
Abstract
The family B DNA polymerase gene of Thermococcus thioreducens, an archaeon recently isolated from the Rainbow hydrothermal vent field, was cloned and its protein product expressed, purified and characterized. The gene was found to encode a 1,311 amino acid chain including an intein sequence of 537 residues. Phylogenetic analysis revealed a predominantly vertical type of inheritance of the intein in the Thermococcales order. Primary sequence analysis of the mature protein (TthiPolB) showed significant sequence conservation among DNA polymerases in this family. The structural fold of TthiPolB was predicted against the known crystallographic structure of a family B DNA polymerase from Thermococcus gorgonarius, allowing regional domain assignments within the TthiPolB sequence. The recombinant TthiPolB was overexpressed in Escherichia coli and purified for biochemical characterization. Compared with other DNA polymerases from the Thermococcales order, TthiPolB was found to have moderate thermal stability and fidelity, and a high extension rate, consistent with an extremely low K m corresponding to the dNTP substrate. TthiPolB performed remarkably well in a wide range of PCR conditions, being faster, more stable and more accurate than many commonly used enzymes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
DNA polymerases play an essential role in the replication and repair of genetic material of all living organisms. They are classified into families according to their sequence similarities. The first four families (Ito and Braithwaite 1991) were defined by homology to Escherichia coli DNA polymerases (family A for Pol I homologs, family B for Pol II homologs, family C for Pol III homologs) and to the eukaryotic DNA polymerase β (family X). Family D was later proposed to account for a new heterodimeric DNA polymerase found in the Euryarchaeota subdomain of archaea (Cann and Ishino 1999). The most recently described DNA polymerase family is the Y family (Ohmori et al. 2001), which consists of phylogenetically related DNA polymerases that are characterized by low fidelity DNA synthesis and their ability to bypass DNA lesions. While bacterial replicative DNA polymerases belong to the C family and eukaryal ones belong to the B family (except for mitochondrial DNA polymerases which are family A), the situation in archaea (which have no family A or C DNA polymerases) is more complex. According to available genome sequences, crenarchaeotes have 2 or 3 family B and no family D DNA polymerases. This implies that chromosomal replication is performed by family B DNA polymerases in the Crenarchaeota kingdom, while euryarchaeotes have a single family B polymerase and a single family D DNA polymerase, both being likely involved in replication (Henneke et al. 2005).
Family B DNA polymerases from hyperthermophilic archaea have attracted considerable interest as PCR enzymes because of their remarkable thermal stability, their proofreading 3′–5′ exonuclease activity and their monomeric nature making them relatively easy to produce. Several have been characterized so far, especially from species belonging to the Thermococcales order such as Pyrococcus furiosus (Lundberg et al. 1991), Pyrococcus abyssi (Dietrich et al. 2002; Gueguen et al. 2001) Thermococcus litoralis (Kong et al. 1993), Thermococcus sp. 9°N-7 (Southworth et al. 1996), Thermococcus aggregans (Niehaus et al. 1997), Thermococcus kodakarensis (Takagi et al. 1997), Thermococcus gorgonarius (Bonch-Osmolovskaya et al. 1996) and Thermococcus fumicolans (Cambon-Bonavita et al. 2000). Family B DNA polymerases from other euryarchaeotes include Methanothermobacter thermoautotrophicus (Kelman et al. 1999) and crenarchaeotes such as Pyrobaculum islandicum (Kahler and Antranikian 2000). In contrast with their high sequence similarities, these DNA polymerases show important variations in their properties such as fidelity, thermal stability, kinetic parameters and processivity, even within the same genus.
A novel hyperthermophilic, obligately sulfur-reducing archaeon, Thermococcus thioreducens (T. thioreducens) was isolated from samples collected at the Rainbow hydrothermal vent site (Mid-Atlantic ridge) and described by our group (Pikuta et al. 2007). Its genomic material was used for the search of a family B DNA polymerase gene. An open reading frame encoding the targeted enzyme was identified, cloned and sequenced. The recombinant DNA polymerase T. thioreducens PolB (TthiPolB) was expressed, purified and characterized. The structural fold of TthiPolB was also predicted against known crystallographic structures of family B DNA polymerases to highlight relationships between functional properties and structural features.
Materials and methods
Strains and culture conditions
Thermococcus thioreducens (ATCC: BAA-394, DSM: 14981) was grown as described (Pikuta et al. 2007). Recombinant plasmids were propagated in E. coli strain TOP10 (Invitrogen, USA) for those based on pCR4-TOPO and strain TOP10F’ (Invitrogen, USA) for those based on pCRT7/CT-TOPO. E. coli strain Rosetta(DE3) (Novagen, USA) was the host for protein expression. All E. coli cultures were grown in Luria–Bertani medium containing 100 mg/l ampicillin (to select for the expression vector) at 37°C with vigorous shaking; chloramphenicol was added to a final concentration of 35 mg/l in expression cultures (to retain the Rosetta phenotype).
Gene cloning
Genomic DNA of T. thioreducens was isolated from 400 mg of fresh biomass through a standard phenol/chloroform extraction followed by ethanol precipitation (Sambrook et al. 1989). Two DNA polymerase gene fragments were amplified by PCR from the genomic DNA using primers pairs (5′-ATGATCCTCGAYACTGACTAC/5′-CTCGCCCTCGTGGTAGAGC and 5′-AGAAGTAYGCGGTGATAGACG/5′-GATGTAGCTTATCACCGTKCC) designed from conserved regions in an alignment of six available family B DNA polymerase sequences from Thermococcales species. A 105 bp linker DNA was amplified from the pCR4-TOPO vector (Invitrogen, USA), using T3 (5′-AATTAACCCTCACTAAAGGG) and T7 (5′-TAATACGACTCACTATAGGG) primers. Genomic DNA from T. thioreducens was digested with Eco RV and ligated to the linker with T4 ligase. Both enzymes were from Promega (USA) and used according to the manufacturer’s instructions. The product was used as a template in nested PCR with DNA polymerase-specific primers (designed from the previously amplified fragments) and linker-specific primers. Amplified fragments were excised from an agarose gel and inserted into a pCR4-TOPO vector for sequencing. The entire coding region of the DNA polymerase gene was then amplified with the high-fidelity DNA polymerase KOD (Novagen, USA) using primers (POLB-1F: 5′-ATGATCCTCGATGCCGACTACATCACCGAG, POLB-1R: 5′-TCACCTCTTTTTCACCTTCAGCCACGCGCC) based on the exact sequence of the gene ends, and cloned into a pCRT7/CT-TOPO (Invitrogen, USA) expression vector. The insert from two clones as well as the PCR product were completely sequenced. An inteinless version of the gene was constructed following the overlap extension method (Horton et al. 1989), amplifying the overlapping fragments from genomic DNA using primers POLB-1F with 5′-GTAGTAACTGTTTGCCAGAATCTTGATGGCGCGTTG and POLB-1R with 5′-TCTGGCAAACAGTTACTACGGCTACTACGGCTACGC, respectively. The inteinless gene was cloned into a pCRT7/CT-TOPO expression vector and its sequence verified.
