
Abstract
Thermophilic bacteria are of great value for industry and research communities. Unfortunately, the cellular processes and mechanisms of these organisms remain largely understudied. In the present study, we investigate how the inactivation of adenylate kinase (AK) affects the adenine nucleotide homeostasis of a gram-positive moderate thermophile, Geobacillus stearothermophilus strain NUB3621-R. AK plays a major role in the adenine nucleotide homeostasis of living cells and has been shown to be essential for the gram-negative mesophile Escherichia coli. To study the role of AK in the maintenance of adenylate energy charge (EC) and cell viability of G. stearothermophilus, we generated a recombinant strain of this organism in which its endogenous gene coding for the essential protein adenylate kinase (AK) has been replaced with the adk gene from the mesophile Bacillus subtilis. PCR, DNA sequencing and Southern analysis were performed to confirm proper gene replacement and preservation of neighboring genes. The highest growing temperature for recombinant cells was almost 20°C lower than for wild-type cells (56 vs. 75°C). This temperature-sensitive phenotype was secondary to heat inactivation of B. subtilis AK, as evidenced by enzyme activity assays and EC measurements. At higher temperatures (65°C), recombinant cells also had lower EC values (0.09) compared to wild-type cells (0.45), which reflects a disruption of adenine nucleotide homeostasis following AK inactivation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thermophilic bacillus species can be isolated from a wide range of environments and are important contaminants in the food industry (Sharp 1992) and a precious source of temperature stable enzymes to biotechnology companies (Vieille et al. 1996). The development of a genetic system for members of this group (Imanaka et al. 1982; Wu and Welker 1989; Vallier and Welker 1990) will allow the study of key cellular processes in gram-positive thermophilic organisms. Geobacillus stearothermophilus NUB3621-R is a moderate thermophilic bacillus with an optimal growth temperature of 65°C and a wide growth range of 45–73°C (Wu and Welker 1989). Modification of Chang and Cohen’s (1989) protoplast/polyethylene glycol protocol developed for Bacillus subtilis enabled Welker and coworkers to transform G. stearothermophilus strain NUB3621-R with plasmid DNA (Wu and Welker 1989) and build a chromosome map for this organism (Vallier and Welker 1990).
We used G. stearothermophilus strain NUB3621-R as a model organism to study the role of adenylate kinase (AK) in adenine nucleotide homeostasis in gram-positive thermophiles. Adenine nucleotide homeostasis dictates the energy levels available to living cells and therefore tight control is crucial to cellular metabolism. Atkinson defined adenylate energy charge (EC) in living cells as EC = ([ATP] + 0.5 [ADP]/([ATP] + [ADP] + [AMP]) (Atkinson 1968).
EC directly measures the amount of energy stored in adenine nucleotide pools accessible for cellular metabolism (Atkinson 1968; Chapman et al. 1971). In gram-negative bacteria, EC values in growing cells have been shown to be regulated by adenylate kinase (AK – E.C. 2.7.4.3), a small, ubiquitous enzyme (Glembotski et al. 1981). AK catalyzes the reaction Mg+2 ATP + AMP ↔ Mg+2 ADP + ADP.
AK is essential for the gram-negative bacteria Escherichia coli (Cousin and Buttin 1969; Glembotski et al. 1981; Haase et al. 1989). E. coli strains expressing temperature-sensitive AK mutations show deficient growth and decreased cell viability at nonpermissive temperatures (Cousin and Buttin 1969; Glembotski et al. 1981; Haase et al. 1989). These phenotypes have been correlated to decreased EC values, suggesting that AK regulation of adenine nucleotide homeostasis is essential to cellular growth and survival in E. coli.
Although adenylate kinase is ubiquitous and its function is well conserved, its primary, tertiary and quaternary structures can vary strikingly among prokaryotes (Glaser et al. 1992; Ferber et al. 1997; Vonrhein et al. 1998; Criswell et al. 2003; Munier-Lehmann et al. 1999). Moreover, in prokaryotic organisms the adk gene can be found in different genomic loci (Kath et al. 1993; Suh et al. 1996; Blattner et al. 1997; Hansmann and Martin 2000), which suggests that adk gene expression may vary among these organisms. The observed differences in AK structure, activity and possibly expression suggest that adenine nucleotide homeostasis is also controlled differently in these organisms and is reflected by widely varying values of EC (Chapman et al. 1971).
In this study, we describe the production of a genetically modified strain of G. stearothermophilus, in which the endogenous adk gene has been replaced with the homolog from the mesophile B. subtilis. The gene replacement was performed through homologous recombination and produced a temperature-sensitive strain of G. stearothermophilus NUB3621-R. The observed temperature-sensitive phenotype was linked to the inactivation of B. subtilis AK at increased temperatures and subsequent failure to maintain high EC levels necessary for growth.
Methods
Bacterial strains, growth conditions, and DNA plasmids
Bacterial strains and DNA plasmids used in this work are listed in Table 1. E. coli cultures were grown in LB medium. G. stearothermophilus NUB3621-R (kind gift of Dr. N. Welker, Northwestern University, Evanston, IL, USA) cultures were grown in a modified LB medium (mLB) (Chen et al. 1986). Alternatively, G. stearothermophilus NUB3621-R cells were cultivated in TS medium (4% tryptone, 0.5% NaCl, supplemented with 0.6 mM MgSO4·7H2O, 0.9 mM CaCl2·2H2O, 0.04 mM FeSO4·7H2O and 1.1 mM nitrilotriacetic acid) (N. Welker, personal communication, Northwestern U., Evanston, IL, USA). Solid medium was prepared by the addition of 15 g/l of agar (Sigma). Tryptone and yeast extracts were purchased from Difco.
