
Abstract
The family B DNA polymerase gene from the archaeon Thermococcus marinus (Tma) contains a long open reading frame of 3,939 bp that encodes 1,312 amino acid residues. The gene is split by one intervening sequence that forms a continuous open reading frame with the two polymerase exteins. In this study, the Tma DNA polymerase gene both with (precursor form) and without (mature form) its intein was expressed in Escherichia coli, purified by heat treatment and HiTrap™ Heparin HP column chromatography and characterized. Primary sequence analysis of the mature Tma polymerase showed high sequence identity with DNA polymerases in the genus Thermococcus. The expressed precursor form was easily spliced during purification steps. The molecular mass of the purified Tma DNA polymerases is about 90 kDa, as estimated by SDS-PAGE. Both Tma DNA polymerases showed the same properties. PCR performed with this enzyme was found to be optimal in the presence of 50 mM Tris–HCl (pH 8.4), 40 mM KCl, 12.5 mM (NH4)2SO4, 2 mM MgCl2, 0.05% Triton X-100 and 0.0075% BSA. Furthermore, long-range PCR and time-saving PCR were performed using various specific ratios of Taq and Tma DNA polymerases (Tma plus DNA polymerase).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
DNA polymerases are best known for their roles in DNA repair, recombination (Kornberg and Baker 1992) and replication, in which they catalyze a new strand of DNA in the 5′ → 3′ direction using an antiparallel DNA strand as a template. The ability of DNA polymerases to copy DNA templates has been exploited in a variety of in vitro reactions for sequencing, amplification, mutation, labeling and recombination of DNA, as well as in several other applications that are fundamental to molecular biology (Reha-Krant 2008). On the discovery of a third domain of life called Archaea (Woese and Fox 1977; Woese et al. 1990), a new field of investigation regarding DNA polymerases opened up. Since then, numerous hyperthermophilic archaea have been shown to contain DNA polymerases viable for commercial use and new applications. The genera Thermococcus and Pyrococcus are strictly hyperthermophile species and contain both native and recombinant enzymes that are among the most hyperthermostable ever known (Takagi et al. 1997; Griffiths et al. 2007; Kong et al. 1993; Mattila et al. 1991; Lundberg et al. 1991; Southworth et al. 1996; Baross and Holden 1996; Perler et al. 1996; Cambon-Bonavita et al. 2000; Marsic et al. 2008; Bult et al. 1996; Saiki et al. 1988). Family B DNA polymerases from the two hyperthermophilic archaea have been studied mainly for biotechnological as well as, to a lesser extent, phylogenetic applications, especially since the discovery of inteins within them (Takagi et al. 1997; Hodges et al. 1992). Inteins, polypeptide products resulting from the transcription and translation of intervening sequences in-frame with their host genes, are removed from the polypeptide precursor by protein splicing (Xu and Perler 1996) and interestingly, most inteins of family B DNA polymerases have both self-splicing and homing endonuclease domains. Recently, split mini-inteins also have been found in Nanoarchaeum equitans family B DNA polymerase along with its trans-splicing ability (Choi et al. 2006). The hyperthermophilic euryarchaeon Thermococcus marinus, has also been isolated from the chimney of deep-sea hydrothermal vent (mid-Atlantic ridge) (Jolivet et al. 2004). In this study, we present novel and detailed data on the cloning and expression of a DNA polymerase gene from hyperthermophilic Thermococcus marinus. A sequence comparison of the encoded DNA polymerase to other archaeal proteins indicates that the enzyme belongs to the family B DNA polymerases. The purified recombinant enzyme was biochemically characterized and its application to PCR was successfully demonstrated.
Materials and methods
Strains and culture conditions
T. marinus (DSM 15227) was obtained from the German Collection of Microorganisms and Cell Cultures (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, DSMZ). The T. marinus cells were grown anaerobically in DSMZ medium 990 as recommended in the DMSZ protocol. Medium containing (per liter) 4.0 g of peptone, 2.0 g of yeast extract, 35.0 g of sea salts (Sigma, USA.), 3.46 g of PIPES, 5 g of elemental sulfur, 0.5 g of NH4Cl, 0.35 g of KH2PO4, 0.2 g of CaCl2, 6.7 mg of FeCl3, 2.9 mg of Na2WO4 and 0.1 mg of resazurin was prepared with pH adjusted to 6.8 at 25°C, and incubated at 100°C for 8 h. The heated medium was filtered through normal filter paper to remove sediments and dispensed into a 120 ml serum bottles (Wheaton Co., USA) containing finely divided sulfur (0.5% w/v) and 100% N2 gas. After sealing the serum bottle, 0.3 ml of 5% Na2S·9H2O was added to the medium to eliminate any traces of oxygen, followed by sterilization at 100°C. The cells were then inoculated with a syringe and incubated anaerobically at 80°C for 24 h.
The E. coli strain DH5α was used for plasmids propagation for DNA sequencing while E. coli strain Rosetta(DE3)pLysS (Stratagene, USA) containing plasmid pET-22b(+) (Novagen, USA) was used for gene expression. E. coli strains were cultivated in Luria-Bertani medium with appropriate antibiotics at 37°C with vigorous shaking.