Sequence determination and analysis
Sequence determinations were performed using an ABI 310 Genetic Analyzer and the DYEnamic ET Terminator Cycle Sequencing Kit (Amersham-Pharmacia, USA). Raw sequences were manually edited with Chromas (http://www.technelysium.com.au/) and assembled in GCG (Accelrys, USA). Codon usage analyses, translations and calculation of molecular weight were performed using the Sequence Manipulation Suite (Stothard 2000). Sequence alignments were performed using ClustalW (Thompson et al. 1994). Phylogenetic analyses were conducted using MEGA version 2.1 (Kumar et al. 2001). Phylogenetic trees were constructed with the neighbor-joining method (Saitou and Nei 1987) with 1,000 replications of bootstrap resampling.
Nucleotide sequence accession numbers
The sequence of the family B DNA polymerase from T. thioreducens described in this study has been deposited in GenBank under accession number [GenBank:EF058197]. The sequence of the PCNA gene described in the exonuclease assay has been deposited under accession number [GenBank:EF058196]. Accession numbers of DNA polymerase sequences from other Thermococcus species mentioned in this study are as follows: T. aggregans, [GenBank:Y13030]; T. fumicolans, [GenBank:Z69882]; T. hydrothermalis, [GenBank:AJ245819]; T. litoralis, [GenBank:M74198]; T. peptonophilus, [GenBank:E13953]; T. sp. 9°N-7, [GenBank:U47108]; T. sp. GE8, [GenBank:AJ250333]; T. kodakarensis, [GenBank:D29671].
Sequence and domain assignments
The functional domains of TthiPolB were identified within its primary sequence by superimposing the homologous residues onto the known crystallographic structure of thermostable family B DNA polymerase (TgoPolB) from Thermococcus gorgonarius [PDB:1tgo]. TthiPolB residues were positioned by chemical based conformational sampling coupled to the usage of energy restraints derived from the force field available in the crystallographic and NMR System (CNS) program suite (Brunger et al. 1998). The resulting detailed model was refined with conjugate gradient minimization with no experimental energy terms used. All backbone atoms of the molecules were fixed while the remaining atoms were minimized for 500 steps. The resulting structure was visualized with PYMOL (http://www.pymol.org).
Expression and purification
A culture of E. coli Rosetta(DE3) harboring the recombinant expression vector containing the inteinless version of the TthiPolB gene was grown in a 10 l fermenter at 37°C. When the optical density at 600 nm reached 0.5, expression was induced by the addition of isopropyl thio-β-d-galactoside (IPTG) at a final concentration of 0.5 mM. After 16 h, cells were harvested by centrifugation. All subsequent steps were performed at 4°C. Cell were resuspended in buffer P (10 mM sodium phosphate, pH 7.0, 100 mM NaCl, 0.1 mM EDTA, 0.5 mM PMSF and 0.5 mM dithiothreitol) and disrupted by sonication (6 cycles of 45 pulses) using a Branson Sonifier 250 (VWR Scientific, USA). Cell debris were removed by centrifugation (12,000g, 20 min). The supernatant was heated for 25 min at 80°C and the precipitate was removed by further centrifugation. The supernatant was loaded into a heparin column (HiTrap Heparin HP, Amersham, USA) that had been pre-equilibrated with buffer P and eluted with a 0.1–1.5 M NaCl linear gradient in buffer P, using an Äkta Explorer FPLC system (Amersham-Pharmacia, USA). Fractions containing DNA polymerase (showing a band of the expected size on a SDS-PAGE gel) were pooled, dialyzed against buffer B (25 mM Bis-Tris-Propane pH 9.5, 0.1 mM EDTA, 50 mM KCl, 0.5 mM PMSF and 0.5 mM dithiothreitol) and applied to an anion-exchange column (HiTrap Q HP, Amersham, USA) that had been pre-equilibrated with the same buffer. Bound proteins were eluted with a 0.0–0.75 M NaCl linear gradient in buffer B. Fractions containing DNA polymerase were pooled, concentrated using a Centricon Plus PL-30 centrifugal filter device (Millipore, USA), applied to a Sephacryl S-200 gel filtration column (Pharmacia, USA) pre-equilibrated with buffer T (50 mM Tris pH 7.5, 0.1 mM EDTA, 50 mM KCl, 0.5 mM PMSF and 0.5 mM dithiothreitol) and eluted with the same buffer. Fractions containing DNA polymerase were pooled and mixed with glycerol and Triton X-100 at a final concentration of 50 and 0.5%, respectively. Aliquots were stored at −20°C in 1.5 ml tubes. Protein concentrations were determined according to Bradford (Bradford 1976), using BSA as a standard.
DNA polymerase activity assay
A non-radioactive, fluorescence-based DNA polymerase activity assay was used which we adapted from Tveit and Kristensen (2001), and which was originally described by Seville et al. (1996). It was based on the specific binding affinity of the dye PicoGreen (Molecular Probes, Invitrogen) with dsDNA. Annealed primer-template was prepared as follows: a solution containing 200 μg/ml M13mp18 ssDNA (Bayou Biolabs, USA) and 170 nM UPlong primer (5′-TTCCCAGTCACGACGTTGTAAAACGACGGCCAGTG) was heated at 70°C for 5 min, cooled to room temperature over 20 min and stored at −20°C until needed. A typical DNA polymerase assay was performed as follows: 1.2 μl of the annealed primer-template was mixed with 2 μl of dNTP solution (at 2.0 mM each) and 1.8 μl of 10× reaction buffer in a final volume of 18 μl in a 500 μl PCR tube. Unless specified otherwise, the 10× reaction buffer consisted of 100 mM Tris-HCl pH 8.25, 600 mM KCl, 20 mM MgCl2 and 1% Triton X-100. After a 1-min equilibration at the assay temperature (74°C unless specified otherwise), the reaction was started by the addition of 2 μl of DNA polymerase solution (diluted in the same reaction buffer). Incubation took place in a thermal cycler (Mastercycler gradient, Eppendorf, USA). The reaction was stopped after a precise amount of time by the addition of 2 μl of 250 mM EDTA, after which the reaction tube was immediately put on ice until all reactions were completed. Reactions were diluted with 150 μl TE (10 mM Tris-HCl pH 7.2, 1 mM EDTA) and transferred into 3.5 ml methacrylate cuvettes (Perfector Scientific, USA), where 1.5 μl of PicoGreen and 850 μl TE were added. Cuvettes were kept in the dark until the fluorescence was measured in a FluoroMax-3 fluorometer (Jobin Yvon, USA). Excitation and emission wavelengths were 485 and 525 nm, respectively. Because of the small volume, a 12-mm platform was placed below the sample in the cuvette compartment in order to get reproducible readings. Fluorescence values were converted into dsDNA mass amounts using a standard curve obtained with known amounts of added dsDNA. Incorporation of dNTPs (in pmol per unit of time) was calculated as the increase in dsDNA amount in pg divided by 649 (average molecular weight of a deoxyribonucleoside monophosphate pair). One unit of DNA polymerase activity was defined as the amount required to incorporate 10 nmol of dNTP into dsDNA in 30 min (extrapolated from 2-min measurements) at 74°C in the reaction buffer described above.
