
Abstract
Misfolded prion protein (PrPSc) is known for its role in fatal neurodegenerative conditions, such as Creutzfeldt–Jakob disease. PrP fragments and their mutants represent important tools in the investigation of the neurotoxic mechanisms and in the evaluation of new compounds that can interfere with the processes involved in neuronal death. Metal-catalyzed oxidation of PrP has been implicated as a trigger for the conformational changes in protein structure, which, in turn, lead to misfolding. Targeting redox-active biometals copper and iron is relevant in the context of protection against the oxidation of biomolecules and the generation of oxidative stress, observed in several conditions and considered an event that might promote sporadic prion diseases as well as other neurodegenerative disorders. In this context, ortho-pyridine aroylhydrazones are of interest, as they can act as moderate tridentate ligands towards divalent metal ions such as copper(II). In the present work, we explore the potentiality of this chemical class as peptide protecting agents against the deleterious metal-catalyzed oxidation in the M112A mutant fragment of human PrP, which mimics relevant structural features that may play an important role in the neurotoxicity observed in prion pathologies. The compounds inhere studied, especially HPCFur, showed an improved stability in aqueous solution compared to our patented lead hydrazone INHHQ, displaying a very interesting protective effect toward the oxidation of methionine and histidine, processes that are related to both physiological and pathological aging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The prion protein (PrP) is known for its role in fatal neurodegenerative conditions, such as the Creutzfeldt–Jakob disease, fatal familial insomnia, Gertsmann–Straussler–Scheinker syndrome, and kuru [1, 2]. The physiological, cellular prion protein, usually denoted as PrPC, is a cell surface glycoprotein highly expressed in the central and peripheral nervous system. PrPC is composed of 209 amino acids, presenting the residues 23–231 from the original translation product that contains 253 amino acids. The peptide 1–22 is cleaved during trafficking and residues 232–253 are replaced by a saccharide moiety [3]. PrPC is divided into two regions of different structural properties. In mammals, the N terminus (until residue 120) consists of unstructured octapeptide PHGGSWGQ repeats that can bind divalent metal ions such as copper(II) [4]. The C terminus (residues 121–231), on the other hand, is composed of three α-helixes and a double-stranded antiparallel β-sheet [5]. The physiological role of PrPC is still unclear, although it is proposed that it is involved in several functions in the central and peripheral nervous systems. Evidences point to its interaction with a variety of binding partners, which suggests that PrPC can modulate some processes such as memory and inflammatory reactions, cellular functions like proliferation and differentiation, as well as controlling a number of transduction signaling pathways [6]. PrPC has also been implicated in cellular copper homeostasis or copper-dependent enzymatic functions [7], although the physiological relevance of PrP binding to this metal remains mostly speculative. On the other hand, the confirmation of such a large number of functions, as described in the literature, seems improbable, highlighting the need for rigorous assessments [8].
In pathology, PrPC misfolds into the insoluble scrapie prion protein (PrPSc), presenting β-sheet rich content and protease resistance [9, 10]. The hypothesis that the infectious feature of such diseases depends solely on this conversion is called “protein-only hypothesis” or, more recently, prion hypothesis [10]. The PrP106–126 fragment has been linked to neurotoxicity and proposed as a model for the study of misfolded PrP [11, 12]. This synthetic peptide can reproduce biophysical and biological characteristics of PrPSc but it is not infectious [13]. PrP106–126 as well as other amyloidogenic PrP-derived peptides constitutes important tools in the investigation of PrP neurotoxic mechanisms and in the evaluation of new compounds that can potentially interfere with the chemico-pathophysiological mechanisms underlying neuronal death.
The use of prion fragments has also shed some light into another relevant process related to copper coordination: the metal-catalyzed oxidation of specific amino acid residues, which may lead to loss or gain of function and can be linked to pathology. In 2001, Requena et al. [14] showed that the Syrian hamster recombinant SHa29–231 protein presented aggregation and precipitation concomitant with copper-mediated oxidation. Since residues of the amino acids histidine and methionine are prone to be oxidized in the presence of copper, the wild-type fragment Ac-SKPKTNMKHM-NH2 and its mutants [15] also constitute a simplified model to study post-translational structural modifications that could be related to the conversion of PrPC into PrPSc. It has been shown that the mutant peptide which does not contain any methionine did not undergo oxidation, but fragmentation of the main-chain. However, in the case of methionine-containing peptides, the main chain was not cleaved, and the oxidation of this residue to methionine sulfoxide occurred. These results reveal that methionine residues of prion protein may play a role as reactive oxygen species (ROS) scavengers and that their presence protects peptides from fragmentation. In all of these cases, Cu2+ ions catalyze the oxidation of methionine to methionine sulfoxide [15]. On the other hand, oxidation of methionine is also not a favorable process, since evidences suggest that it might be a trigger for conformational changes in PrP’s structure [16, 17], which, in turn, can lead to protein misfolding. This reaction can occur in vivo, either because of physiological (normal) aging or at the site of inflammation, where large amounts of oxidants are present [18].