Transformation of bacteria with plasmid DNA and NUB3621-R:ThEV selection
Chemically competent E. coli was transformed by the heat shock method (Hanahan et al. 1991). Transformation of G. stearothermophilus NUB3621-R protoplasts with p12ThEV plasmid DNA was performed according to the protocol developed by Wu and Welker (1989). To cure the plasmid from recombinant cells, transformants were grown in liquid TS cultures without antibiotics. Cell samples were diluted and spread on solid TS media containing chloramphenicol (7 mg/l). Cells growing on chloramphenicol were then replica-plated to TS plates containing either tetracycline (5 mg/l) or chloramphenicol. The tetracycline plates were incubated at 65°C and the chloramphenicol plates at 50°C overnight. Colonies growing on chloramphenicol but not on tetracycline containing media were selected for PCR and Southern blot screening.
Highest growth temperature determination for NUB3621-R:ThEV cells
NUB3621-R:ThEV cells were grown in solid TS media complemented with chloramphenicol (7 mg/l). After 16 h at 50°C, cells were streaked onto solid TS media complemented with chloramphenicol (7 mg/l) and incubated at various temperatures in a Fisherbrand Scientific Isotemp Forced Air Incubator 600 (± 0.1°C). Plates were screened for colony growth after 48 h.
DNA manipulation and sequencing
NUB3621-R genomic DNA was isolated using QIAGEN’s genomic-tip 20/G kit and used as template for PCR reactions with degenerate primers for the ribosomal genes rplO and rpmJ with primers L15_deg_F (5′-CGGGATCCGGAGTTCGTCCGT GGTTYGARGGNGG-3′) and L36_deg_R (5′-CGGGATCCTGTCGAATCACTTTGCAYTTNTCRC-3′). The PCR product was sequenced by Lone Star Labs (Houston, TX, USA), using primers SecYDEgF (5′-CCGGAATTCATHCCNGTNATHTTYGCN-3′) and MAPDEgR (5′-GGGAAGCTTRTT NACCATNGGYTCDAT-3′). The sequence information (gene bank # AY729037) was used to design primers to amplify NUB3621-R secY, adk and map genes. All PCR reactions were performed on a Primus PCR system (MWG Biotech) using the Expand Long Template PCR System (Roche Applied Science).
Plasmid DNA was isolated with QUIAGEN’s Plasmid Maxi Kit. Restriction enzymes from Promega and New England BioLabs were routinely used according to the manufacturer’s instructions. DNA ligation reactions were performed overnight at 20°C using Promega’s T4 DNA Ligase enzyme.
Southern blots
Approximately 5.0 μg of total DNA isolated from NUB3621-R and NUB3621-R:ThEV cells was treated with 10 units of EcoRI and HindIII at 37°C overnight and 2 ng of p12ThEV was also similarly treated. The DNA samples were then separated on a 0.6% agarose gel. Transfer of DNA to Byodine B membrane (Pall Life sciences) was performed according to the manufacturer’s instructions. The same nylon membrane was used for hybridization with different probes. Hybridization and stripping procedures followed the membrane manufacturer’s protocol. DNA probes were generated using the PCR DIG Probe synthesis kit and were detected using the DIG Nucleic Acid Detection Kit (Roche Applied Science), according to the manufacturer’s instructions.
Southern blot probes
The probes used in the southern blots were amplified from plasmid DNA with the following primers: B. subtilis adk, adk_f and adk_r; G. stearothermophilus NUB3621-R adk, Nub_ADK_F_NdeI and Nub_ADK_R_BamHI; tet gene, Tet_ATG_F_EcoRI and Tet_STOP_R_EcoRI; cat gene, dCATf and RC6.
Oligonucleotide primers
The oligonucleotide primers used in this work were purchased from Integrated DNA Technologies (Coralville, IA, USA) and MWG Biotech (High Point, NC, USA). Oligonucleotide primers used to amplify secY and map genes from genomic DNA isolated from NUB3621-R were NUB_LSecY_F_SacI (5′-CCG AGC TCG GTT TTA ACA GCC GGA ACG-3′), Rec_SecY_R (CTG CAG TTC GGG ATC CCC TCG CTC CCC CTC AG), NUB_LMap_R_SphI (ACA TGC ATG CAG TGT TAC GAA AAA CTG AAT GAA CAC C 3′), Rec_Map_F (5′-GGA TCC CGA ACT GCA TGA GCT TCT CGG AGG-3′). Primers dCATf (5′-GCT CTA GAC ACT TTA GAT AAA AAT TTA GGA GGC-3′) and RC6 (5′-TTT CTG CAG TTA TAA AAG CCA GTC ATT AGG-3′) were used to amplify the cat gene from plasmid pPW15(7) (39). Primers adk_F (5′-CCG CGG ATC CAT GAA CTT AGT CTT AAT GGG-3′) and adk_r (5′-GCT CTA GAT CAT TTT TTT AAT CCT CCA AG-3′) were used to amplify the B. subtilis adk gene from the pETADK vector. Primer WAC (5′-GGT TTT ATT CAG GCG CTT GG-3′) was used, in combination with RC6, to confirm through PCR the homologous recombination event. Primers for Southern blots were Nub_ADK_F_NdeI (5′-GGA ATT CCA TAT GAA TTT AGT ACT AAT GGG TTT GC-3′); Nub_ADK_R_BamHI (5′-CGG GAT CCT TAT TGT AAT CCT CCG AGA AGC-3′); Tet_ATG_F_EcoRI (5′-CGA ATT CCC TAT TCA CAA TCG AAT TTA CGA CAC AAC-3′); Tet_STOP_R_EcoRI (5′-GGA ATT CCC CTT TGA GAA TGT TTA TAT ACA TTC AAG G-3′).