In the case when DNA polymerase activity was measured as a function of temperature, MOPS buffer was used instead of Tris in order to minimize pH variations with temperature. When relative DNA polymerase activity was measured as a function of buffer type and concentration, the pH of buffers were adjusted at room temperature so that they would all be 7.5 at 72°C, as calculated from the temperature coefficients (Tris: −0.028, Hepes: −0.014, MOPS: −0.011, phosphate: −0.003 pH unit per °C). In addition, Triton X-100 was not included in the buffers used in these experiments.
Structural thermostability
Thermal stability of the DNA polymerase structure was investigated through circular dichroism measurements as a function of time during incubation at 95°C. DNA polymerase solution was dialyzed overnight against sodium phosphate buffer pH 8.9 and diluted to a final concentration of 50 μg/ml in the same buffer. A volume of 2.4 ml was used in a 10 mm cylindrical quartz cell (Starna Cells, USA). Circular dichroism at 200–250 nm (10 reads per points) was measured at regular time intervals during a total of 35 h, on an OLIS RSM 1000 CD spectrophotometer (Olis, USA).
Functional thermostability
Functional thermostability was evaluated by measuring residual DNA polymerase activity after various pre-incubation times at 95°C. A solution of TthiPolB at 17 μg/ml in the assay reaction buffer described above was incubated at 95°C for over 3 h. Samples were taken at various times and cooled to room temperature, before being diluted ten times in reaction buffer. DNA polymerase activity was measured as described with four replicates per time point using 2 μl of the diluted DNA polymerase per reaction. The assay temperature was 72°C. Taq polymerase (New England Biolabs, USA) was used as a control in parallel under the same conditions except for the reaction buffer, which was the one provided by the manufacturer.
Fidelity assay
The error rate generated by TthiPolB was measured using a modified version of the yeast-based PCR fidelity assay described by Flaman et al. (1995), in which the reporter gene HIS3 was replaced by the ADE2 gene. Wild-type TP53 cDNA was PCR amplified using TthiPolB. The PCR product was co-transformed with a linearized expression vector into a TP53 transcriptional activity reporter strain, and cloned in vivo by homologous recombination. After selection of colonies expressing the TP53 protein, white colonies expressed the wild type protein whereas red colonies expressed mutant proteins (Flaman et al. 1994). Therefore, the percentage of red colonies was used to calculate the error rate per nucleotide per cycle (ER) using the following formula: ER = % red colonies/(d × del × 100), where d is the amount of effective doublings and del is the effective target size (542 bp). The amount of effective doublings was calculated using the equation 2d = (amount of PCR product)/(amount of starting target). PCR amplifications were performed in a 50 μl volume using 200 μM dNTP, 1.5 mM MgCl2, 10 mM Tris-HCl pH 9.0 (at 25°C), 50 mM KCl, 0.1% Triton-X100, 50 pmol of P3 and P4 primers (Flaman et al. 1994) and 13 pg of input DNA. The cycling profile consisted of 30 cycles of 94°C 20 s, 62°C 30 s and 72°C 2 min, preceded by 3 min at 95°C and followed by 5 min at 72°C. The fidelity assay was performed in parallel with Taq polymerase (ABgene, France) as a control.
Exonuclease assay
The substrate for the exonuclease assay was a 750 bp long double-stranded DNA fragment encoding the T. thioreducens PCNA, amplified by PCR using TthiPolB, separately with PTO-modified primers and unmodified primers. The primers (forward: 5′-ATGCCGTTCGAGATAGTTTTTGATG, reverse: 5′-TCAGTCCTCGACGCGCGGGGCGAGG) were synthesized by MWG Biotech (USA). The PTO modification was on the first three nucleotides of the modified primers. The PCR products were purified using GeneClean Turbo (Qbiogene, USA) in order to remove free primers. Exonuclease assay was performed as follows: 120 ng of substrate (purified PCR product) was mixed with 1.8 μl of 10× reaction buffer (same buffer as in DNA polymerase assay) in a final volume of 18 μl in a 500 μl PCR tube. When indicated, the mixture also contained 2 nmol each dNTP. After a 1-min pre-incubation at 74°C, the reaction was started by the addition of 2 μl of DNA polymerase solution in reaction buffer, corresponding to 3.4 ng or 0.5 U TthiPolB. For the negative control, 2 μl of buffer alone were added under the same conditions. The reaction was stopped in the same way as in the DNA polymerase activity assay, and the dsDNA concentration was measured using PicoGreen as described. All assays were performed in triplicates.
Determination of apparent Michaelis–Menten constants
DNA polymerase activity assays were performed as described in four replicates for each data point and three incubation times per condition. For the determination of the K m corresponding to the DNA substrate, the reaction mix contained a variable amount of primed template (0.5–10.0 nM in the final reaction). For the determination of the K m corresponding to the dNTP substrate, the reaction mix contained dNTP concentrations ranging from 1 to 200 μM each. The DNA polymerase amount was 1.7 ng per reaction and 1.13 ng per reaction, respectively for the DNA parameter and the dNTP parameter experiments. The apparent K m parameters were determined from a Lineweaver–Burk plot (Lineweaver and Burk 1934).
Extension rate
Extension rate was determined by applying the DNA polymerase activity assay described above to monitor the extent of conversion of the M13 template into dsDNA under a large excess of DNA polymerase. A template concentration of 1 nM instead of 6 nM was used with polymerase to template molar ratios of 2–50, incubation times from 0 to 5 min and four replicates per data point.
Polymerase chain reaction
Unless specified otherwise, PCR using TthiPolB was performed as follows. Reaction mixture typically contained 10% of the 10× reaction buffer described above, 200 μM each dNTP, 600 nM each primer, a variable amount of template and between 140 and 270 ng/ml DNA polymerase (corresponding to 1 to 2 units per 50 μl reaction). The cycling profile was: initial denaturation, 2 min at 95°C; 30 cycles of 15 s denaturation at 95°C, 20 s annealing at 50–65°C and 30–60 s extension at 72°C; final extension, 3 min at 72°C.