Over the last years, we have been exploring the moderate chelating abilities of aroylhydrazones in the modulation of abnormal metal–protein interactions in the context of diseases related to protein misfolding and aggregation [19,20,21]. In such disorders, often called aggregopathies, a dyshomeostasis of physiological metals, such as copper, iron, and zinc, is present [22,23,24]. These metals are able to accelerate the rate of oligomerization of the proteins involved in pathology, as well as increase the toxicity of the aggregates [23, 25,26,27]. Furthermore, targeting copper and iron is relevant in the context of protection against the oxidation of biomolecules and generation of oxidative stress, which is widely observed in these conditions and considered an event that might lead to sporadic prion diseases and other neurodegenerative diseases [28, 29].
The ortho-pyridine aroylhydrazones can act as tridentate ligands toward divalent metal ions such as copper(II) and zinc(II) through the Npy, N, O system. Coordination of biometals by the HPCIH ligand (pyridine-2-carboxaldehyde isonicotinoyl hydrazone) has been extensively studied in the context of different diseases such as Alzheimer’s [21] and iron overload-related Friedrich ataxia [30]. Another structure-related ligand of interest is HPCFur (pyridine-2-carboxaldehyde 2-furoyl hydrazone, Scheme 1). Substitution of the para-pyridine moiety for a five-membered heterocyclic aromatic ring—2 furoyl—results in smaller, more soluble compounds. Moreover, furan-derived molecules usually present improved pharmacokinetic characteristics, such as optimized solubility and a good bioavailability. This group is widely employed in a variety of approved drugs [31]. The aroylhydrazone HPCFur is not a new compound, and its synthesis, characterization, and coordination properties have already been explored [30, 32,33,34].

Chemical structures of the aroylhydrazones. a HPCIH and b HPCFur. Donor atoms involved in the tridentate coordination are highlighted. Hydrogens are numbered according to NMR assignment
In the present work, we explore the potentiality of these two ortho-pyridine aroylhydrazones as peptide protecting agents toward the deleterious metal-catalyzed oxidation effects in a mutant fragment of human PrP (Ac-SKPKTNMKHA-NH2, abbreviated dMKHA—Scheme 2), which mimics some relevant structural features that may play an important role in the neurotoxicity observed in prion pathologies.

Structure of the mutant human prion protein PrP103–112 fragment Ac-SKPKTNMKHA-NH2 (dMKHA)
Materials and methods
Chemicals and solutions
Apart from pyridine-2-carboxaldehyde, an oily colorless liquid that requires distillation immediately before use, all chemicals and solvents were purchased from commercial sources in the highest available purity and employed as received.
Stock solution of copper(II) nitrate was prepared from analytical grade reagent and its concentration was measured gravimetrically via oxinate precipitation.
Hydrazones’ syntheses
The syntheses and characterization of both ligands have already been described [30, 35]. Preparation of HPCIH was performed according to Richardson et al. [36]. This ligand was recently widely characterized, both in solution and in solid state by our research group [21]. The synthesis of HPCFur was performed similarly.
Peptide’s synthesis
dMKHA was assembled using the solid-phase peptide synthesis strategy on a Liberty 1 Peptide Synthesizer. The procedure for the synthesis and purification of the peptide has already been reported in a previous publication [15].
Instrumentation, chemical characterization, and in vitro studies
The melting points of the organic ligands were determined in triplicate in a Fisatom apparatus, model 431. Thermogravimetry was done in a Pyris 1 TGA PerkinElmer analyzer, under nitrogen atmosphere, between 20 and 900 °C, at a heating rate of 10 °C min−1. Infrared spectra were acquired in the mid-infrared region (4000–450 cm−1) in a 100 FT-IR PerkinElmer absorption spectrophotometer. Samples were prepared in KBr pellets. UV–visible spectra were recorded on a Lambda 35 PerkinElmer molecular absorption equipment in the 200–800 nm range. The ‘spectroscopy room’ temperature was kept at 17 °C at all times. 1.0 cm quartz cuvettes were employed. Solutions were prepared at low concentrations (50 µM) to fit into the Lambert–Beer law limit. For the stability in 1% DMSO/water, the initial scanning spectrum was obtained immediately after dilution in water and other spectra were acquired at regular intervals until the system reached equilibrium, i.e., until there were no further spectral changes.