DNA manipulation and sequence analysis
DNA manipulations and isolation of genomic DNA of T. marinus were both performed using standard procedures, as described by Sambrook and Russell (2001) and Robb (1995), respectively. Restriction enzymes and other modifying enzymes were purchased from Takara Bio Inc., (Japan) and RexGene Biotech Co., Ltd. (Korea). Small-scale preparation of plasmid DNA from E. coli cells was performed using a plasmid mini-prep kit (Qiagen, Germany). The nucleotide sequences of the purified PCR product and subcloned DNAs were determined by sequencing at Macrogen (Korea) and compared with known proteins in the database using the BLAST sequence comparison program. Nucleotide and deduced amino acid sequence analysis was performed using the SeqMan and EditSeq (DNAStar, Inc.) software. The MultiAlign program (Corpet 1988) was used for multiple sequence alignment between functionally related proteins.
Isolation of DNA polymerase gene
A fragment containing the Tma DNA polymerase gene was amplified from T. marinus genomic DNA by using the degenerate primer pair (forward mixed primer, 5′-NANTACGACATACCCTTNGC-3′ and the reverse mixed primer, 5′-AACCTGGTTCTCNATNTAGTA-3′) predicted from the conserved amino acid sequences EYDIPFA and YYIENQV within the family B DNA polymerases Vent DNA polymerase (GenBank accession no. P30317), Tzi DNA polymerase (GenBank accession no. ABD14868) and Pfu DNA polymerase (GenBank accession no. D12983). The PCR reactions were performed in a total volume of 50 μl containing the following PCR mixture: PyroAce® DNA polymerase buffer, 0.25 mM dNTP, 20 pmol of each primer, 1 μl of Tma genomic DNA and 5 U of PyroAce® DNA polymerase (RexGene Biotech Co., Ltd., Korea). The PCR reaction conditions were as follows: 94°C, 3 min (one initial denaturation step); 94°C, 1 min; 47°C, 1 min; 72°C, 3 min for 30 cycles, and one additional cycle at 72°C for 10 min using Palm-Cycler (Corbett Life Science, Australia). The amplified products were approximately 3.3 kb long and sequencing of the fragment showed high homology to the sequences of other archaeal family B DNA polymerases in the GenBank databases and corresponded to the expected partial Tma DNA polymerase gene.
A DNA Walking SpeedUp ™ Premix Kit (Seegene, Korea) was employed in the amplification of the 5′- and 3′- unknown flanking regions from the conserved sequences of the partial Tma DNA polymerase gene. Two specific antisense primers (Tma N-1, 5′-GAAGGTCAATCTTCTTCCAGGT-3′ and Tma N-2, 5′-AATCATCTCCTTCTCGGTCGAA-3′) and two sense primers (Tma C-1, 5′-GTAATCCACGAGCAGATAACGC-3′ and Tma C-2, 5′-GAGAAGCTCGTAATCCACGAG-3′) synthesized on the basis of the known sequences between the conserved regions were used to capture unknown target sites along with DNA Walking DW-ACP™ and DW-ACPN™ primers. In following the manufacturer’s recommended protocols, the 5′ end region of the Tma DNA polymerase gene was amplified initially with the Tma N-2 and DW-ACP™ primers, under the following PCR conditions: one initial denaturation step (94°C, 3 min) and 30 cycles of amplification (94°C, 1 min; 57°C, 1 min; 72°C, 3 min). This PCR product was used as the template in the second round of PCR, which utilized Tma N-1 and DW-ACPN™ as primers. The second PCR-amplified product, which measured approximately 1.2 kb, was sequenced and then confirmed to contain the start codon. The 3′- downstream region of the Tma DNA polymerase gene was amplified using the sense Tma C-1 and DW-ACPTM primers under the same PCR conditions as listed above. The amplified product was used as the template in the second round of PCR, which utilized Tma C-2 and DW-ACPN™ as primers. The product was approximately 1.2 kb long and was confirmed to contain the stop codon by sequencing.
Sequence analysis
Sequence alignment of the euryarchea family B DNA polymerases was performed with Basic BLAST program using National Center for Biotechnology Information (NCBI) databases and Vector NTI program (Invitogen, USA). For molecular phylogenetic analysis, the tentative phylogenetic tree was constructed with the neighbor-joining method using MEGA4.0 (Tamura et al. 2007). The obtained tree topologies were subjected to maximum likelihood analysis using PhyML program (Guindon et al. 2005) with the WAG amino acid substitution model. The robustness of each clustering of branches was assessed by the bootstrap method with 1000 replicates.