Results and discussion
Cloning and sequence analysis of a family B DNA polymerase gene
A comparison of family B DNA polymerase sequences from the Thermococcales order revealed a remarkable high similarity at several conserved regions, including at the nucleotide level, allowing the design of PCR primers with limited degeneracy. Two non-overlapping fragments, respectively, 450 and 330 base pairs (bp) long, were amplified with such primers, and their sequence confirmed that they belonged to a family B DNA polymerase gene. An approach similar to the vectorette PCR technique (Riley et al. 1990), yet simpler, was devised to determine the ends of the gene sequence. A linker was designed from a region of the pCR4-TOPO vector that could easily be amplified using readily available T3 and T7 primers and which was verified not to correspond to any archaeal genomic sequence. Digested genomic DNA from T. thioreducens was then ligated with this linker and used as a template for PCR using DNA polymerase-specific primers designed from the previously amplified fragments and linker-specific primers. The resulting amplified fragments were determined to contain sequences corresponding to the DNA polymerase gene termini. The fragments encoded 131 and 182 amino acid residues of the N- and the C-termini, respectively, as well as additional upstream and downstream sequences. The entire gene, amplified using primers designed from the newly determined gene terminal sequences, was found to be 3,936 bp long and to encode a 1,311 amino acid (aa) polypeptide chain (Fig. 1a).

Sequence and structure overview of TthiPolB. The complete sequence of the mature TthiPolB (774 aa) is shown (a) with the removed intein fragment (537 aa) (b). The residues that are highly conserved in hypothermophilic family B DNA polymerases are highlighted in large letters. There are five domains and subdomains that include the N-terminal (M1-E130, F327-A368), exonuclease (G131-F326), palm subdomain (P369-I449, A501-F588), fingers (P450-Y500) and thumb (F589-R775) regions. The three-dimensional structure of TgoPolB, which has 91% sequence identity to TthiPolB, was used to predict the tertiary folding of TthiPolB (c)
Alignments with homologous DNA polymerase gene sequences revealed the presence of an intein gene within the coding sequence, encoding a 537 aa protein and splitting the DNA polymerase sequence into a 491 aa N-terminal and a 283 aa C-terminal fragments (Fig. 1b). Inteins are “selfish” genetic elements that are post-translationally excised from a protein precursor through a self-catalytic protein splicing process (Perler 2002; Pietrokovski 2001). First discovered in yeast (Hirata et al. 1990; Kane et al. 1990), over 300 have been identified so far in all domains of life (http://www.neb.com/neb/inteins.html). In addition to their splicing domains, most have a homing endonuclease domain that confers them potential mobility. Archaeal family B DNA polymerase genes have three known specific insertion sites for intein sequences designated pol-a, pol-b and pol-c. The majority of archaeal DNA polymerase genes sequenced so far include one to three intein elements (Fig. 2a). According to InBase nomenclature, the intein within the T. thioreducens family B DNA polymerase belongs to the “DNA polymerase motif B” group, typified by the prototype allele Tli Pol-1, and should be designated Tthi Pol. Among Thermococcus DNA polymerases that have been studied so far, TthiPolB is the only one with a single intein at the pol-b insertion site. Tthi Pol’s closest homologue, with 77.1% identity, is Thy Pol-1 from T. hydrothermalis, which has a second intein at the pol-c insertion site. A phylogenetic tree illustrating the position of Tthi Pol among other known inteins of the DNA polymerase motif B group is shown in Fig. 2b. A comparison with the phylogenetic tree of family B DNA polymerases belonging to the Thermococcales order (Fig. 2c) suggests a predominantly vertical type of inheritance of the motif B inteins. Indeed, with the exception of Pyrococcus sp. GB-D, the tree topology is remarkably conserved between the DNA polymerases and the motif B inteins, suggesting a common intein ancestor for the Thermococcales order and multiple loss events (affecting T. fumicolans, T. peptonophilus, T. sp. 9°N-7 among others). The position of P. GB-D in the intein tree suggests a recent cross-generic reinvasion from a Thermococcus species by horizontal transmission. We compared codon usage between the nucleotide sequences encoding the motif B intein and the mature polymerase in both T. TthiPolB and P. GB-D, and found no significant difference in T. thioreducens but noticeable differences in GB-D (data not shown), which further argues in favor of a recent mobility event in the case of P. GB-D. Those observations are consistent with the model of recurrent invasion and extinction of selfish genes proposed by Goddard and Burt (1999), with the particularity of a remarkably stable fixation and no evidence of interspecific lateral transfer within the Thermococcus genus.

Molecular phylogeny of TthiPolB and its intein. The distribution of inteins in Thermococcus DNA polymerase precursor polypeptide chains is shown (a), with black and white elements representing extein and intein sequences, respectively. The numbers indicate sizes in amino acid residues and the length of each element is proportional to its actual size. Lines represent alignment gaps. The reduced size of T. aggregans’ third intein is due to the absence of the internal endonuclease domain that is present in other species. The apparent shorter size of T. hydrothermalis’ N-terminus is due to the unavailability of the complete gene sequence. The phylogenetic tree of the DNA polymerase motif B group archaeal inteins (b) is based on 442 common amino acid sites. Intein names, as appearing in the InBase database, are indicated in parentheses. The scale bar represents 1 inferred substitution per 10 residues. Tthi Pol’s closest homologue is Thy Pol-1, with 77.1% identity. The phylogenetic dendrogram of mature family B DNA polymerase sequences in the Thermococcales order (c) is based on 737 common amino acid sites. Accession numbers (NCBI protein database except for T. peptonophilus: GenBank) are indicated in parentheses. The scale bar represents 1 inferred substitution per 50 residues. Both trees were built using the Neighbor-Joining method with the Poisson Correction distance model. Numbers at nodes represent bootstrap values (1,000 replications)
The phylogenetic tree illustrating the relatedness between family B DNA polymerases (inteinless amino acid sequences) in the Thermococcales order (Fig. 2c) shows TthiPolB grouped together with most Thermococcus DNA polymerases. Interestingly, as it was also observed with 16 s rRNA-based phylogenetic analysis (Pikuta et al. 2007), T. litoralis (Kong et al. 1993) and T. aggregans (formerly T. sp. TY) (Niehaus et al. 1997) sequences seem as distant from other Thermococcus sequences as they are from the Pyrococcus genus. TthiPolB appears closely clustered with T. hydrothermalis (Querellou, unpublished) and T. 9°N-7 (Southworth et al. 1996) family B DNA polymerases, which are also among the closest homologues together with T. GE8 (Querellou, unpublished), according to pairwise comparisons of 737 common amino acid sites, with 92.4, 93.4 and 92.5% identity, respectively. Alignments of complete protein sequences (not available for T. hydrothermalis) show 92.9% identity with T. 9°N-7 and 92.3% with T. GE8. The mature form of TthiPolB is 774 amino acid residues long and has a calculated molecular weight of 89.98 kDa. TthiPolB will be discussed from here on as the mature T. thioreducens family B DNA polymerase without its intein sequence. The amino acid sequences corresponding to the functional domains are shown against the known crystallographic structure of family B DNA polymerase from Thermococcus gorgonarius [PDB:1tgo] (Hopfner et al. 1999), designated as TgoPolB from here on, that shares 91.0% sequence identity with TthiPolB (Fig. 1c).