Total adenine nucleotide extraction
Wild-type and recombinant NUB3621-R cells were cultivated in liquid TS media containing rifamycin (5 mg/l) in a New Brunswick Scientific BF3000 Benchtop fermenter system. Cells were grown at 50°C up to an O.D.590 of 0.5. A 50 ml aliquot of cell culture was then transferred to a 250-ml Erlenmeyer and placed into a New Brunswick Innova 4230 shaker set at 65°C and 255 rpm for the specified amount of time. For the transient increase in temperature, cells were kept in the fermenter throughout the duration of the experiment. Cell culture samples (1.0 ml, in triplicates) were transferred to 9.0 ml of boiling Tris buffer (100 mM Tris acetate pH 7.75, 4 mM EDTA). The samples were kept at 100°C for 2 min. Samples were then chilled on ice, centrifuged (2 min at 5,000 rpm at 4°C) and the supernatant transferred to a new tube. Growth medium samples were obtained by passing 10 ml of cell culture through a 0.2-μm filter (Millipore) and treated as described above. Total adenine nucleotide samples were kept on ice until assayed with the luciferase activity assay. Two acid extraction methods (Swedes et al. 1975; Kahru et al. 1990) were also employed.
Total protein extraction
Total protein was extracted as described previously (Swedes et al. 1975). An amount of 750 μl of cell culture was added to 750 μl of an ice cold 10% HClO4 solution. Samples were then centrifuged (10 min, 14,000 rpm) and the pellet was brought back into solution with 100 μl of 50 mM phosphate buffer pH 7.3. Protein concentrations were estimated using the Bio-Rad protein Assay (Bio-Rad).
Sample preparation and the luciferase activity assay
Sample preparation for the determination of ATP, ADP and AMP concentrations was carried out as described by Chapman and colleagues (1971). ATP concentrations were determined by means of the luciferase reaction. Firefly (Photinus pyralis) luciferase (1.13.12.7) and luciferin (D-[-]-2-(6′-hydroxy-benzothiazolyl)-Δ 2 -thiazoline-4-carboxylic acid) were purchased from Roche Applied Science. Sample preparation was performed according to the manufacturer’s instructions. An ATP sample of 50 µl was placed into a Greiner 96-well plate. Fifty microliters of luciferase solution (50 mM Tris acetate, pH 7.75, 2 mM EDTA, 60 mM DTT, 0.01% (w/v) BSA, 10 mM magnesium acetate, 35 μM D-luciferin and 0.05 μg of luciferase) was added to the samples and the plate was immediately placed on an Alpha Innotech Fluorchem 5500. The signal was integrated from 1 to 60 s and the ATP concentrations estimated through comparison with an ATP standard curve. AMP and ADP were converted to ATP (Chapman et al. 1971) prior to the luciferase experiments and their concentrations estimated as follows: [AMP] = ([ATP] + [ADP] + [AMP]) – ([ATP] + [ADP]); and [ADP] = ([ATP] + [ADP]) – [ATP]. All measurements were performed in triplicate.
Purification of AK
Adenylate kinase was purified from natural abundance as described in Barzu and Michelson (1983) (Barzu and Michelson 1983). Wild-type and recombinant NUB3621-R cells were grown for one night at 50°C and 200 rpm in 2 l of mLB medium. Cells were harvested by centrifugation (10 min at 5,000 rpm) and brought back into solution by addition of 20 ml of buffer A (50 mM Tris–HCl, pH 7.4, 1 mM MgCl2). Cells were kept on ice and lysed by sonication on a Branson Sonifier 250 (5×30 s pulses at 70% duty cycle and an output control of 7). Cell debris was removed by two rounds of centrifugation at 20,000 rpm for 20 min. The crude extract was filtered through a 0.2-μm filter (Millipore) and then applied in a Blue Sepharose 6 Fast Flow column (9.0×1.2 cm) (Amersham Biosciences) equilibrated in buffer A. The column was washed with buffer A until no protein could be detected in the flow through. AK was eluted by the addition of 0.5 mM of AP5A. Fractions containing ADK activity were concentrated using Vivaspin concentrators (10,000 MWCO—Vivascience) and applied to a HiLoad 16/60 Superdex 200 (Amersham Biosciences) column equilibrated in buffer B (Buffer A plus 100 mM NaCl). Fractions showing ADK activity were concentrated using Vivaspin concentrators (10,000 MWCO—Vivascience). Enzyme concentrations were estimated using the Bio-Rad Protein Assay. The enzyme was stored at −80°C.
AK activity assays
Adenylate kinase activity in the direction of ADP production was determined by an end point coupled assay. 45 μl of reaction buffer (25 mM phosphate buffer, pH 7.3, 5 mM MgCl2, 65 mM KCl, 1 mM DTT, 2 mM ATP) with 5 nM of NUB3621-R or NUB3621-R:ThEV AK was kept at indicated temperatures for 3 min in a Primus PCR system (MWG Biotech). The reaction was started by addition of 5 μl of a 20 mM AMP solution prepared in reaction buffer. A sample of reaction buffer without addition of AK (blank) was treated in the same manner as a control for AMP and ATP degradation at various temperatures. After 2 min, the reaction was stopped by adding 50 μl of an ice cold solution of 2 mM P1, P5 -di(adenosine-5′)pentaphosphate (Ap5A). The reaction mix was kept on ice. The secondary reaction was started by transferring 90 μl of the primary reaction to 710 μl of the NADH buffer (25 mM phosphate buffer, pH 7.3, 5 mM MgCl2, 65 mM KCl, 1 mM DTT, 1.0 mM phosphoenolpyruvate, 10 µm/ml of lactate dehydrogenase and 10 μm/ml of pyruvate kinase). The secondary reaction was allowed to proceed to completion at room temperature. ADP production by AK at various temperatures was estimated by subtracting the amount of NADH consumed from the NADH concentration found for the blank reaction.