Subcloning for expression and purification
Two primers based on the nucleotide sequence of the Tma DNA polymerase gene were synthesized to allow the whole Tma DNA polymerase gene containing its intein to be expressed. The 5′ (N-terminal) primer, TmaN, was of the sequence 5′-CTGTAGTCATATGATTCTCGATACCGACTGCATC-3′ and contained a unique NdeI site (underlined) that includes the ATG translation initiation codon. The 3′ (C-terminal) primer, TmaC, was 5′-NNNNNAAGCTTCACTTCTTTCCCTTCGGC-3′, which matches the C-terminal sequence and has a unique HindIII site (underlined) added. The PCR mixture was the same as described above except for the primers. The PCR reaction consisted of 30 cycles of 94°C for 1 min, 58°C for 1 min and 72°C for 3 min, preceded by 3 min at 94°C and followed by 10 min at 72°C. The amplified fragment containing the whole Tma DNA polymerase gene was digested with NdeI and HindIII, purified from 0.8% low melting agarose gel and ligated into the expression vector pET-22b(+) (Novagen, USA), which had been digested with the same enzymes. The resultant expression plasmid was named pTMAP.
To express the genetically mature form of Tma DNA polymerase without its intein from the enzyme gene, an expression plasmid was constructed using overlapping PCR (Ho et al. 1989). The regions encoding the N-terminal extein portion (sense primer, TmaN and antisense primer, TmaEx1 [5′-CGTAGTAGCCGTAGTAACTGTTCGCCAGGATTTTGATAG-3′]) and the C-terminal extein portion (sense primer, TmaEx2 [5′-CTATCAAAATCCTGGCGAACAGTTACTACGGCTACTACGG-3′] and antisense primer, TmaC) of Tma DNA polymerase were amplified separately using primers containing the overlapping sequences. Each of the amplified products purified from low melting point agarose were then used as templates for a consecutive PCR reaction using the primers TmaN and TmaC. The 2.4 kb amplified product was digested with NdeI and HindIII and then ligated into the expression vector pET-22b(+), which had been digested with the same enzymes. The resulting expression plasmid was named pTMAM.
Expression plasmids were transformed into E. coli Rosetta(DE3)pLysS cells and overexpression of the Tma polymerase gene was induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) at the mid-exponential growth phase (O.D600 0.6), followed by an 8 h incubation at 37°C. The cells were harvested by centrifugation (5,590×g at 4°C for 20 min) and resuspended in buffer A (50 mM Tris–HCl buffer (pH 8.0) containing 0.1 M KCl and 15% glycerol). The cells were disrupted by sonication and a crude enzyme sample was treated at 80°C for 30 min after centrifugation (18,700×g at 4°C for 15 min). The resulting supernatant was applied to a HiTrap™ Heparin HP column (GE Healthcare) equilibrated with buffer A (Kim et al. 2007). The column was washed and bound proteins were eluted with a KCl gradient (0–1000 mM) in buffer A. The peak fractions containing the Tma DNA polymerase were dialyzed against storage buffer (20 mM Tris–HCl (pH 7.4), 0.1 mM EDTA, 0.1% Tween 20, 0.5% Nonidet P40, 0.1 M KCl and 50% glycerol), and stored at −20°C. Protein concentration was determined by the method by Lowry et al. (1951) with bovine serum albumin (BSA) as a standard. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed with 10% polyacrylamide gel, as previously described by Laemmli (1970).
DNA polymerase activity assay
The DNA polymerase activity of the purified enzyme was measured as described by Choi et al. (2006). The basic reaction mixture (50 μl) contained 50 mM Tris–HCl (pH 7.5), 14 mM MgCl2, 80 mM KCl, 1 mM 2-mercaptoethanol, 100 μM each of dATP, dCTP and dGTP, 10 μM dTTP, 0.5 μCi of [methyl-3H]thymidine 5′-triphosphate (30 Ci/mmol, GE Healthcare, code No. TRK354), 1.25 μg of activated calf thymus DNA and enzyme solution. One unit of Tma DNA polymerase is defined as the amount of polymerase that incorporates 10 pmol of [3H]TTP into an acid-insoluble product at 75°C in 10 min.
Exonuclease activity assay
Measurement of exonuclease activity was performed as previously described (Choi et al. 2006; Song et al. 2007). Briefly, preparation of the 3′-end-labeled DNA substrate required end filling of pBluescript SK-DNA linearized with NotI using Klenow fragments in the presence of [α-32P]dCTP (3000 Ci/mmol, GE Healthcare, code No. AA0005). Labeling was followed by purification of the DNA substrate by gel filtration and ethanol precipitation. The exonuclease activity of the purified Tma DNA polymerase was analyzed in the basic reaction mixture (50 μl) containing 50 mM Tris–HCl (pH 7.5), 14 mM MgCl2, 80 mM KCl, 0.01% BSA and end-labeled DNA substrate at 75°C for 10 min in the presence and absence of dNTPs. The reaction was stopped by adding 1 ml of 5% (w/v) trichloroacetic acid in the presence of BSA as a carrier. After centrifugation, the supernatant was withdrawn and its radioactivity was counted.
Optimization of PCR amplification
Oligonucleotide primers that anneal to λ DNA (Sanger et al. 1982) were synthesized for the PCR assays. The sequences of the primers are presented in Table 1. PCR buffer optimization experiments were performed with 0.25 U of Tma DNA polymerase in a 50 μl reaction mixture containing 0.2 pmol each of the primers anchor-λ F and λ-2R, 250 μM dNTPs and 23 ng of λ DNA as a template. The reaction buffer conditions are indicated in the corresponding figure legends. PCR was conducted as follows: 2 min at 94°C; 25 cycles of 20 s at 94°C, and 60 s at 72°C.