Structure prediction
While work to pursue the three-dimensional structure of TthiPolB by X-ray crystallography is in progress, the primary sequence of TthiPolB was aligned and modeled against the crystallographic structure of TgoPolB to predict the protein’s tertiary structure. Among the family B DNA polymerase structures determined to date, TthiPolB has the highest sequence identity to that of TgoPolB. We have therefore assumed that the three-dimensional structure of TthiPolB would share very similar topological and structural features with TgoPolB (Hopfner et al. 1999). The projected structural domains within the mature TthiPolB enzyme include the N-terminal domain (M1-E130, F327-A368), the exonuclease (3′–5′) domain (G131-F326), the palm (P369-I449, A501-F588), fingers (P450-Y500) and thumb (F589-R775) (Fig. 1c). These are the general structural characteristics of family B DNA polymerases and examples of these are readily observed in the structures of gp43 from bacteriophage RB69 (Wang et al. 1997), 9°N-7 pol from Thermococcus sp. 9°N-7 (Rodriguez et al. 2000) and KOD polymerase from T. kodakarensis (Hashimoto et al. 2001). Moreover, the exonuclease and palm domains share structural similarities and have comparable active sites to those of family A DNA polymerases and they both show metal dependence for exonuclease and replication mechanisms (Hopfner et al. 1999; Wang et al. 1997).
Expression and purification
Failed attempts to express TthiPolB in common E. coli hosts suggested sensitivity to codon bias, as a comparison of codon usage in the TthiPolB gene and in the E. coli genome (Nakamura et al. 2000) revealed significant differences. Consequently, a Rosetta strain, carrying a plasmid providing the host with an additional supply of rare tRNA genes (argU, ileX, glyT, leuW and proL) was used for expression. Recombinant TthiPolB could then be expressed at high levels.
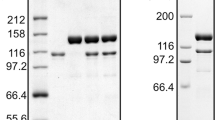
TthiPolB was purified to >95% purity, as estimated by SDS-PAGE analysis, after a heat treatment followed by three chromatographic steps: heparin, anion-exchange and gel filtration. Approximately 20 mg of purified TthiPolB can be routinely obtained per liter of culture with a specific activity of 146 U/μg. The specific activity is remarkably high compared to other reports on archaeal family B DNA polymerase purification [66 U/μg with Pyrococcus abyssi (Dietrich et al. 2002), 25 U/μg with Thermococcus sp. 9°N-7 (Southworth et al. 1996)] and consistent with a higher extension rate (see below), although some of the differences can probably be attributed to differences in purity and to the way units are defined and measured.
Biochemical properties
The enzymatic activity of TthiPolB was measured under various conditions of pH, temperature and medium composition (Fig. 3), using a DNA polymerase assay adapted from Tveit and Kristensen (2001). More than 80% of maximal activity was obtained between pH 7.8 and 9.0, measured at 25°C, which corresponds to pH 6.5–7.7 at 72°C when Tris is used as a buffer (Fig. 3a). In contrast with T. thioreducens’ optimal growth temperature of 82–85°C and upper growth limit of 94°C (Pikuta et al. 2007), maximal DNA polymerase activity was observed between 72 and 76°C, above which activity decreased abruptly (Fig. 3b). This is probably the result of thermal denaturation of the DNA substrate rather than the enzyme itself. At temperatures above the apparent optimum for activity, most primer molecules are free rather than annealed to the M13 template, which prevents determination of the actual optimal temperature.

Effects of pH, temperature, salts and buffers on TthiPolB activity. Relative DNA polymerase activity was measured as a function of pH (a), temperature (b), potassium and sodium chloride concentration (c), magnesium chloride concentration (d) and buffer type and concentration (e). Activity assays were carried out at 72°C as described except for the varied reaction condition and the absence of Triton X-100. MOPS buffer instead of Tris was used in experiment b because of its lower temperature coefficient, in order to minimize pH variation with temperature. Buffer pH were adjusted at room temperature so they would all reach the value of 7.5 at 72°C, as calculated from their temperature coefficients. Error bars represent standard deviation calculated from four replicates
The enzyme was active under a wide range of salt concentrations, and showed a slight preference for potassium chloride over sodium chloride (Fig. 3c). Over 80% of maximal activity was obtained with KCl concentrations of 50–80 mM, with a maximum at 60 mM. Magnesium ions are known cofactors for DNA polymerases. As expected, no activity was detected in the absence of magnesium. Activity was maximal between 1.5 and 3.0 mM magnesium chloride (Fig. 3d).
The effects of buffer type and concentration on DNA polymerase activity were examined (Fig. 3e). Tris was found to allow the highest activity. The enzyme was 20% less active in HEPES and MOPS and 50% less active in phosphate buffer at concentrations of 10 mM. In all cases, activity decreased with increased buffer concentration. The decrease was stronger in the case of phosphate, possibly because of a reduced availability of magnesium ions through complexation with phosphate. The same experiment was done with decreased concentrations of potassium chloride while the buffer concentration was increased, in order to minimize ionic strength fluctuations, and gave very similar results (data not shown), suggesting that even Tris has an inhibitory effect on DNA polymerase activity and may not be the ideal buffer for this DNA polymerase, although it was the best among the ones we tested.
The DNA polymerase activity was also measured with other additives commonly used in DNA polymerase buffers. The presence of bovine serum albumin (tested at 0–200 μg/ml) or ammonium sulfate (tested at 0–20 mM) in the Tris-containing reaction buffer did not have any significant effect on activity, while addition of Triton X-100 at a concentration of 0.1% had a notable effect, increasing DNA polymerase activity by as much as 30% (data not shown).
Thermal stability
Thermal stability of TthiPolB was investigated from both a structural and a functional point of view. Circular dichroism (CD) spectroscopy was used to detect structural changes during incubation at 95°C, at the wavelengths typically used to monitor β-sheets (212 nm, Fig. 4a) and α-helices (222 nm, Fig. 4b). The data obtained suggest an extreme structural stability at this temperature, with no detectable change during the first 20 h and an apparent half-life of 22 h. Interestingly, evaluation of functional thermostability as the measure of residual DNA polymerase activity after incubation at 95°C showed a quite different result (Fig. 4c), with a half-life at that temperature of 125–130 min, similar to published values for other thermostable DNA polymerases (Table 1). The result obtained with Taq as a control further validated the method used, giving a half-life of 45 min, consistent with the manufacturer’s claim of 45 min at 94°C (New England Biolabs). The absence of correlation between structural and functional thermal stability observations suggests that the activity loss of TthiPolB with time may result from local irreversible changes affecting the active site rather than protein denaturation on a global scale.

