second column of SP-Sepharose (SP) fast flow resin. The final protein eluted from the phenyl-Sepharose (PS) matrix is essentially free of any ATP-dependent nuclease as indicated by the identical digestion pattern observed in the activity assay performed with and without ATP (Fig. A2). The monomeric enzyme has a molecular weight of about 20 kDa (marked by an arrow in Fig. 2a), which was also confirmed by the mass spectrometric data (Fig. A11).

TfoI enzyme purification. a 15% silver-stained SDS–PAGE with pooled fractions containing active TfoI sample from each step of the protein purification is shown here. The arrow marks the position of TfoI. The lane labels indicate whole cell lysate as crude; clarified cell lysate after sonication as lysate; and pooled active fractions from Q-Sepharose, SP-Sepharose, Affi-Gel Blue gel, and phenyl-Sepharose columns as QS, SP, AG, and PS, respectively. TfoI enzyme activity in the pooled active fractions from each purification step using Lambda DNA as a substrate is shown in (b)
The absence of any other DNases in the purified TfoI was further confirmed by performing the overdigestion assay. Unaltered band pattern observed for the Lambda DNA digested with 30-fold excess of enzyme compared with that of cleavage with one unit of the enzyme indicates both absence of non-specific nuclease contamination as well as absence of any star activity of TfoI under the test conditions (Fig. 3a). The ligation of fragments generated by TfoI digestion of Lambda DNA and an identical digestion profile observed for the substrate DNA (Lambda DNA) in the ‘cut’ and ‘re-cut’ samples in the cut-ligate-cut assay, indicates that the 5′- and the 3′-termini of the digested products are intact, i.e., the final purified enzyme is free of any nucleases and phosphatases (Fig. 3b, A3).

Quality control assays for TfoI purity. a Overdigestion of a microg of the Lambda DNA with upto 30 units of pure TfoI for 16 h, and b cut-ligate-re-cut assay of the Lambda DNA with the purified enzyme was performed to assess the purity of purified TfoI. Digestion of Lambda DNA with one unit and 16 units of the enzyme for 1 h served as control in the overdigestion assay in (a)
Determination of TfoI recognition and cleavage site
Analysis of digestion patterns obtained on TfoI digestion of different phage DNA and plasmid DNA using the REBpredictor program suggested that TfoI recognizes 5′–TTAA–3′ as a target sequence (Fig. 4, A4). The largest sized band of about 1.6 kb obtained from digesting pBR322 was subjected to DNA sequencing to determine the sequence at the cleavage site. The –TTAA– DNA sequence is a recognition sequence of the known REases, such as MseI and its isoschizomers. We, therefore, also sequenced the same fragment of pBR322 plasmid digested with MseI. The sequence at the fragment ends for both TfoI (Fig. A5) and MseI (data not shown) was identical, indicating that the recognition site of TfoI is the same as that of MseI, i.e., TfoI is an isoschizomer of MseI. The 4-base –TTAA– sequence as the recognition site was reconfirmed by digestion of a PCR fragment containing a single –TTAA–site generated from the pBR322 plasmid DNA, and sequencing of both fragments (Fig. 5). However, the DNA sequencing data posed an ambiguity regarding the cleavage site due to the presence of a false peak of adenine base at the 3′-end in the chromatogram (marked by ‘a’ in Figs. A5, 5), likely due to the non-template dependent terminal nucleotide addition activity of AmpliTaq polymerase used for DNA sequencing (Hu 1993; Samuelson et al. 2004).