Results
Characterization of the adk gene in NUB3621-R genome
As the thermophilic Bacilli group is phylogenetically diverse (Studholme et al. 1999), we determined the location and sequence of the adk gene and its flanking regions in the NUB3621-R genome. Based on the sequence information for strain S10 [Bacillus (Geobacillus) stearothermophilus Genome Sequencing Project http://www.genome.ou.edu/bstearo.html], we designed degenerate primers (L15_deg_F and L36_deg_R) to amplify the region in between the rplO and the rpmJ genes from NUB3621-R genomic DNA. Sequencing the region confirmed the location of adk between secY and map in NUB3621-R (gene bank #AY729037) and that the NUB3621-R adk gene is 73% identical to B. subtilis strain 168 adk (79% at the protein level) and 77% identical to G. stearothermophilus S10 adk (85% at the protein level).
Construction of the homologous recombination plasmid
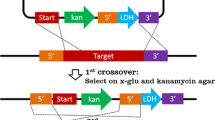
To study the impact of AK inactivation in the adenine nucleotide homeostasis of a gram-positive thermophile, we designed a homologous recombination plasmid (Fig. 1a) to knock out NUB3621-R’s endogenous adk gene and replace it with the B. subtilis adk gene, without major disturbances to the S10- spc-alpha region. A promoterless marker gene was also introduced, so that true recombinants could be readily screened.

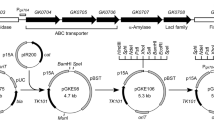
Homologous recombination cassette and p12ThEV construct. a Schematic representation of the homologous recombination cassette showing its gene organization: secY pre-protein translocase secY subunit; adkB.sb B. subtilis adenylate kinase; cat chloramphenicol acetyl-transferase; map methionine aminopeptidase; infA initiation factor IF-I. Relevant restriction sites are also shown. b p12ThEV construct showing the homologous recombination cassette (shown in a) and genes conferring antibiotic resistance to ampicillin (amp) and tetracycline (tet). c 1.0% agarose gel showing PCR products amplified from p12ThEV plasmid with oligonucleotide primers for secY (lane 1), B. subtilis adk (lane 2), cat (lane 3) and map/infA (lane 4) fragments found in the homologous recombination cassette. Lane 5 Molecular weight marker (1.0 μg of λ DNA treated with Pst I). Genes are depicted as boxes and RBSs as black bars. Approximate molecular weights are also shown
Based on the genome sequence of G. stearothermophilus NUB3621-R, we designed primers (NUB_LSecy_F_SacI, Rec_SecY_R, NUB_LMap_R_SphI and Rec_Map_F) to PCR amplify the last 1.0 kilobase pairs (kb) of the secY gene and a 1.0-kb region downstream from adk, consisting of the entire map gene and the 5′ portion of the infA gene. The putative ribosome binding sites found in this region (black boxes in Fig. 1a) were kept to minimize perturbations of the original gene organization and expression. We also designed primers to PCR amplify the cat gene (dCATf and RC6) from pPW15(7) (Wei and Stewart 1993) and the adk gene (adk_f and adk_r) from pETADK plasmid. All PCR fragments were sequentially cloned into pUC19 (Yanisch-Perron et al. 1985; Lin-Chao et al. 1992). The homologous recombination cassette was also cloned into an E. coli/G. stearothermophilus shuttle vector, pSTE12 (Narumi et al. 1992), resulting in the p12ThEV construct (Fig. 1b). The p12ThEV plasmid confers ampicillin resistance in E. coli and tetracycline in G. stearothermophilus. It also carries a promoterless copy of the cat gene.
Recombinant strain isolation and characterization
In order to build a genetically stable recombinant strain, we had to verify that the gene replacement had taken place through a unique double homologous recombination event and that the p12ThEV plasmid had been lost. True recombinants were cells that underwent homologous recombination and no longer carried the p12ThEV plasmid. In these cells, adk from B. subtilis should be stably expressed. We also expected to observe a temperature-sensitive phenotype for the recombinant cells, since B. subtilis AK has a denaturation temperature (Tm) of 50.7°C (Glaser et al. 1992). Furthermore, recombinant cells should be chloramphenicol resistant. The stable introduction of the homologous recombination cassette allows expression of the promoterless cat gene to be driven by the endogenous promoter sequences found in the S10- spc-alpha region. Recombinant cells cured for p12ThEV plasmid should also be tetracycline sensitive.
Cells with the expected phenotype for a true recombinant were isolated after extensive selection using replica plates complemented with tetracycline or chloramphenicol and the incubation at different temperatures. The temperature-sensitive, chloramphenicol-resistant and tetracycline-sensitive cells were called NUB3621-R:ThEV. We also determined the highest growing temperature for NUB3621-R:ThEV cells to be 56°C.
We performed PCR, DNA sequencing and Southern blot analysis to confirm that the isolated recombinant strain had, in fact, the correct genotype. NUB3621-R genomic DNA sequencing revealed the presence of HindIII and EcoRI recognition sites in the vicinity of the homologous recombination cassette. We used these enzymes to digest total DNA isolated from NUB3621-R and NUB3621-R:ThEV cells. The same nylon membrane containing NUB3621-R, NUB3621-R:ThEV and plasmid p12ThEV DNA treated with EcoRI and HindIII was probed for the presence of B. subtilis adk, cat and tet genes. As shown in Fig. 3a, the adk probe recognized a single DNA fragment in the recombinant strain (lane 2) and a fragment of the same size in the plasmid DNA control (lane 3). A similar result was obtained when cat was used as a probe (Fig. 3b, lanes 2 and 3). The tet probe recognized a fragment present in the plasmid, but failed to recognize any fragments in the recombinant strain sample, indicating that NUB3621-R:ThEV cells do not carry the p12ThEV plasmid. Wild type NUB3621-R genomic DNA was not recognized by any of the utilized probes.