Long-range PCR and time-saving PCR
A slightly modified method (Choi et al. 2008) was employed to mix ratios ranging from 10:0 to 0:10 of Taq and Tma DNA polymerases based on enzyme units (total 0.25 U/50 μl). Fragments of 10 kb were amplified using anchor-λ F and λ-10R primers (Table 1) along with Taq buffer (PCR buffer; TaKaRa Bio Inc.). PCR amplification was carried out in 30 cycles of 94°C for 20 s and 72°C for 10 min. For confirmation of DNA polymerases extension efficiency, a set of primers (Table 1) was used to amplify DNA fragments 2, 5, 8, 10, 12 and 15 kb in size from the λ DNA. The DNA fragments were amplified using Tma, Tma plus and other commercialized DNA polymerases, including Taq (TaKaRa Bio Inc.) and Pfu (Promega) DNA polymerases. Long-range PCR was performed at 94°C for 1 min; 30 cycles of 94°C for 20 s and 72°C for target length/2 min. Time-saving PCR (2 kb in target size) using a single set of anchor-λ F and λ-2R primers consisted of an initial denaturation of 1 min at 94°C, 30 cycles of 20 s at 94°C, and 5, 10, 20, 40, 60 and 80 s at 72°C. The PCR mixture contained template DNAs (which consists of 23 ng of λ DNA), 10 pmol of each primer, 0.25 U of each DNA polymerase, 1× optimized buffer or the buffer supplied by the manufacturer and 0.25 mM of dNTPs in 50 μl. PCR products were electrophoresed on 0.8% standard agarose gel.
PCR fidelity assay
The pJR-lacZ expression vector, which contains the entire lacZ gene, was used as a template to examine the mutation frequency and evaluate the overall fidelity in PCR (Song et al. 2007; Choi et al. 2008). An 832 bp fragment containing the 5′ region of the lacZ gene was amplified using the following primers: Lac-B, 5′-NNNNGGATCCAATGATAGATCCCGTCGTTTTAC-3′ and Lac-C, 5′-NNNNATCGATAATTTCACCGCCGAAAGGCGC-3′ (BamHI and ClaI sites, respectively, are underlined). PCR was performed using Tma, Tma plus, Taq and Pfu DNA polymerases in the presence of manufacturer’s buffers. All other parameters remained constant, including the dNTP content, primers, template concentration, PCR cycling parameters and the number of PCR cycles performed. Thirty cycles of PCR were carried out as follows: 94°C for 30 s, 60°C for 30 s and 72°C for 1 min. PCR products were treated with restriction enzymes BamHI and ClaI, ligated into appropriate restriction sites within pJR-lacZ and then transformed into E. coli XL1-Blue. Colorless and light blue colonies were counted as mutated plasmid. The mutation frequency (mf) was calculated as the ratio of number of colonies with mutated plasmids to the total number of colonies. Error rates (ER) were calculated using the equation ER = mf/(bp × d), where mf is the mutation frequency, bp is the number of detectable sites in the region of lacZ (832 bp) and d is the number of template doublings (Lundberg et al. 1991; Cline et al. 1996).
Results and discussion
Isolation of the Tma DNA polymerase gene
To isolate the Tma DNA polymerase gene, the conserved amino acid sequence found in archaeal family B DNA polymerases was used to synthesize two degenerate primers. These degenerate primers were used in a PCR reaction to amplify a DNA fragment of about 3.3 kb in length from T. marinus genomic DNA, which was subsequently used as a basic sequence to obtain the whole DNA polymerase gene from T. marinus via primer walking PCR. The nucleotide sequence of the entire open reading frame for the DNA polymerase was determined and consisted of 3,939 bases coding for a protein with 1,312 amino acid residues (Fig. 1a). The molecular mass of the protein derived from this amino acid sequence was 152.38 kDa, which is much larger than expected for a family B DNA polymerase. Alignments with homologous DNA polymerase gene sequences revealed the presence of an intein gene within the coding sequence, encoding a 537 intervening amino acid region and splitting the DNA polymerase sequence into regions consisting of 491 amino acid N-terminal and a 284 amino acid C-terminal ends (Fig. 1a). The DNA polymerase gene without intein-coding region is 2,328 nucleotides long and encodes 775 amino acid residues (Fig. 1b, c). This amino acid sequence gave a protein with a molecular mass of 90.000 kDa, consistent with the average molecular mass of archaeal family B DNA polymerases. The majority of known archaeal family B DNA polymerase inteins contain two conserved sequence motifs characteristic of the LAGLIDADG endonuclease family, dodecapeptide (DOD) motifs, in their central region (Pietrokovski 1998). These intein sequences are inserted in the family B DNA polymerase genes at three conserved sites, pol-a, pol-b and pol-c, in motifs II, III and I of the DNA polymerases, respectively (Dalgaard et al. 1997). The intein from the Tma DNA polymerase gene was inserted into motif III of the DNA polymerase (pol-b). The nucleotide sequence of the whole Tma DNA polymerase gene containing the intein was deposited in the GenBank with the accession number FJ556902.