TfoI digests of various DNA substrates. Digestion pattern obtained upon TfoI digestion of different phage DNA (Lambda, λ; PhiX174, φX174; M13), plasmids (pBR322 and pUC18) and a pBR322 PCR fragment containing a single –TTAA– site as DNA substrate. The labels ‘−’ and ‘+’ indicate samples with and without the added enzyme, respectively

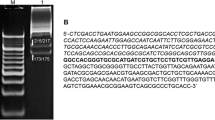
Determination of TfoI cleavage site. a DNA fragment (base positions 871 to 1915) of pBR322 plasmid amplified (using primers 871F and 1915R, ‘-’ in b) containing a single –TTAA– sequence at position 1720 (in bold and underlined) was used as a substrate to determine TfoI cleavage site. The enzyme digest (cut) yielded two fragments (b) which were subjected to DNA sequencing using primers 1500F and 1915R. The predicted sequence above the chromatogram refers to the sequence with –T^TAA– as the TfoI cleavage pattern. Template-independent addition of adenine base by AmpliTaq® used in the sequencing reaction is marked as ‘a’ in the predicted sequence
MseI and NdeI enzymes are known to produce compatible restriction overhangs. Therefore, to resolve if the cleavage site of TfoI is same or different from that of MseI, the end compatibility of TfoI with NdeI was checked (Fig. 6a). A successful ligation of the TfoI-digested pET29b(+) fragment with the NdeI-digested pET28a(+) established that TfoI produces DNA ends that are compatible with those produced by NdeI. The DNA sequencing data of the transformed plasmid obtained after ligation (Fig. 6b) conclusively show that TfoI cleaves between the two thymines in its recognition sequence (5′–T^TAA–3′).

TfoI and NdeI create compatible cohesive ends. The directional cloning approach followed to test end compatibility of TfoI with NdeI enzyme is outlined in (a). The TfoI-XhoI digested fragment (158–311) of pET29b(+) was cloned into NdeI-XhoI digested pET28a(+) vector, and the plasmid isolated from the transformants were DNA sequenced. Successful ligation of the NdeI/TfoI ends, marked by a dotted box, leads to loss of both NdeI and TfoI sites at the ligation site. b Chromatogram of the DNA sequence of a transformant obtained from the cloning described in (a). The predicted sequence above the chromatogram has the insert segment underlined and marked in bold. The ligation site at the NdeI/TfoI end is boxed by a dotted line
Enzyme characterization
Excess of enzyme with prolonged incubation time resulted in complete digestion of substrate DNA indicating that TfoI functions under a broad range of reaction conditions including buffer composition and temperature (data not shown). To determine the optimal buffer and temperature, the enzyme activity was assayed for digestion of one µg of DNA substrate with one unit of the enzyme in one hour. TfoI is observed to be optimally active in the CutSmart NEbuffer and B, G and O buffers from Thermo Scientific (Fig. A6). Of the various tested divalent metal ions, Mg+ 2 and Co+ 2 showed as preferred cofactors, while no activity is observed in the presence of Ca+ 2 ions (Fig. 7). Higher concentrations of Mg+ 2 ions (75 mM and above) is inhibitory to TfoI activity (Fig. A7a). The enzyme is fully functional up to 200 mM NaCl, and shows considerable activity even at high salt concentrations (0.5 M NaCl). However, NaCl is not required for the REase activity (Fig. A7b). TfoI functions best in the temperature range of 55–70 °C (Fig. 8a, A8). In the absence of substrate DNA, TfoI is inactivated after 30 min incubation at 80 °C (Fig. 8b). Under the conditions used in our experiment, heat treatment of TfoI at 80 °C for 20 min in the presence of DNA also led to inactivation of the enzyme (Fig. A9). The banding pattern of TfoI digests of pBR322 plasmid in the absence and presence of 15% glycerol (Fig. A10a) or 10% DMSO (Fig. A10b) remained identical. The enzyme activity is also observed to be unaffected by up to 5 mM concentration of disulfide reducing agents such as DTT and β-ME (Fig. A10c).