Thermal stability of TthiPolB. Structural thermostability was monitored by circular dichroism, measured as a function of incubation time at 95°C. The unfolding of β-sheets (a) and α-helices (b) was recorded at 212 and 222 nm, respectively. There were no apparent structural changes until about 20 h. Thermal stability of TthiPolB’s DNA polymerase activity was also measured (c). TthiPolB and Taq DNA polymerase residual activities were measured and compared at 72°C as a function of pre-incubation time at 95°C. Error bars represent standard deviation calculated from four replicates. The half-life values of TthiPolB and Taq were 125–130 min and 45 min, respectively
Fidelity
The fidelity of TthiPolB was compared to that of Taq polymerase using a yeast-based PCR fidelity assay (Flaman et al. 1994). The error rate exhibited by TthiPolB was 1.4 × 10−5 errors per nucleotide and per replication event, while it was 2.4 × 10−5 for Taq polymerase. In other words, TthiPolB has a 1.7 times higher fidelity than Taq polymerase. This value is similar to those obtained with other archaeal DNA polymerases Tfu from T. fumicolans (Cambon-Bonavita et al. 2000) and Deep Vent from Pyrococcus sp. GDB (Flaman et al. 1994) using the same assay under similar conditions, giving error rates of 1.6 × 10−5 and 1.2 × 10−5, respectively. However, the fidelity of TthiPolB does not match that of Pfu (from P. furiosus), which is reported to exhibit an error rate that is almost an order of magnitude lower than Taq (Cline et al. 1996; Flaman et al. 1994).
Exonuclease activity
Exonuclease assays commonly use radiolabeled templates and measure either the release of radioactive nucleotides from double-stranded DNA (dsDNA) or the decrease in size of end-labeled oligonucleotides through autoradiography (Kong et al. 1993; Blöndal et al. 2001; Hogrefe et al. 2001). In this study, a non-radioactive approach was chosen and an assay was designed based on the one used for DNA polymerase activity determination (Tveit and Kristensen 2001) where variation in dsDNA concentration is measured using a fluorescent dye. An exonuclease-resistant template was used to discriminate between 3′–5′ and 5′–3′ directionality. Phosphorothioate (PTO) modified oligonucleotides in which a non-bridging oxygen is replaced by a sulfur in the phosphate backbone have been shown to confer resistance to exonucleolytic activity (Kunkel et al. 1981; Putney et al. 1981). For this reason, PTO-modified oligonucleotides have been used in gene therapy (Agrawal et al. 1988) and even in PCR to improve specificity by preventing primer degradation by proof-reading DNA polymerases (de Noronha and Mullins 1992; Skerra 1992). In our assay, a DNA sequence was amplified using 5′-PTO modified primers, generating a double-stranded fragment that was resistant to 5′–3′ exonucleolytic degradation while being susceptible to 3′–5′degradation. The same sequence was amplified by unmodified primers, generating an unprotected double-stranded molecule. We anticipated that in the absence of dNTP, a decrease of dsDNA concentration over time would indicate the presence of exonuclease activity, and would demonstrate 3′–5′ directionality when the 5′-PTO protected substrate is used. In the presence of dNTP, any 3′–5′ exonuclease activity would be masked by the presence of the polymerase activity, therefore a decrease in dsDNA concentration with the unprotected substrate could only be the result of a 5′–3′ exonuclease activity, while the PTO-modified substrate would serve as a negative control. The limitation of our method is that quantitation of 3′–5′ exonuclease activity cannot be performed in the presence of dNTP, preventing the determination of an accurate polymerase to exonuclease activity ratio under physiological conditions.
The results of our exonuclease assay are shown in Fig. 5. In the absence of dNTP, exonuclease activity is demonstrated by the decrease in dsDNA concentration comparatively to the control. The same level of decrease when the template is 5′-protected indicates that the directionality of the exonuclease activity is 3′–5′ (Fig. 5a). We calculated the degradation rate to be approximately two nucleotides per second per enzyme molecule under the assay conditions.

Exonuclease activity assay. Changes in dsDNA amounts were measured as a function of incubation time at 72°C in the absence (a) and presence (b) of dNTP. Experiments using TthiPolB are indicated with filled squares. Negative controls with no DNA polymerase present are shown as open squares. Solid lines correspond to unmodified template while dashed lines correspond to 5′-PTO modified template. Error bars represent standard deviation calculated from three replicates. The decrease in dsDNA content (expressed in amount of base pairs destroyed) in the absence of dNTP in the reaction medium (a) indicates exonuclease activity. The same level of activity with 5′-PTO modified (exonuclease-resistant) template suggests a 3′–5′ directionality of the exonuclease activity. In the presence of dNTP (b), an increase in dsDNA content (expressed in base pairs created) is observed, indicating an unexpected DNA synthesis, partially inhibited when the template is 5′-PTO modified
In the presence of dNTP, the assay showed a surprising increase in dsDNA concentration with the unprotected and a somewhat smaller increase with the PTO-protected templates, while the negative control containing no DNA polymerase did not show any significant change (Fig. 5b). Since no family B DNA polymerase was ever reported to exhibit a 5′–3′ exonuclease activity so far, we expected to see no variation in dsDNA level over time. The hypothesis that the intriguing increase in dsDNA concentration could be explained by the extension of partially extended chains in the PCR product could be dismissed because nucleotide incorporation was an order of magnitude slower than when a regular primed template was present. In addition, during the course of the experiment, the amount of dsDNA increased by 80%, which would indicate that as much as half of the PCR product would have had to be single-stranded at the start of the experiment, a highly unlikely scenario considering the small size of the amplified sequence and the long extension times used during its amplification. An extensive investigation of this phenomenon was outside the scope of this report and will be the object of future studies. However, it is strongly suspected that the observed result is due to the extension of self-primed 3′ ends. Because of the high assay temperature, the DNA present is probably in dynamic equilibrium between different secondary structures and the 3′ ends can form transient hairpins which can become templates for regular DNA polymerizing activity. Extension of such priming sites requires simultaneous strand displacement, which would explain the lower extension rate.
While the exonuclease assay described here allowed the demonstration of a 3′–5′ exonuclease activity in TthiPolB, no conclusion could be drawn on a possible 5′–3′ activity because of the unexpected DNA synthesis that was observed (a decrease in dsDNA amount with the unprotected template and no variation with the PTO-protected template, both in presence of dNTP, would have indicated the presence of a 5′–3′ exonuclease activity). However, such an activity in TthiPolB is very unlikely because of its homology to family B DNA polymerases with known structures (Hashimoto et al. 2001; Hopfner et al. 1999; Rodriguez et al. 2000). In contrast with family A DNA polymerases which contain both 5′–3′ and 3′–5′ exonuclease domains, known family B DNA polymerases only have a single exonuclease domain of 3′–5′ directionality.
Kinetic parameters
Apparent Michaelis constants were determined for each of the two substrates, annealed primer-template DNA and dNTP, in the presence of an excess of the other substrate (Fig. 6a, b). The K m for the DNA parameter, 0.66 ± 0.02 nM, was an order of magnitude higher than that of other archaeal family B DNA polymerases while being significantly lower than that of Taq polymerase (Table 1). However, the K m for the dNTP parameter, 10.5 ± 1.5 μM each, was lower than any published constant for a thermophilic DNA polymerase.