DNA sequencing analysis of the region between secY and map genes confirmed the insertion of the B. subtilis adk and cat genes in the genome of NUB3621-R:ThEV cells. It also showed that none of the genes in this region had any mutations.
ADK activity in NUB3621-R:ThEV cells
Insertion of the recombination cassette into the genome should drive the expression of B. subtilis AK at comparable levels found for NUB3621-R AK in wild-type cells. Purification of AK from natural abundance in wild-type and recombinant cells confirmed that both bacterial strains express AK in similar amounts (Table 2). Previous experiments have shown that B. subtilis AK has a T m of 50.1°C, 25°C lower than its counterpart from G. stearothermophilus (Glaser et al. 1992). So it was expected that the AK enzyme isolated from NUB3621-R:ThEV should lose activity at a significantly lower temperature than wild-type NUB3621-R AK. Activity assays performed to estimate the rate of ADP generation from ATP and AMP by AK at different temperatures showed that, as expected, AK purified from NUB3621-R:ThEV cells is more temperature sensitive than AK isolated from NUB3621-R wild-type cells (Fig. 4). NUB3621:ThEV AK reached its maximum activity at 60°C and was dramatically reduced at higher temperatures. In contrast, AK isolated from wild-type NUB3621-R cells showed an activity maximum at 75°C (Fig. 4).
Adenylate levels in NUB3621-R:ThEV cells
The physiological role of AK is to maintain the balance of adenine nucleotide levels. The ratios between different adenine nucleotides are directly reflected in the cell’s EC, which can be used to measure a cell’s energy pool (Atkinson 1968). Thus, in order to relate the observed temperature-sensitive phenotype of NUB3621-R:ThEV cells with the physiological role of AK, we measured the adenylate EC of both wild-type and NUB3621-R:ThEV cells under various growth conditions. In order to estimate EC, we had to extract total adenine nucleotide from cells. Although two other extraction methods were attempted, we found that for NUB3621-R cells the boiling buffer method performed the best. As can be seen in Table 3, both NUB3621-R:ThEV and wild-type cells had similar energy charges at 50°C. EC values for wild-type cells kept at 65°C for 30 min reached 0.45 and were maintained at high levels even after 1 h at 65°C (EC = 0.41). Total protein levels also increased for wild-type NUB3621-R cells kept at 65°C (Table 3). Total protein levels found for NUB3621-R:ThEV cells kept at 65°C for 30 min were somewhat increased when compared to cells growing at 50°C (1.40 mg/ml of culture vs. 1.07 mg/ml of culture), but fell after 60 min at 65°C (0.61 mg/ml of culture). High concentrations of adenine nucleotides found in the media prohibited accurate EC measurements for NUB3621-R:ThEV cells kept at 65°C for extended periods of time. In contrast, these high background levels were not observed for wild-type cells kept at 65°C.
To investigate the capacity of NUB3621-R:ThEV cells to recover EC, we also investigated the changes in EC after a transient increase in temperature. Cells growing at 50°C were kept at 65°C for 20 min and the temperature lowered to 50°C again. The EC for NUB3621-R:ThEV and wild-type cells was measured before temperature elevation, 20 min after the temperature elevation to 65°C, and 20 min after the temperature was returned to 50°C. As can be seen in Table 3, wild-type cells showed an increase in EC to 0.43 after 20 min at 65°C. Twenty minutes after the temperature was returned to 50°C, EC values of the wild type cells were similar (0.22) to its initial value of 0.20. Total protein levels for wild-type cells also increased after the temperature was raised to 65°C (1.91 mg/ml of culture), and, after 20 min at 50°C, decreased to levels similar to the initial ones. NUB3621-R:ThEV EC decrease from 0.16 at 50°C to 0.09 after 20 min at 65°C. EC values could not be accurately measured after the cells were returned to 50°C due to the high concentrations of adenine nucleotides found in the media. Total protein levels for NUB3621-R:ThEV cells remained constant throughout the experiment (Table 3). NUB3621-R:ThEV’s cell viability was also investigated after a transient increase in temperature. NUB3621-R:ThEV and wild-type NUB3621-R cell aliquots for every experimental condition described above were serially diluted and spread on solid media. The plates were then incubated for 24 h at 50°C. For wild-type cells, no significant decrease in cell viability could be observed after incubation at the high temperatures. On the other hand, only 0.001% of NUB3621-R:ThEV cells treated in similar fashion were able to grow on solid media after 24 h at 50°C.
Discussion
We replaced the endogenous G. stearothermophilus adk gene with that of B. subtilis adk using homologous recombination. Recombinant NUB3621-R:ThEV cells did not show any noticeable phenotype other than the inability to grow at elevated temperatures (Fig. 2). This suggests that the homologous recombination event did not disturb the expression or organization of neighboring essential genes secY, map and infA (Shiba et al. 1984; Chang et al. 1989; Breitling et al. 1994; Cummings and Hershey 1994). Our results from PCR and Southern blot analysis have shown that NUB3621-R:ThEV cells carry a single, stable genomic copy of B. subtilis adk (Fig. 3).