Divalent metal ion dependence of DNA cleavage by TfoI. The plasmid pBR322 DNA digested with TfoI in presence of different divalent metal ions indicates preference of Mg+ 2 and Co+ 2 as a cofactor for endonuclease activity. REase activity assay set in the Thermo Scientific buffer B (B) serves as a positive control, and ‘−’ refers to the uncut plasmid

Effect of temperature on TfoI activity. a Temperature dependence of TfoI cleaving Lambda DNA indicates optimum temperature range as 55–70 °C for its activity. b TfoI is heat inactivated at 80 °C. The pBR322 plasmid DNA was used in the heat inactivation assay and TfoI activity at 55, 65 and 80 °C without pre-incubation served as controls. The partial digestion of the pBR322 plasmid observed at 80 °C is due to the digestion of the DNA that occurs before the enzyme is fully heat denatured. The undigested DNA is marked by the lane labeled as ‘−’
TfoI RM gene cluster identification in the T. fonticaldi PL17 genome
The whole genome shotgun project of T. fonticaldi PL17 is deposited at DDBJ/ENA/GenBank, accession number LZDH00000000. The T. fonticaldi PL17 genome annotation using NCBI PGAP, RAST and GLIMMER did not yield any gene as a Type II REase. However, two genes were marked as methylase (MTase) subunit (YeeA) of a Type II restriction enzyme and methylation subunit of a Type III restriction–modification system by RAST and GLIMMER annotation (and not by PGAP). The protein sequences for these two MTases were subjected to protein BLAST analysis. The protein corresponding to the MTase subunit annotated as Type III MTase present on contig 6 [location: 462674-461760 (-ve strand)] shows 53.9% sequence identity with the MseI MTase (Micrococcus sp. NEB 446). Analysis of the neighboring sequence of this gene yielded an ORF downstream [location: 461759-461213 (-ve strand)] which when subjected to BLAST analysis shows significant sequence identity with other –TTAA– recognizing REases of the Type II RM systems; 47.22, 62.22, and 64.64% with MseI, RspRSORF124P, and Tam77409Ip, respectively (Fig. 9). The sequence analysis data thus clearly suggest that the two-gene cluster on contig 6 [location: 462,674 − 461,213 (-ve strand)] codes for a –TTAA– recognizing Type II RM system. It should be mentioned here that ORF analysis of the 3 kb DNA sequence encompassing the annotated YeeA Type II MTase gene in all six reading frames indicates that there was no sequence that showed similarity to known REases.

Comparison of TfoI sequence with known orthologous REases. Multiple sequence alignment of the deduced amino acid sequence of putative TfoI (TfoIp) with the protein sequence of other –TTAA– cleaving REases from Micrococcus sp. NEB 446 (MseI), Roseiflexus species RS-1 [RspRSORF124P] (RspRS-1p) and Thermus amyloliquefaciens YIM 77,409 (Tam77409Ip) using T-Coffee (Di Tommaso et al. 2011). The conserved residues among the sequences are highlighted; with 100% conserved residues (asterisk) in white on black background and similar amino acid residues are depicted as white on grey background. The 33 residue extension at the N-terminus of Tam77409Ip protein is not included in the alignment analysis. The final letter ‘p’ in the enzyme name indicates that the protein sequence is of a putative enzyme
Discussion
TfoI from T. fonticaldi PL17 is a first of the restriction enzymes from the Tepidimonas genus. It is 4-base cutter, cleaving within the palindromic sequence of 5′–TTAA–3′ between the two thymine bases to generate a 2-base 5′-overhang (5′–T^TAA–3′) and requires a divalent metal ion as the only cofactor for activity. Therefore, TfoI can be classified as a Type IIP REase. Although the PGAP, RAST, and GLIMMER gene annotation algorithms did not mark the presence of a complete RM system in T. fonticaldi PL17 genome, protein sequence analysis of the MTase subunits of RM system allowed us to fish out the TfoI RM gene cluster. The gene located at position 461759-461213 of contig 6 in the negative strand of the genome codes for putative-TfoI REase encoding a protein of 181 amino acid residues. Four-step purification of the native enzyme yielded functionally pure protein free of any potential nucleases as indicated by the quality control assays known for restriction enzymes. The pure monomeric protein on an SDS–PAGE revealed a band between 15 and 25 kDa bands of the molecular weight ladder. Mass spectrometry of the sample indicated a mass of 20.696 kDa (Fig. A11), which is in agreement with the calculated molecular weight of 20.694 kDa for the putative monomeric protein from the gene sequence.