Amino acid sequence and phylogenetic relationship of Tma DNA polymerase. a Precursor map of Tma DNA polymerase included two exteins and one intein. The number of nucleotides and amino acids are indicated on the upper side and lower side, respectively. b Amino acid sequence of mature Tma DNA polymerase consisted of only two exteins with the removed intein region. The homing site (lacking the intein) is indicated as V. c Amino acid sequence of Tma intein. d Phylogenetic relationship of Tma DNA polymerase with other euryarchea family B DNA polymerases. The unrooted tree was constructed by MEGA4.0 and PhyML program. GenBank accession numbers and PDB accession numbers are indicated in parentheses. The scale bar represents 1 inferred substitution per 50 residues. Bootstrap support was assessed by 1000 repetition resampling of the data set and construction of the phylogeny
Similarity analysis of the Tma DNA polymerase amino acid sequence
The deduced amino acid sequence of the mature Tma DNA polymerase, which lacks the intein region, was aligned and compared with high similar sequences from archaeal family B DNA polymerases (data not shown). Amino acid sequence alignment revealed that Tma DNA polymerase contains all of the motifs, which are highly conserved among archaeal family B DNA polymerases, including the three 3′ → 5′ exonuclease motifs (Blanco et al. 1991), the six 5′ → 3′ polymerase motifs (Braithwaite and Ito 1993) and the DNA-binding motif Y-G(G/A) (Truniger et al. 1996). The presence of highly conserved motifs in Tma DNA polymerase suggests that this enzyme possesses 3′ → 5′ exonuclease activity and 5′ → 3′ polymerase activity.
The close phylogenetic relationship among the family B DNA polymerases, excluding the intein region, was corroborated (Fig. 1d) with MEGA4.0 program on the basis of the known amino acid sequences of the DNA polymerases in the Thermococcales order from the NCBI databases. Basic BLAST and Vector NTI analysis also indicated that the amino acid sequence of the mature form of T. marinus DNA polymerase showed a high degree of similarity to the Thermococcales order DNA polymerses: Thermococcus sp. 9°N-7 (95.4% identity), Thermococcus sp. GE8 (94.3% identity), Thermococcus kodakarensis (93% identity), Thermococcus gorgonarius (92.6% identity), Thermococcus thioreducens (91.5% identity), Pyrococcus sp. GE23 (81.4% identity), Pyrococcus abyssi GE5 (81.4% identity), Pyrococcus horikoshii OT3 (81% identity), Pyrococcus abyssi (80.8% identity), Pyrococcus furiosus (79.8% identity), Thermococcus litoralis (78% identity) and Thermococcus aggregans (77% identity).
Gene expression and purification of Tma DNA polymerase
The precursor (originating from pTMAP) and mature (originating from pTMAM) forms of the Tma DNA polymerase expressed in E. coli Rosetta(DE3)pLysS cells (3.0 g each) were purified to near homogeneity, respectively.
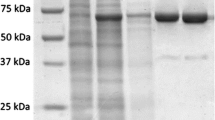
The total activities were determined to be 3,619.41 unit (originating from pTMAP) and 106,409.62 unit (originating from pTMAM) based on the heat treatment (after heat treatment of sonicated extracts at 80°C for 30 min), and the specific activities of the purified enzymes were determined to be 868.52 U mg−1 (originating from pTMAP) and 1000.12 U mg−1 (originating from pTMAM). The purification of the enzymes was monitored by SDS-PAGE (Fig. 2). The SDS-PAGE revealed a single protein band of about 90 kDa, which was consistent with the calculated molecular mass of 90,000.60 Da derived from the 775 residue amino acid sequence. We furthermore attempted to purify the precursor form of Tma DNA polymerase from the sonicated extract of E. coli cells containing pTMAP in order to research protein splicing. Interestingly, however, this approach failed because the expressed precursor form (152.38 kDa) (Fig. 2a, lane 2) was easily spliced during sonication or heat treatment at 80°C into the mature Tma DNA polymerase (90 kDa) and intein (62 kDa) (Fig. 2a, lane 3). A comparison between the expression levels of pTMAP and pTMAM showed that the level of pTMAM was approximately 30 times higher than the pTMAP. This result indicated that the sequence of the intein region may actually interfere with Tma DNA polymerase gene expression.