Temperature-sensitive phenotype of NUB3621-R:ThEV cells. Recombinant NUB3621-R:ThEV and wild-type NUB3621-R cells were streaked on TS solid media supplemented with the indicated antibiotics. Plates were kept 24 h at the indicated temperatures. Wild-type NUB3621-R cells are rifamycin resistant and grow in the temperature range of 45–75°C. Following gene replacement of its endogenous adk gene with plasmid p12hEV recombinant cells should be rifamycin and chloramphenicol resistant and have a temperature sensitive phenotype

Southern blot analysis of NUB3621-R:ThEV genomic DNA. a Total DNA from NUB3621-R wild-type cells (WT); total DNA from NUB3621-R:ThEV cells (Rec) and plasmid p12ThEV control DNA (Pl) were treated with HindIII and EcoRI, separated on an 0.6% agarose gel and transferred to a nylon membrane. The membrane was then hybridized with DIG labeled probes for B. subtilis adk, cat or tet genes (depicted as boxes on top). b Schematic representation of NUB3621-R:ThEV genome after gene replacement. Genes are depicted as boxes and RBSs as black bars. The approximate locations of relevant restriction sites are also shown
Expression of adk in gram-positive bacteria is driven by two promoter regions found in the S10- spc-alpha cluster (Suh et al. 1996; Li et al. 1997). Since the B. subtilis adk gene in NUB3621:ThEV cells uses the same promoter in the same operon context as wild-type adk, we expected B. subtilis adk gene expression levels to be comparable. Purification of AK from NUB3621-R and NUB3621-R:ThEV cells showed that there is no significant difference in the amount of enzyme expressed by these two strains (Table 2). Furthermore, analysis of chloramphenicol acetyl-transferase expression through activity assays showed that the promoterless cat gene found just downstream from B. subtilis adk in the homologous recombination cassette was constitutively expressed in NUB3621-R:ThEV cells and was not influenced by the presence of chloramphenicol in the media (results not shown). Thus, we are confident that expression of the genes introduced by the homologous recombination cassette was driven by the endogenous promoter sequences found in the S10- spc-alpha region.
The maximum growing temperature tolerated by NUB3621-R:ThEV cells was 56°C. In previous studies, B. subtilis AK has been shown to have a T m of 50.7°C (without substrate) (Glaser et al. 1992). The fact that NUB3621-R:ThEV cells were able to grow at almost 5°C higher than the measured T m for B. subtilis AK was unexpected, but consistent with our enzyme activity results (Fig. 4). We also employed circular dichroism to follow the temperature induced denaturation of AKs isolated from wild-type and NUB3621-R:ThEV cells (data not shown). The CD experiments show that NUB3621-R:ThEV AK is, indeed, less stable than the wild-type enzyme and corroborate our kinetics data. The biochemical characterization of NUB3621-R AK will be the subject of a following study.

Temperature profiles for the activity of adenylate kinase isolated from wild-type NUB3621-R (solid squares) and recombinant NUB3621-R:ThEV (open circles) cells. AK was incubated at indicated temperatures for 5 min. Substrates AMP and ATP were added to the enzyme mixture and the reaction allowed to proceed for 3 min at indicated temperatures. The reaction was stopped by the addition of Ap5A. ADP production was estimated by the degradation of NADH through a coupled assay containing phosphoenolpyruvate, pyruvate kinase and lactate dehydrogenase. Curves shown are the average of three independent experiments. Error bars are shown for the standard deviations
Atkinson (1968) suggested using the adenylate energy charge as a direct measurement of the metabolic state of a cell (Atkinson 1968). EC has been shown to correlate well with cell growth and protein production for several organisms (Chapman et al. 1971; Swedes et al. 1975; Glembotski et al. 1981; Kahru et al. 1982). In order to investigate the metabolic attributes of NUB3621-R:ThEV cells, we determined their EC values under several growth conditions. Both wild-type and NUB3621-R:ThEV cells had low EC values when grown at 50°C (Table 3, footnote a). The low metabolic state for both cell lines when grown at 50°C was reflected in the slower doubling times and lower total protein concentrations (Table 3, footnote a). Wild-type cells at their optimal growth temperature, 65°C, displayed an increase in their energy charge values. More importantly, higher EC values were accompanied by faster doubling times and total protein concentrations (Table 3, footnote a). The transient increase in temperature experiment also showed that, EC values for NUB3621-R decreased 20 min after the cells were brought back from 65 to 50°C (Table 3, footnote b). Thus for G. stearothermophilus NUB3621-R, energy charge values correlated well with the metabolic state of the cells.
As AK have been shown to regulate cellular adenine nucleotide homeostasis in other organisms (Glembotski et al. 1981), we expected a decrease in EC values for NUB3621-R:ThEV cells kept at nonpermissive temperatures when compared to wild-type cells. As expected, in the transient increase in temperature experiment the energy charge values were decreased in NUB3621-R:ThEV after 20 min at 65°C (Table 3, footnote b). Moreover, we could not rescue the original EC values found at 50°C by returning the cells to permissive temperatures (Table 3, footnote b). Recombinant NUB3621-R:ThEV cell viability was also dramatically affected when cells were kept for 20 min at 65°C. Low EC values and cell-viability found for NUB3621-R:ThEV cells kept at 65°C were not accompanied by a dramatic decrease in optical density or total protein concentration, which seems to rule out cell lysis at high temperatures. Rather, such low EC and cell viability values may reflect a low metabolic state from which the cells cannot be rescued, suggesting that once the adenylate energy charge decreased beyond a critical threshold value, viability cannot be restored. The low EC values and cell viability found for NUB3621-R:ThEV at 65°C correlated well with the temperature inactivation of AK (Fig. 4). A similar result wass also observed for a temperature-sensitive adenylate kinase mutant strain of E. coli (Glembotski et al. 1981). These E. coli cells are less viable and show lower EC values after having been kept for 90 min at nonpermissive temperatures (Glembotski et al. 1981).