T. fonticaldi PL17 is a slightly thermophilic bacterium with 55 °C as optimum growth temperature which is in agreement with the temperature of the source hot spring from where it is isolated. Nevertheless, the organism grows in a temperature range of 37–60 °C under the laboratory conditions. Interestingly, the enzyme TfoI from T. fonticaldi PL17 is also found to be functional in a broad range of reaction conditions, including temperature between 37–70 °C and salt concentration. Such a phenotype may be considered beneficial, since the temperature of the Polok hot springs are reported to show seasonal variations, below 60–75 °C (Das et al. 2012). TfoI, like most REases uses Mg2+ as an obligatory cofactor for activity. In addition, Co2+ can equally well substitute for Mg2+ as a cofactor, while the enzyme activity is weak in the presence of Mn+ 2. Even though TfoI cleaves DNA at 70 °C, it is inactivated at 80 °C. Incubation of TfoI at 80 °C leads to irreversible heat denaturation of the enzyme and no residual activity is observed when a DNA substrate is digested with the 80 °C-preheated TfoI (Fig. A9). Unaltered activity of TfoI in high concentrations of glycerol and DMSO indicate that there is no detectable star activity of TfoI (Wei et al. 2008). Disulfide reducing agents did not have any effect on TfoI activity suggesting that the protein is unlikely to contain disulfide bond(s) of any structural consequence.
REase is a component of the restriction–modification system in an organism. The production of TfoI by T. fonticaldi PL17 hints at the presence of a methyltransferase in this bacterium, which is also supported by the presence of a gene similar to MseI MTase in its genome. The T. fonticaldi PL17 genomic DNA (gDNA) subjected to digestion with purified TfoI and Tru1I (an isoschizomer) clearly indicates gDNA protection from cleavage (Fig. 10), revealing the presence of a functional TfoI restriction–modification unit. T. fonticaldi PL17 has a 69.5% GC-rich genome, and is likely to contain fewer AT-rich sites. Therefore, genomic DNA of two other organisms, P. aeruginosa (high GC content, 66% (Labaer et al. 2004)) and E. coli BL21(DE3) (GC content, 50.8% (Jeong et al. 2009)), were also subjected to TfoI and Tru1I. A 4-base cutter, HaeIII, cleaving at –GG^CC– sites was used as yet another control to test the GC-rich genomic DNA sensitivity. As evident from Fig. 10, while BL21(DE3) gDNA shows a smear with both types of cutters, a similarly GC-rich genome of P. aeruginosa shows smearing with TfoI and Tru1I. As expected, no intact gDNA is observed for both P. aeruginosa and T. fonticaldi PL17 digestion with HaeIII. These data show that high GC content alone is not sufficient to protect gDNA from (A + T)-rich recognition sequence cutting REases. Thus, T. fonticaldi PL17 harbors a complete functional Type II restriction–modification system for TfoI, and this is a first report of an RM system in the Tepidimonas genus.

Determination of T. fonticaldi P17 genomic DNA sensitivity to TfoI. Both the –TTAA– cleaving enzymes (Tru1I and TfoI) do not digest the T. fonticaldi P17 genomic DNA. Restriction digestion of P. aeruginosa MTCC1934, and E. coli BL21(DE3) genomic DNA were set as controls for TfoI and Tru1I activity, while HaeIII, a GC-cutter, served as an REase control with a different specificity. The lane labeled as ‘−’ marks undigested genomic DNA
The widely used –TTAA– cutter MseI produced by Micrococcus species (Morgan 1988) has other known commercially available isoschizomers, such as RspRSII (Park et al. 2014), SaqAI from Salinibacterium aquaticus RFL1, Tru9I from Meiothermus ruber 9, and Tru1I from Meiothermus ruber RFL1 (the genus name Meiothermus mentioned here is as per the re-classification of Thermus ruber (Nobre et al. 1996)). While MseI and SaqAI are mesophilic enzymes, the other three isoschizomers are thermophilic enzymes with optimal activity at 60 °C for RspRS II or 65 °C for M. ruber enzymes. TfoI from T. fontacaldi PL17 is a new isoschizomer of MseI, and differs from its thermophilic counterparts in terms of exhibiting activity over a broad temperature range.