SDS-PAGE analysis of Tma DNA polymerase purification. Electrophoresis was performed on a vertical gel of 10% polyacrylamide and the gel shown was stained with Coomassie brilliant blue R-250. a Purification of Tma DNA polymerase derived from pTMAP. b Purification of Tma DNA polymerase derived from pTMAM. Lane M high-molecular mass markers, lane 1 sonicated extract of uninduced cells, lane 2 sonicated extract of induced cells, lane 3 heat treatment, lane 4 HiTrap™ Heparin HP column chromatography
Characterization of Tma DNA polymerase
The optimum pH and temperature for Tma DNA polymerase activity were 7.0 in Tris–HCl buffer (Fig. 3a) and 75°C (Fig. 3b), respectively. The thermostability of Tma DNA polymerase (0.05 μg/μl concentration) was tested by measuring the decrease in activity after preincubation at two different temperatures, 94 and 99°C. The half life of the enzyme was found to be about 2 h at 94°C and 45 min at 99°C (Fig. 3c). KCl and (NH4)2SO4 when added affected the activity of Tma DNA polymerase, with optimal concentrations of 10 and 17.5 mM, respectively (Fig. 3d, e). The DNA polymerase was demonstrated to be highly dependent on MgCl2 in the range of 0–20 mM (Fig. 3f), with maximal activity at 14 mM MgCl2 and no detectable activity in the absence of MgCl2. The biochemical parameters of Tma DNA polymerase compared with those of Taq and Pfu DNA polymerases is presented in Table 2.

Characterization of Tma DNA polymerase. a Effect of pH on Tma DNA polymerase activity: 50 mM MOPS-NaOH, Tma DNA polymerase derived from pTMAP (open square), 50 mM Tris–HCl, Tma DNA polymerase derived from pTMAP (open circle), 50 mM MOPS-NaOH, Tma DNA polymerase derived from pTMAM (filled triangle), 50 mM Tris–HCl, Tma DNA polymerase derived from pTMAM (filled circle). All pH values of buffers were as measured at 75°C. b Effect of temperature on Tma DNA polymerase activity. c Purified Tma DNA polymerase was incubated separately at 94°C (filled circle) and 99°C (filled triangle). Aliquots of the mixture were removed at intervals of up to 8 h and quenched in ice. The residual activity of the quenched samples was measured in the basic reaction mixture. d Effects of KCl on Tma DNA polymerase activity. e Effect of (NH4)2SO4 on Tma DNA polymerase activity. f Effects of MgCl2 on Tma DNA polymerase activity. c–f Experiments were performed using Tma DNA polymerase derived from pTMAM only. Error bars represent standard deviation calculated from three replicates
Exonuclease activity and PCR fidelity of Tma DNA polymerase
The presence of 3′ → 5′ exonuclease activity in DNA polymerase confers proofreading capability, enhances fidelity (Kahler and Antranikian 2000) and can remove mispaired bases up to five nucleotides after the misincorporation (Hamilton et al. 2001). The presence of 5′ → 3′ exonuclease activity in DNA polymerase can replace mismatched base pairs with the correct nucleotide. Exonuclease activity of Tma DNA polymerase was checked by assaying the 32P-labeled product released from an end-labeled DNA substrate. Tma DNA polymerase released about 85% 32P from the 3′-end of the substrate DNA in the absence of dNTPs within 10 min (Fig. 4). This demonstrates that Tma DNA polymerase possesses 3′ → 5′ exonuclease activity, a result consistent with its similarity to the amino acid sequence of archaeal family B DNA polymerases, which are known to possess associated 3′ → 5′ exonuclease activity. The importance of proofreading activity has been demonstrated for Pfu and Thermococcus litoralis (Vent) DNA polymerases, which exhibit 5-fold and 7- to 40-fold increases in error rate, respectively, when 3′ → 5′ exonuclease activity is inactivated (Mattila et al. 1991; Cline et al. 1996).

Exonuclease activities of Tma DNA polymerase. 3′ → 5′ exonuclease activity was measured in the absence (open circle) or presence (filled circle) of dNTPs. Activity was calculated as the amount of supernatant radioactivity/total radioactivity. Error bars represent standard deviation calculated from three replicates
Comparisons between Tma and Tma plus DNA polymerase and either Taq or Pfu DNA polymerase were performed to measure their fidelity in PCR. Tma DNA polymerase showed high fidelity with an error rate of 0.46 × 10−5 and Tma plus DNA polymerase exhibited an error rate of 0.79 × 10−5 in the Taq buffer. Error rates of DNA polymerases increased in the following order: Pfu (0.43 × 10−5) < Tma (0.46 × 10−5) < Tma plus (0.79 × 10−5) < Taq (1.77 × 10−5) (Table 3). The fidelity of Tma DNA polymerase was approximately fourfold better than Taq DNA polymerase and roughly the same when compared to Pfu DNA polymerase. Tma plus DNA polymerase also had superior fidelity to Taq DNA polymerase.
Application of Tma DNA polymerase to PCR
The optimal buffer for PCR with Tma DNA polymerase was determined. According to duplicate experiments, the optimal buffer for PCR with Tma DNA polymerase consisted of 50 mM Tris–HCl (pH 8.4), 40 mM KCl, 12.5 mM (NH4)2SO4, 2 mM MgCl2, 0.05% Triton X-100, 0.0075% BSA.
Application to long-range PCR
The most successful protocols for long-range PCR combine modified PCR buffers with a two-polymerase system to provide optimum levels of both processive polymerase activity and proofreading activity (Barnes 1994; Cline et al. 1996).