Unfortunately, high background levels of adenine nucleotides found in the media did not allow accurate determinations of EC values to be made on NUB3621-R:ThEV cells kept at 65°C for extended periods of time (Table 3). For E. coli cultures, up to 15% of the total adenine nucleotides have been found in the media (Glembotski et al. 1981) and this value is considerably higher, up to 35%, for an adenine-requiring mutant of E. coli growing under an adenine-starvation regime (Swedes et al. 1975). This suggests that, when E. coli cells are grown under stressful conditions, more adenine nucleotides are found in the media. The optimal temperature for wild-type G. stearothermophilus NUB3621-R is 65°C. At 50°C, the cell doubling time and EC values are significantly lower, while adenylate concentrations in the media increase, suggesting cell stress. Recombinant NUB3621-R:ThEV cells are also under stress at 50°C, but for the opposite reason. For NUB3621-R:ThEV cells, 50°C is fairly close to the nonpermissive temperature of 56°C, and thus doubling time and EC values decrease and medium adenylate levels rise concomitantly with rising temperature and stress.
At least one example exists of AK function being complemented by the overexpression of a double mutant mouse guanylate kinase in E. coli (Stolworthy and Black 2001). We are confident that no similar complementation mechanism is taking place in NUB3621-R:ThEV. The low EC values observed for NUB3621-R:ThEV cells growing at 65°C coincides with the temperature inactivation of NUB3621-R:ThEV AK (Fig. 4) and the strain’s observed temperature-sensitive phenotype (Fig. 2). Collectively, these results suggest that in NUB3621-R cells, AK is essential for maintaining the high cellular ATP concentrations required for growth and that no other enzyme complements AK’s function in cellular adenine nucleotide homeostasis in NUB3621-R cells.
Even though AK belongs to a diverse family of enzymes (Glaser et al. 1992; Ferber et al. 1997; Vonrhein et al. 1998; Munier-Lehmann et al. 1999), its function has been shown to be essential for cell viability in organisms as diverse as E. coli (Glembotski et al. 1981), a gram-negative, mesophilic organism and the eukaryotic fission yeast Schizosaccharomyces pombe (Konrad 1993). The results described in this work demonstrate that no other gene complements AK’s function in the phosphotransfer network of the gram-positive thermophile bacillus NUB3621-R. Moreover, an active AK is essential for maintaining high adenylate energy charge and cell viability in this organism.
To our knowledge, this is the first report of gene replacement in moderate thermophilic bacilli. Replacing the gene of interest with a mesophilic counterpart in NUB3621-R may prove useful in understanding key cellular processes in this important group of organisms.
Abbreviations
- adk :
-
Adenylate kinase gene
- AK:
-
Adenylate kinase protein
- Ap5A:
-
P1, P5 -Di(adenosine-5′)pentaphosphate
- cat :
-
Chloramphenicol acetyl-transferase gene
- EC:
-
Energy charge
- infA :
-
Initiation factor IF-I gene
- secY:
-
Pre-protein translocase secY subunit gene
- map :
-
Methionine aminopeptidase gene
- RBS:
-
Ribosomal binding site
References
Atkinson DE (1968) The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 7:4030–4034
Barzu O, Michelson S (1983) Simple and fast purification of Escherichia coli adenylate kinase. FEBS Lett 153:280–284
Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y (1997) The complete genome sequence of Escherichia coli K-12. Science 277:1453–1474
Breitling R, Schlott B, Behnke D (1994) Modulation of the spc operon affects growth and protein secretion in Bacillus subtilis. J Basic Microbiol 34:145–155
Chang S, Cohen SN (1989) High frequency transformation of Bacillus subtilis protoplasts by plasmid DNA. Mol Gen Genet 168:111–115
Chang SY, McGary EC, Chang S (1989) Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J Bacteriol 171:4071–4072
Chapman AG, Fall L, Atkinson DE (1971) Adenylate energy charge in Escherichia coli during growth and starvation. J Bacteriol 108:1072–1086
Chen ZF, Wojcik SF, Welker NE (1986) Genetic analysis of Bacillus stearothermophilus by protoplast fusion. J Bacteriol 165:994–1001
Cousin D, Buttin G (1969) Mutants thermosensibles d‘ Escherichia coli K12. III – Une mutation lêtale d’ E. coli affectant l’activité de l’adénylate-kinase. Ann Inst Pasteur (Paris) 117:612–630
Criswell AR, Bae E, Stec B, Konisky J, Phillips GN Jr (2003) Structures of thermophilic and mesophilic adenylate kinases from the genus Methanococcus. J Mol Biol 330:1087–1099
Cummings HS, Hershey JW (1994) Translation initiation factor IF1 is essential for cell viability in Escherichia coli. J Bacteriol 176:198–205
Ferber DM, Haney PJ, Berk H, Lynn D, Konisky J (1997) The adenylate kinase genes of M. voltae, M. thermolithotrophicus, M. jannaschii, and M. igneus define a new family of adenylate kinases. Gene 185:239–244
Glaser P, Presecan E, Delepierre M, Surewicz WK, Mantsch HH, Barzu O, Gilles AM (1992) Zinc, a novel structural element found in the family of bacterial adenylate kinases. Biochemistry 31:3038–3043