It is noteworthy that of the seven reported –TTAA– cutters in the REBASE along with TfoI (this study), a site-specific endonuclease with identical recognition sequence is produced by Gram-positive mesophiles (MseI and SaqAI), Gram-negative moderate thermophiles (RspRSII, Tru9I, Tru1I, and TfoI) and a phage (SphSshM2 from Synechococcus phage S-ShM2). Such an observation may indicate that this REase is acquired by horizontal gene transfer. This idea also gains support from the fact T. fonticaldi PL17, a 69.5% GC rich organism, carries the TfoI RM-encoding gene cluster with a GC content of 42.6% which is surrounded by ORFs with greater than 65% GC content.
TfoI being a 4-base cutter or a frequent cutter is not a suitable REase for routine gene cloning. However, TfoI may be used for limited random DNA fragmentation and coupled with its property to generate cohesive ends suitable for ligation at a compatible site (for, e.g., NdeI), it can be used in preparing genomic DNA libraries. In addition, similar to the widely accepted application of MseI and its isoschizomers in AFLP (Amplified Fragment Length Polymorphisms) analysis (Vos et al. 1995; Vuylsteke et al. 2007) for the identification of genetic variation among closely related species (Zhao et al. 2006), karyotyping of chromosomes (Ludena et al. 1991) or in the study of genomic repetitive sequences, such as teleomere sequences (Vaquero-Sedas and Vega-Palas 2012), TfoI is an alternative isoschizomer of MseI in such analyses.
References
Agarkova IV, Dunigan DD, Van Etten JL (2006) Virion-associated restriction endonucleases of chloroviruses. J Virol 80:8114–8123. doi:10.1128/JVI.00486-06
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genom 9:75. doi:10.1186/1471-2164-9-75
Bickle TA, Kruger DH (1993) Biology of DNA restriction. Microbiol Rev 57:434–450
Chaturvedi D, Chakravorty M (2003) Restriction–modification system in bacteriophage MB78. Biochem Biophys Res Commun 303:884–890
Das S, Sherpa MT, Sachdeva S, Thakur N (2012) Hot springs of Sikkim (Tatopani): a socio medical conjuncture which amalgamates religion, faith, traditional belief and tourism. Asian Acad Res J Social Sci Hum 1:80–93
Delcher AL, Harmon D, Kasif S, White O, Salzberg SL (1999) Improved microbial gene identification with GLIMMER. Nucl Acids Res 27:4636–4641
Dempsey RM, Carroll D, Kong H, Higgins L, Keane CT, Coleman DC (2005) Sau42I, a BcgI-like restriction–modification system encoded by the Staphylococcus aureus quadruple-converting phage Phi42. Microbiol 151:1301–1311. doi:10.1099/mic.0.27646-0
Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucl Acids Res 39:W13–W17. doi:10.1093/nar/gkr245
Hadi SM, Bachi B, Iida S, Bickle TA (1983) DNA restriction–modification enzymes of phage P1 and plasmid p15B. Subunit functions and structural homologies. J Mol Biol 165:19–34
Hu G (1993) DNA polymerase-catalyzed addition of nontemplated extra nucleotides to the 3′ end of a DNA fragment. DNA Cell Biol 12:763–770. doi:10.1089/dna.1993.12.763
Jeong H, Barbe V, Lee CH, Vallenet D, Yu DS, Choi S-H, Couloux A, Lee S-W, Yoon SH, Cattolico L (2009) Genome sequences of Escherichia coli B strains REL606 and BL21 (DE3). J Mol Biol 394:644–652
Joshi A, Siddiqi JZ, Rao GR, Chakravorty M (1982) MB78, a virulent bacteriophage of Salmonella typhimurium. J Virol 41:1038–1043
Kim O-S, Cho Y-J, Lee K, Yoon S-H, Kim M, Na H, Park S-C, Jeon YS, Lee J-H, Yi H (2012) Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62:716–721. doi:10.1099/ijs.0.038075-0