The efficiency of DNA fragment amplification was evaluated as the ratio of Taq DNA polymerase to Tma DNA polymerase, varied from 10:0 to 0:10. DNA amplification was most efficient at a ratio of 8:2 (Fig. 5). We named this enzyme mixture with Taq (2 U) and Tma (0.5 U) DNA polymerase as Tma plus DNA polymerase. To investigate the extension efficiency of the Tma plus DNA polymerase, PCR was conducted using various fragment sizes. Taq DNA polymerase was barely capable of amplifying the λ DNA fragment of 10 kb and incapable of amplification with longer fragments (Fig. 6a). On the contrary, Tma plus DNA polymerase enhanced efficiency in an even manner; the 15 kb fragment could be successfully amplified using 23 ng of λ DNA as a template (Fig. 6a). Long PCR-based approaches have been used to directly amplify and subsequently sequence the mitochondrial (mt) genome for parasitic helminthes (Hu et al. 2002; Burger et al. 2007). Tma plus DNA polymerase might be useful in long-range DNA amplification and various PCR-based applications.

Optimal ratio of DNA polymerase mixture. A target of 10 kb was replicated using the mixture of DNA polymerases (Taq:Tma), of which concentration ratios were varied as indicated. Lane M 1 kb DNA ladder

Comparison of PCR amplification of Tma, Tma plus, Taq, and Pfu DNA polymerase. a Application to long-range PCR. Target product sizes (kb) are indicated above each lane. b Application to time-saving PCR. Elongation times used for PCR amplification of 2 kb fragment are indicated above each lane. Tma (lanes 1–6); Tma plus (lanes 7–12); Taq (lanes 13–18); Pfu (lanes 19–24) DNA polymerases. Lane M 1 kb DNA ladder
Application to time-saving PCR
Thermostable DNA polymerases are mainly used for the in vitro amplification of DNA fragments and DNA sequence determination (Perler et al. 1996). Even though Taq DNA polymerase has been widely used in PCR, its high error rate contributes to unexpected mutations that occur in amplicons. Therefore, several family B DNA polymerases were isolated from the hyperthermophilic archaea in order to minimize any misincorporations caused in PCR. Among them, Pfu DNA polymerase was found to possess high proofreading activity, but its poor amplification rate made it difficult to replace Taq DNA polymerase. Tma DNA polymerase replicated 2 kb template at 5 s, but Taq DNA polymerase needed more than 40 s for duplication in two-step PCR (Fig. 6b). In this study, we have confirmed that Tma DNA polymerase has superior PCR efficiency in spite of its 3′ → 5′ exonuclease activity.
References
Barnes W (1994) PCR amplification of up to 35-kb DNA with high fidelity and high yield from λ bacteriophage templates. Proc Natl Acad Sci USA 91:2216–2220
Baross J, Holden J (1996) Overview of hyperthermophiles and their heat-shock proteins. Adv Protein Chem 48:1–34
Blanco L, Bernad A, Blasco M, Salad M (1991) A general structure for DNA-dependent DNA polymerases. Gene 100:27–38
Braithwaite D, Ito J (1993) Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res 21:787
Bult C, White O, Olsen G, Zhou L, Fleischmann R, Sutton G, Blake J, FitzGerald L, Clayton R, Gocayne J (1996) Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 273:1058
Burger G, Lavrov D, Forget L, Lang B (2007) Sequencing complete mitochondrial and plastid genomes. Nat Protoc 2:603
Cambon-Bonavita M, Schmitt P, Zieger M, Flaman J, Lesongeur F, Gérard R, Bindel D, Frisch N, Lakkis Z, Dupret D (2000) Cloning, expression, and characterization of DNA polymerase I from the hyperthermophilic archaea Thermococcus fumicolans. Extremophiles 4:215–225
Choi J, Nam K, Min B, Kim S, Söll D, Kwon ST (2006) Protein trans-splicing and characterization of a split family b-type DNA polymerase from the hyperthermophilic archaeal parasite Nanoarchaeum equitans. J Mol Biol 356:1093–1106
Choi J, Song J, Nam K, Lee J, Bae H, Kim G, Sun Y, Kwon ST (2008) Unique substrate spectrum and PCR application of Nanoarchaeum equitans family B DNA polymerase. Appl Environ Microbiol 74:6563–6569
Cline J, Braman JC, Hogrefe HH (1996) PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res 24:3546–3551
Corpet F (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890
Dalgaard JZ, Klar AJ, Moser MJ, Holley WR, Chatterjee A, Mian IS (1997) Statistical modeling and analysis of the LAGLIDADG family of site-specific endonucleases and identification of an intein that encodes a site-specific endonuclease of the HNH family. Nucleic Acids Res 25:4626–4638
Griffiths K, Nayak S, Park K, Mandelman D, Modrell B, Lee J, Ng B, Gibbs M, Bergquist P (2007) New high fidelity polymerases from Thermococcus species. Protein Expr Purif 52:19–30
Guindon S, Lethiec F, Duroux P, Gascuel O (2005) PHYML Online: a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res 33:W557–W559