Glembotski CC, Chapman AG, Atkinson DE (1981) Adenylate energy charge in Escherichia coli CR341T28 and properties of heat-sensitive adenylate kinase. J Bacteriol 145:1374–1385
Haase GH, Brune M, Reinstein J, Pai EF, Pingoud A, Wittinghofer A (1989) Adenylate kinases from thermosensitive Escherichia coli strains. J Mol Biol 207:151–162
Hanahan D, Jessee J, Bloom FR (1991) Plasmid transformation of Escherichia coli and other bacteria. Methods Enzymol 204:63–113
Hansmann S, Martin W (2000) Phylogeny of 33 ribosomal and six other proteins encoded in an ancient gene cluster that is conserved across prokaryotic genomes: influence of excluding poorly alignable sites from analysis. Int J Syst Evol Microbiol 50(Pt 4):1655–1663
Imanaka T, Fujii M, Aramori I, Aiba S (1982) Transformation of Bacillus stearothermophilus with plasmid DNA and characterization of shuttle vector plasmids between Bacillus stearothermophilus and Bacillus subtilis. J Bacteriol 149:824–830
Kahru A, Liiders M, Vanatalu K, Vilu R (1982) Adenylate energy charge during batch culture of Thermoactinomyces vulgaris 42. Arch Microbiol 133:142–144
Kahru A, Vilu R (1990) Role of adenine nucleotides in the regulation of bacterial energy metabolism: theoretical problems and experimental pitfalls. Microbios 62:83–92
Kath T, Schmid R, Schafer G (1993) Identification, cloning, and expression of the gene for adenylate kinase from the thermoacidophilic archaebacterium Sulfolobus acidocaldarius. Arch Biochem Biophys 307:405–410
Konrad M (1993) Molecular analysis of the essential gene for adenylate kinase from the fission yeast Schizosaccharomyces pombe. J Biol Chem 268:11326–11334
Li X, Lindahl L, Sha Y, Zengel JM (1997) Analysis of the Bacillus subtilis S10 ribosomal protein gene cluster identifies two promoters that may be responsible for transcription of the entire 15-kilobase S10-spc-alpha cluster. J Bacteriol 179:7046–7054
Lin-Chao S, Chen WT, Wong TT (1992) High copy number of the pUC plasmid results from a Rom/Rop-suppressible point mutation in RNA II. Mol Microbiol 6:3385–3393
Munier-Lehmann H, Burlacu-Miron S, Craescu CT, Mantsch HH, Schultz CP (1999) A new subfamily of short bacterial adenylate kinases with the Mycobacterium tuberculosis enzyme as a model: a predictive and experimental study. Proteins 36:238–248
Narumi I Sawakami, K., Nakamoto, S., Nakayama, N., Yanagisawa, T., Takahashi, N., Kihara, H (1992) A newly isolated Bacillus stearothermophilus K1041 and its transformation by electroporation. Biotechnol Tech 6:83–86
Sharp RJ, Riley PW, White D (1992) Heterotrophic thermophilic bacilli. In: Kristjansson JK (ed) Thermophilic bacteria. CRC Press, Boca Raton, pp 19–50
Shiba K, Ito K, Yura T, Cerretti DP (1984) A defined mutation in the protein export gene within the spc ribosomal protein operon of Escherichia coli: isolation and characterization of a new temperature-sensitive secY mutant. EMBO J 3:631–635
Stolworthy TS, Black ME (2001) The mouse guanylate kinase double mutant E72Q/D103N is a functional adenylate kinase. Protein Eng 14:903–909
Studholme DJ, Jackson RA, Leak DJ (1999) Phylogenetic analysis of transformable strains of thermophilic Bacillus species. FEMS Microbiol Lett 172:85–90
Suh JW, Boylan SA, Oh SH, Price CW (1996) Genetic and transcriptional organization of the Bacillus subtilis spc-alpha region. Gene 169:17–23
Swedes JS, Sedo RJ, Atkinson DE (1975) Relation of growth and protein synthesis to the adenylate energy charge in an adenine-requiring mutant of Escherichia coli. J Biol Chem 250:6930–6938
Vallier H, Welker NE (1990) Genetic map of the Bacillus stearothermophilus NUB36 chromosome. J Bacteriol 172:793–801
Vieille C, Burdette DS, Zeikus JG (1996) Thermozymes. Biotechnol Annu Rev 2:1–83
Vonrhein C, Bonisch H, Schafer G, Schulz GE (1998) The structure of a trimeric archaeal adenylate kinase. J Mol Biol 282:167–179
Wei P, Stewart CR (1993) A cytotoxic early gene of Bacillus subtilis bacteriophage SPO1. J Bacteriol 175:7887–7900
Wu LJ, Welker NE (1989) Protoplast transformation of Bacillus stearothermophilus NUB36 by plasmid DNA. J Gen Microbiol 135:1315–1324
Yanisch-Perron C, Vieira J, Messing J (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119
Acknowledgements
This work was in part funded by a grant from the National Science Foundation (NSF/MCB - 0212417). R.C. is the recipient of a training fellowship from the W.M. Keck Foundation to the Gulf Coast Consortia through the Keck Center for Computational and Structural Biology. The authors would like to thank the MacKenzie Lab (Rice University, Houston, TX) for equipment, helpful discussions and suggestions. The authors are indebted to Dr. N. Welker (Northwestern University, Evanston, IL, USA) for help with G. stearothermophilus NUB3621-R transformation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G. Antranikian
The authors would like to dedicate this paper to the memory of Dr. Neil Welker
Rights and permissions
About this article
Cite this article
Couñago, R., Shamoo, Y. Gene replacement of adenylate kinase in the gram-positive thermophile Geobacillus stearothermophilus disrupts adenine nucleotide homeostasis and reduces cell viability. Extremophiles 9, 135–144 (2005). https://doi.org/10.1007/s00792-004-0428-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-004-0428-x