Labaer J, Qiu Q, Anumanthan A, Mar W, Zuo D, Murthy TV, Taycher H, Halleck A, Hainsworth E, Lory S, Brizuela L (2004) The Pseudomonas aeruginosa PA01 gene collection. Genome Res 14:2190–2200. doi:10.1101/gr.2482804
Loenen WA, Dryden DT, Raleigh EA, Wilson GG, Murray NE (2014) Highlights of the DNA cutters: a short history of the restriction enzymes. Nucl Acids Res 42:3–19. doi:10.1093/nar/gkt990
Ludena P, Sentis C, De Cabo SF, Velazquez M, Fernandez-Piqueras J (1991) Visualization of R-bands in human metaphase chromosomes by the restriction endonuclease MseI. Cytogenet Cell Genet 57:82–86. DOI:10.1159/000133119 doi
Morgan RD (1988) Mse I, a unique restriction endonuclease from Micrococcus species which recognizes 5′ T/TAA 3′. Nucl Acids Res 16:3104. doi:10.1093/nar/16.7.3104
Nobre MF, Truper HG, Costa. MSD (1996) Transfer of Thermus ruber (Loginova et al. 1984), Themus silvanus (Tenreiro et al. 1999, and Themus chliarophilus (Tenreiro et al. 1995) to Meiothermus gen. nov. as Meiothermus ruber comb. nov., Meiothermus silvanus comb. nov., and Meiothermus chliarophilus comb. nov., respectively, and emendation of the genus Thermus. Int J Syst Bacteriol 46:604–606
Oliveira PH, Touchon M, Rocha EP (2014) The interplay of restriction–modification systems with mobile genetic elements and their prokaryotic hosts. Nucl Acids Res 42:10618–10631. doi:10.1093/nar/gku734
Park YJ, Nishikawa T, Matsushima K, Minami M, Nemoto K (2014) A rapid and reliable PCR-restriction fragment length polymorphism (RFLP) marker for the identification of Amaranthus cruentus species. Breed Sci 64:422–426. doi:10.1270/jsbbs.64.422
Pingoud A, Wilson GG, Wende W (2014) Type II restriction endonucleases—a historical perspective and more. Nucl Acids Res 42:7489–7527. doi:10.1093/nar/gku447
Raleigh EA, Brooks JE (1998) Restriction modification systems: where they are and what they do. In: de Brujin FJ, Lupski JR, Weinstock GM (eds) Bacterial genomes. Chapman and Hall, New York, pp 78–92. doi:10.1007/978-1-4615-6369-3_8
Rao DN, Dryden DT, Bheemanaik S (2014) Type III restriction–modification enzymes: a historical perspective. Nucl Acids Res 42:45–55. doi:10.1093/nar/gkt616
Reasoner DJ, Geldreich EE (1985) A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol 49:1–7
Roberts RJ (2005) How restriction enzymes became the workhorses of molecular biology. Proc Natl Acad Sci USA 102:5905–5908. doi:10.1073/pnas.0500923102
Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev S, Dryden DT, Dybvig K, Firman K, Gromova ES, Gumport RI, Halford SE, Hattman S, Heitman J, Hornby DP, Janulaitis A, Jeltsch A, Josephsen J, Kiss A, Klaenhammer TR, Kobayashi I, Kong H, Kruger DH, Lacks S, Marinus MG, Miyahara M, Morgan RD, Murray NE, Nagaraja V, Piekarowicz A, Pingoud A, Raleigh E, Rao DN, Reich N, Repin VE, Selker EU, Shaw PC, Stein DC, Stoddard BL, Szybalski W, Trautner TA, Van Etten JL, Vitor JM, Wilson GG, Xu SY (2003) A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucl Acids Res 31:1805–1812. doi:10.1093/nar/gkg274
Roberts RJ, Vincze T, Posfai J, Macelis D (2015) REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucl Acids Res 43:D298–D299. doi:10.1093/nar/gku1046
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Samuelson JC, Zhu Z, Xu SY (2004) The isolation of strand-specific nicking endonucleases from a randomized SapI expression library. Nucl Acids Res 32:3661–3671. doi:10.1093/nar/gkh674