Hamilton S, Farchaus J, Davis M (2001) DNA polymerases as engines for biotechnology. BioTechniques 31:370–383
Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59
Hodges R, Perler F, Noren C, Jack W (1992) Protein splicing removes intervening sequences in an archaea DNA polymerase. Nucleic Acids Res 20:6153–6157
Hu M, Chilton N, Gasser R (2002) Long PCR-based amplification of the entire mitochondrial genome from single parasitic nematodes. Mol Cell 16:261–267
Jolivet E, Corre E, L’Haridon S, Forterre P, Prieur D (2004) Thermococcus marinus sp. nov. and Thermococcus radiotolerans sp. nov., two hyperthermophilic archaea from deep-sea hydrothermal vents that resist ionizing radiation. Extremophiles 8:219–227
Kahler M, Antranikian G (2000) Cloning and characterization of a family B DNA polymerase from the hyperthermophilic crenarchaeon Pyrobaculum islandicum. J Bacteriol 182:655–663
Kim Y, Lee H, Bae S, Jeon J, Lim J, Cho Y, Nam K, Kang S, Kim S, Kwon ST (2007) Cloning, purification, and characterization of a new DNA polymerase from a hyperthermophilic archaeon, Thermococcus sp. NA1. J Biol Chem 17:1090–1097
Kong H, Kucera R, Jack W (1993) Characterization of a DNA polymerase from the hyperthermophile archaea Thermococcus litoralis: vent DNA polymerase, steady state kinetics, thermal stability, processivity, strand displacement, and exonuclease activities. J Biol Chem 268:1965–1975
Kornberg A, Baker TA (1992) DNA replication, 2nd edn. W.H. Freeman and company, NY
Laemmli U (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lowry O, Rosebrough N, Farr A, Randall R (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Lundberg K, Shoemaker D, Adams M, Short J, Sorge J, Mathur E (1991) High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene 108:1–6
Marsic D, Flaman J, Ng J (2008) New DNA polymerase from the hyperthermophilic marine archaeon Thermococcus thioreducens. Extremophiles 12:775–788
Mattila P, Korpela J, Tenkanen T, Pitkaenen K (1991) Fidelity of DNA synthesis by the Thermococcus litoralis DNA polymerase: an extremely heat-stable enzyme with proofreading activity. Nucleic Acids Res 19:4967–4973
Perler F, Kumar S, Kong H (1996) Thermostable DNA polymerases. Adv Protein Chem 48:377–435
Pietrokovski S (1998) Modular organization of inteins and C-terminal autocatalytic domains. Protein Sci 7:64–71
Reha-Krant L (2008) Recent patents of gene sequences relative to DNA polymerases. Recent pat DNA Gene Seq 2:145–163
Robb F (1995) Archaea: a laboratory manual. Cold Spring Harbor, New York
Saiki R, Gelfand D, Stoffel S, Scharf S, Higuchi R, Horn G, Mullis K, Erlich H (1988) Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239:487–491
Sambrook J, Russell D (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor, New York
Sanger F, Coulson A, Hong G, Hill D, Petersen G (1982) Nucleotide sequence of bacteriophage lambda DNA. J Mol Biol 162:729–773
Song J, Choi J, Kim T, Seo M, Lee M, Kim H, Kwon ST (2007) Characterization and PCR performance of a family B-type DNA polymerase from the hyperthermophilic crenarchaeon Staphylothermus marinus. Enzyme Microb Technol 40:1475–1483
Southworth MW, Kong H, Kucera RB, Ware J, Jannasch HW, Perler FB (1996) Cloning of thermostable DNA polymerases from hyperthermophilic marine Archaea with emphasis on Thermococcus sp. 9°N-7 and mutations affecting 39–59 exonuclease activity. Proc Natl Acad Sci USA 93:5281–5285
Takagi M, Nishioka M, Kakihara H, Kitabayashi M, Inoue H, Kawakami B, Oka M, Imanaka T (1997) Characterization of DNA polymerase from Pyrococcus sp. Strain KOD1 and its application to PCR. Appl Environ Microbiol 63:4504–4510
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596
Truniger V, Lazaro J, Salas M, Blanco L (1996) A DNA-binding motif coordinating synthesis and degradation in proofreading DNA polymerases. EMBO J 15:3430–3441
Woese CR, Fox GE (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA 74:5088–5090
Woese CR, Kandler O, Wheelis M (1990) Towards a natural system of organisms: proposal for the domains archaea, bacteria, and eucarya. Proc Natl Acad Sci USA 87:4576–4579
Xu M-Q, Perler F (1996) The mechanism of protein splicing and its modulation by mutation. EMBO J 15:5146–5153
Acknowledgments
This work was supported by the Marine and Extreme Genome Research Center Program of the Ministry of Land, Transportation and Maritime Affairs, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. Matsunaga.
Rights and permissions
About this article
Cite this article
Bae, H., Kim, K.P., Lee, J.I. et al. Characterization of DNA polymerase from the hyperthermophilic archaeon Thermococcus marinus and its application to PCR. Extremophiles 13, 657–667 (2009). https://doi.org/10.1007/s00792-009-0248-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-009-0248-0