Determination of TthiPolB’s kinetic parameters. Double-reciprocal (Lineweaver–Burk) plots were used to determine apparent Michaelis constants K m DNA (a) and K dNTP m (b). Polymerization rates were measured as a function of DNA (a) and dNTP (b) concentrations under optimal reaction conditions and an excess of the other substrate. Extension rate of TthiPolB was measured (c). Complete replication of the circular M13mp18 template was defined as 100% template extension. Relative template extensions were measured with different molar ratios of TthiPolB to DNA template, which values are indicated in the symbol legend. Within 1 min of incubation, 80% of the single-stranded template was converted into dsDNA, under conditions of large excess of DNA polymerase
As opposed to the determination of K m values, which does not require knowledge of enzyme concentration nor even precise activity measurements (relative activity can suffice), it is crucial to have a high precision in both the knowledge of enzyme molar concentration and the ability to measure absolute activity values in order to determine the k cat accurately from Michaelis–Menten kinetic experiments. We devised an experiment that would allow a more reliable estimation of the k cat by being completely independent from a precise knowledge of enzyme or DNA concentration. When a template of known size (the primed M13 template has a 7,215 nucleotide long single-stranded region) is incubated with excess amounts of polymerase and variation of dsDNA concentration is measured over time, the concentration of dsDNA is expected to reach a plateau corresponding to the conversion of all available single stranded DNA (ssDNA) into dsDNA. The height of the plateau should be independent of polymerase concentration, while the slope of the initial part of the curve should increase with increasing enzyme concentration until reaching a maximum corresponding to the polymerase’s maximal extension rate (in other words, the k cat). Under these conditions, the extent of conversion from ssDNA to dsDNA can reasonably be expressed in terms of template length rather than template concentration. The experimental results (Fig. 6c) indicate that at least 80% of the template 7,215 nucleotides were converted into dsDNA after 1 min, which corresponds to an extension rate of 96 nucleotides per second. TthiPolB is therefore among the fastest thermophilic DNA polymerases described so far (Table 1).
PCR performance
TthiPolB was tested successfully in various PCR applications using templates such as genomic DNA, plasmid DNA, PCR products, E. coli colonies and yeast colonies. It performed remarkably well in cloning applications using degenerate primers including the RSO-PCR technique (Sarkar et al. 1993; Weber et al. 1998), even when commercial DNA polymerases such as Taq or KOD failed to generate PCR products. Examples of PCR amplification are shown in Fig. 7.

PCR with TthiPolB. DNA fragments of various sizes were PCR amplified with TthiPolB using genomic DNA (lanes 1–2 and 4–7) and plasmid DNA (lane 3) as a template. A total of 5 μl of each product were analyzed on a 0.8% agarose gel stained with ethidium bromide and visualized under UV illumination. Lane M: 200 bp DNA ladder. Fragment sizes for lanes 1–7: 604, 904, 993, 1,504, 2,225, 2,784 and 3,047 bp, respectively. PCR conditions were as described in the main text. Extension time: 30 s (lanes 1–4), 90 s (lanes 5–7)
Conclusion
The family B DNA polymerase gene of T. thioreducens encodes a 774 aa DNA polymerase and a 537 aa intein. The DNA polymerase is most active in 10 mM Tris pH 7.8–9.0 (measured at 25°C), 60 mM KCl, 1.5–3.0 mM MgCl2, 0.1% Triton X-100 and at temperatures between 72 and 76°C (under laboratory conditions). It has a proofreading 3′–5′ exonuclease activity. Its fidelity is 1.7 times higher than that of Taq polymerase and within the range of other DNA polymerases from the Thermococcus genus while being significantly lower than the high-fidelity polymerase Pfu. Its thermal stability is somewhat lower than that of other archaeal DNA polymerases but higher than that of Taq polymerase. The apparent Michaelis constant for the dNTP parameter is the lowest among published values for thermostable DNA polymerases and the extension rate is among the highest. TthiPolB appears to be more comparable to Taq than to most commercially available archaeal DNA polymerases in terms of robustness, versatility and reliability, while exhibiting higher extension rate, fidelity and thermal stability. It could therefore advantageously replace Taq in most PCR applications. Some of the properties of TthiPolB are summarized in Table 1 and compared with published results for other thermostable DNA polymerases.
References
Agrawal S, Goodchild J, Civeira MP, Thornton AH, Sarin PS, Zamecnik PD (1988) Oligodeoxynucleoside phophoramidates and phosphorothioates as inhibitiors of human immunodeficiency virus. Proc Natl Acad Sci USA 85:7079–7083
Blöndal T, Thorbjarnardóttir SH, Kieleczawa J, Hjörleifsdóttir S, Kristjánsson JK, Einarsson JM, Eggertsson G (2001) Cloning, sequence analysis and functional characterization of DNA polymerase I from the thermophilic eubacterium Rhodothermus marinus. Biotechnol Appl Biochem 34:37–45
Bonch-Osmolovskaya E, Svetlichny V, Ankenbauer W, Schmitz-Agheguian G, Angerer B, Ebenbichler C, Laue F (1996) Eur Patent Appl EP 0 834 570 A1
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram qualities of protein using the principle of protein-dye binding. Anal Biochem 72:248–254
Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM et al (1998) Crystallography & NMR System: a new solftware suite for macromolecular structure determination. Acta Cryst D 54:905–921
Cambon-Bonavita MA, Schmitt P, Zeiger M, Flaman JM, Lesongeur F, Raguenes G, Bindel D, Frisch N, Lakkis Z, Dupret D, Barbier G, Querellou J (2000) Cloning, expression, and characterization of DNA polymerase I from the hyperthermophilic archaea Thermococcus fumicolans. Extremophiles 4:215–225
Cann IKO, Ishino Y (1999) Archael DNA replication: identifying the pieces to solve a puzzle. Genetics 152:1249–1267
Cline J, Braman JC, Hogrefe HH (1996) PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res 15:3546–3551
Dietrich J, Schmitt P, Zeiger M, Preve B, Rolland JL, Chaabihi H, Gueguen Y (2002) PCR performance of the highly thermostable proof-reading B-type DNA polymerase from Pyrococcus abyssi. FEMS Microbiol Lett 217:89–94
Flaman JM, Frebourg T, Moreau V, Charbonnier F, Martin C, Ishioka C, Friend SH, Iggo R (1994) A rapid PCR fidelity assay. Nucleic Acids Res 22:3259–3260
Flaman JM, Frebourg T, Moreau V, Charbonnier F, Martin C, Chappuis P, Sappino AP, Limacher IM, Bron L, Benhattar J et al (1995) A simple functional assay for screening cell lines, blood and tumors. Proc Natl Acad Sci USA 92:3963–3967
Goddard MR, Burt A (1999) Recurrent invasion and extinction of a selfish gene. Proc Natl Acad Sci USA 96:13880–13885