Sharma P, Kumar R, Capalash N (2013) Restriction enzymes from thermophiles. In: Satyanarayana T, Littlechild J, Kawarabayasi Y (eds) Thermophilic microbes in environmental and industrial biotechnology: biotechnology of thermophiles. Springer, pp 611–647. doi: 10.1007/978-94-007-5899-5_23
Szekeres M, Szmidt AE, Torok I (1983) Evidence for a restriction/modification-like system in Anacystis nidulans infected by cyanophage AS-1. Eur J Biochem 131:137–141
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J (2016) NCBI prokaryotic genome annotation pipeline. Nucl Acids Res 44:6614–6624. doi:10.1093/nar/gkw569
Thakur N, Das S, Sherpa MN, Ranjan R (2013) GPS mapping and physical description of Polok, Borong and Reshi Tatopani—hot springs of Sikkim. JIARM 10:367–647
Vaquero-Sedas MI, Vega-Palas MA (2012) The restriction endonuclease Tru9I is a useful tool to analyze telomere sequences separately from interstitial telomeric sequences in Arabidopsis thaliana. AJMB 2:242–244. doi:10.4236/ajmb.2012.23025
Vasu K, Nagaraja V (2013) Diverse functions of restriction–modification systems in addition to cellular defense. Microbiol Mol Biol Revs 77:53–72. doi:10.1128/MMBR.00044-12
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M et al (1995) AFLP: a new technique for DNA fingerprinting. Nucl Acids Res 23:4407–4414. doi:10.1093/nar/23.21.4407
Vuylsteke M, Peleman JD, van Eijk MJ (2007) AFLP technology for DNA fingerprinting. Nat Protoc 2:1387–1398. doi:10.1038/nprot.2007.175
Wang Y, Qian PY (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PloS One 4:e7401. doi:10.1371/journal.pone.0007401
Wei H, Therrien C, Blanchard A, Guan S, Zhu Z (2008) The fidelity index provides a systematic quantitation of star activity of DNA restriction endonucleases. Nucleic Acids Res 36:e50. doi:10.1093/nar/gkn182
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Xia YN, Van Etten JL (1986) DNA methyltransferase induced by PBCV-1 virus infection of a Chlorella-like green alga. Mol Cell Biol 6:1440–1445
Xia YN, Burbank DE, Uher L, Rabussay D, Van Etten JL (1986) Restriction endonuclease activity induced by PBCV-1 virus infection of a Chlorella-like green alga. Mol Cell Biol 6:1430–1439
Zhao H, Bughrara SS, Oliveira JA (2006) Genetic diversity in colonial bentgrass (Agrostis capillaris L.) revealed by EcoRI-MseI and PstI-MseI AFLP markers. Genome 49:328–335. doi:10.1139/g05-113
Acknowledgements
This work was supported by the financial assistance from CSIR-IMTECH (BSC0402, BSC0120 to AKP; OLP0091 to BK) and Department of Biotechnology-India (BT/PR7368/INF/22/177/2012 to AKP and GAP0096 to BK) funding to BK. The authors are thankful to Dr. Rakshak Kumar for sample collection, and acknowledge the support from the Sikkim State Council of Science and Technology and the Department of Forest, Govt. of Sikkim, in sample collection. RK is supported by the research fellowship scheme of University Grants Commission, India. BK is a recipient of the Department of Biotechnology-Ramalingaswami re-entry fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Huang.
The GenBank accession number for the 16S rRNA gene sequence and the whole genome shotgun sequencing data of the strain Tepidimonas fonticaldi PL17 is KF206381 and LZDH00000000, respectively.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, R., Pinnaka, A.K. & Krishnan, B. TfoI produced by Tepidimonas fonticaldi PL17, a moderate thermophilic bacterium, is an isoschizomer of MseI. Extremophiles 21, 523–535 (2017). https://doi.org/10.1007/s00792-017-0922-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-017-0922-6