Gueguen Y, Rolland JL, Lecompte O, Azam P, Le Romancer G, Flament D, Raffin JP, Dietrich J (2001) Characterization of two DNA polymerases from the hyperthermophilic euyarchaeon Pyrococcus abyssi. Eur J Biochem 268:5961–5969
Hashimoto H, Nishioka M, Fujiwara S, Takagi M, Imanaka T, Inoue T, Kai Y (2001) Crystal structure of DNA polymerase from hyperthermophilic archaeon Pyrococcus kodakaraensis KOD1. J Mol Biol 306:469–477
Henneke G, Flament D, Hübscher U, Querellou J, Raffin JP (2005) The hyperthermophilic eurychaeota Pyrococcus abyssi likely requires the two DNA polymerases D and B for DNA replication. J Mol Biol 350:53–64
Hirata R, Ohsumi Y, Nakano A, Kawasaki H, Suzuki K, Anraku Y (1990) Molecular structure of gene, VMA1, encoding the catalytic subunit of H (+)-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J Biol Chem 265:6726–6733
Hogrefe HH, Cline J, Lovejoy AE, Nielson KB (2001) DNA polymerases from hyperthermophiles. Methods Enzymol 334:91–116
Hopfner KP, Eichinger A, Engh RA, Laue F, Angerer B (1999) Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc Natl Acad Sci USA 96:3600–3605
Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68
Ito J, Braithwaite DK (1991) Compilation and alignment of DNA polymerase sequences. Nucleic Acids Res 19:4045–4057
Kahler M, Antranikian G (2000) Cloning and characterization of a family B DNA polymerase from the hyperthermophilic crenarchaeon Pyrobavulum islandicum. J Bacteriol 182:655–663
Kane PM, Yamashiro CT, Wolczyk DF, Neff N, Goebl M, Stevens TH (1990) Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H(+)-adenosine triphosphatase. Science 250:651–657
Kelman Z, Pietrokovski S, Hurwitz J (1999) Isolation and characterization of a split B-type DNApolymerase from the archaeon Methanobacterium thermoautotrophicum deltaH. J Biol Chem 274:28751–28761
Kong H, Kucera RB, Jack WE (1993) Characterization of a DNA polymerase from the hyperthermophile archaea Thermococcus litoralis. Vent DNA polymerase, steady state kinetics, thermal stability, processivity, strand displacement, and exonuclease activities. J Biol Chem 268:1965–1975
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244–1245
Kunkel TA, Eckstein F, Mildvan AS, Koplitz RM, Loeb LA (1981) Deoxynucleoside [1-thio] triphosphates prevent proofreading during in vitro DNA synthesis. Proc Natl Acad Sci USA 78:6734–6738
Lineweaver H, Burk D (1934) The determination of enzyme dissociation constants. J Am Chem Soc 56:658–666
Lundberg KS, Shoemaker DD, Adams MW, Short JM, Soge JA, Mathur EJ (1991) High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene 108:1–6
Nakamura Y, Gojobori T, Ikemura T (2000) Codon usage tabulated from the international DNA sequence databases: status for the year 2000. Nucleic Acids Res 28:292
Niehaus F, Frey B, Antranikian G (1997) Cloning and characterization of a thermostable alphaa-DNA polymerase from the hyperthermophilic archaeon Thermococcus sp. TY. Gene 204:153–158
de Noronha CM, Mullins JI (1992) Amplimers with 3′-terminal phosphorothioate linkages resist degradation by vent polymerase and reduce Taq polymerase mispriming. PCR Methods 2:131–136
Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kundel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S et al (2001) The Y-family of DNA polymerases. Mol Cell 8:7–8
Perler FB (2002) InBase: the intein database. Nucleic Acids Res 30:383–384
Pietrokovski S (2001) Intein spread and extinction in evolution. Trends Genet 17:465–472
Pikuta EV, Marsic D, Itoh T, Bej AK, Tang J, Whitman WB, Ng JD, Garriott OK, Hoover RB (2007) Thermococcus thioreducens sp. nov., a novel hyperthermophilic, obligately sulfur-reducing archaeon from a deep-sea hydrothermal vent. Int J Syst Evol Microbiol 57:1612–1618
Putney SD, Benkovic SJ, Schimmel PR (1981) A DNA fragment with an alpha-phosphorothiate nucleotide at one end is assymmetrically blocked from digestion by exonuclease III and can be replicated in vivo. Proc Natl Acad Sci USA 78:7350–7354
Riley J, Butler R, Ogilvie D, Finniear R, Jenner D, Powell S, Anand R, Smith JC, Markham AF (1990) A novel, rapid method for the isolation of terminal sequences from yeast artificial chromosome (YAC) clones. Nucleic Acids Res 18:2887–2890
Rodriguez AC, Park HW, Mao C, Beese LS (2000) Crystal structure of a pol alpha family DNA polymerase from the hyperthermophilic archaeon Thermococcus sp. 9° N-7. J Mol Biol 299:447–462
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual , 2nd edn. Cold Spring Harbor, NY
Sarkar G, Turner RT, Bolander ME (1993) Restriction-site PCR: a direct method of unknown sequence retrieval adjacent to a known locus by using universal primers. PCR Methods Appl 2:18–322
Seville M, West AB, Cull AG, McHenry CS (1996) Fluorometeric assay for DNA polymerases and reverse transcriptases. BioTechniques 21:664–672
Skerra A (1992) Phosphorothioate primers improve the amplification of DNA sequences by DNA polymerases with proofreading activity. Nucleic Acids Res 20:3551–3554
Southworth MW, Kong H, Kucera RB, Ware J, Jannasch HW, Perler FB (1996) Cloning of thermostable DNA polymerases from hyperthermophilic marine Archaea with emphasis on Thermococcus sp. 9° N-7 and mutations affecting 3′–5′ exonuclease activity. Proc Natl Acad Sci USA 93:5281–5285
Stothard P (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 28:1102–1104
Takagi M, Nishioka M, Kakihara H, Kitabayashi M, Inoue H, Kawakami B, Oka M, Imanaka T (1997) Charaterization of DNA polymerase from Pyrococcus sp. strain KOD1 and its application to PCR. Appl Environ Microbiol 63:4504–4510
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tveit H, Kristensen T (2001) Fluorescence-based DNA polymerase assay. Anal Biochem 289:96–98
Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA (1997) Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell 89:1087–1099
Weber KL, Bolander ME, Sarkar G (1998) Rapid acquisition of unknown DNA sequence adjacent to a known segment by multiplex restriction site PCR. BioTechniques 25:415–419
Acknowledgments
We express our gratitude to Jim Hudson for his generous contribution to our structural genomics program. This project was funded by NSF STTR-0611274 and the National Aeronautics and Space Administration (NASA) Consortium for Materials Development on Space (CMDS) (NCC8-243). We are also grateful to Dr. Elena Pikuta for providing T. thioreducens biomass, Leland Cseke for helping with sequencing and to Tom Kristensen for valuable discussion and advice concerning the DNA polymerase assay. Special thanks go to Owen Garriott for originating this study by collecting hydrothermal vent samples and for his continuous support and guidance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. Matsunaga.
Rights and permissions
About this article
Cite this article
Marsic, D., Flaman, JM. & Ng, J.D. New DNA polymerase from the hyperthermophilic marine archaeon Thermococcus thioreducens . Extremophiles 12, 775–788 (2008). https://doi.org/10.1007/s00792-008-0181-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-008-0181-7