For the oxidation studies, reaction mixtures containing 1.0 mM dMKHA at 1:1 and 0.1:1 metal-to-peptide molar ratios were incubated at 25 °C for different time periods. The pH was adjusted to 7.4. The reaction started with the addition of 1% (w/w) hydrogen peroxide solution, which was freshly prepared, and was stopped by the addition of Na2EDTA at peptide-to-Na2EDTA ratio 1:5. In the case of reaction mixtures containing ascorbic acid, the peptide-to-ascorbic acid molar ratio was 1:20. The reaction process was monitored by RP-HPLC at different time points. Aroylhydrazones were prepared in a water/acetonitrile mixture (50% v/v).
For the isolation of the oxidized products, the samples were examined through analytical RP-HPLC using a Jasco instrument, equipped with a JascoMD-2010 plus multiwavelength detector. The oxidized products were conducted on a Teknokroma Europa Protein C18 (250 × 4.6 mm, 300 Å, 5 μm) at a flow rate of 1 mL min−1, with the absorbance monitored at 222 nm. Mobile phases were water (A) and acetonitrile (B) containing 0.1% TFA. Gradient: 0.0–3.0–9.0–24.0–30.0 min, and 100–100–92–92–100% water containing 0.1% TFA for the peptide.
A MicroTOF-Q type Qq-TOF MS instrument (Bruker Daltonik) was used for the MS and MS/MS measurements in positive ion mode. The instrument was equipped with an electrospray ion source, and the spray voltage employed was 4 kV. N2 was used as drying gas. The drying temperature was 200 °C and the flow rate was 4.0 L min−1 (8.0 L min−1 HPLC–MS) using the same method described above. For the MS/MS experiments, N2 was used as the collision gas. The pressure in the collision cell was determined to be 1.2 × 10−2 mbar. The precursor ions for MS/MS were selected with an isolation width of 5 m/z units. The mass spectra were recorded by means of a digitizer at a sampling rate of 2 GHz. The mass spectra were calibrated externally using the exact masses of clusters [(NaTFA)n+ Na]+ generated from the electrosprayed solution of sodium trifluoroacetate (NaTFA). Spectra were evaluated with the DataAnalysis 3.4 software from Bruker. The sample solutions were introduced directly into the ESI source with a syringe pump (Cole-Parmer Ins. Co., Vernon Hills, IL, USA) at a 3 μL min−1 flow rate.
NMR measurements regarding the peptide’s signal assignment were performed in an 800 MHz Bruker Avance III spectrometer with a 3 mm CP TCI 800S7 H-C/N-D-03 Z cryoprobe. All the experiments were done at 5 °C, to decrease the HN exchange rate with the solvent. Peptide’s interaction with copper(II) ions, as well as its oxidation by the Cu2+/H2O2 system, was followed by NMR, at 25 °C, in a Bruker UltraShield 400 MHz magnet connected to a Bruker Avance III HD console, using 3 mm NMR tubes from Hilgenberg and a QXI 400S1 H/P-C/N-D-05 Z probehead. The latter configuration was also employed to characterize and assign the small molecule HPCFur, using 5 mm NMR tubes instead. For the measurements, dMKHA·4TFA (MM = 1638.47 g mol−1) was dissolved in 500 µL of HEPES buffer (50 mM, 100 mM NaCl, pH 7.4). 40 µL of D2O and 10 µL of a 6.0 mM solution of the reference compound 3-(trimethylsilyl)-2,2,3,3-tetradeuteropropionic acid (TSP-d4) were then added, attaining a final dMKHA concentration equal to 5.0 mM. The buffer HEPES was chosen to maintain the pH at a physiological value since its interaction with metal ions can be considered very weak, being a suitable choice for complexation studies [37]. From this initial solution, aliquots of 180 µL were transferred to the 3 mm NMR tubes. Small volumes of stock solutions (0.055 or 0.55 M) of copper(II) chloride in D2O were added for the interaction studies. For the oxidation evaluation, only the 0.055 M stock solution was employed, since higher copper(II) concentrations prevented the observation of the signals of interest due to its strong paramagnetic effect. The reaction started with the addition of 6% (w/w) hydrogen peroxide. Stock solution of HPCFur (50 mM) was prepared in methanol instead of acetonitrile due to the overlapping of this latter solvent’s residual signal with that of ε methyl hydrogens from the methionine residue. Water suppression was achieved through the WATERGATE pulse sequence. 2D TOCSY experiments were carried out using a MLEV17-based sequence with a mixing time of 80 ms. 2D NOESY experiments were done employing a mixing time of 600 ms. Chemical shifts were calibrated by the signal of the internal reference TSP (− 0.083 ppm for hydrogen [38] and − 0.12 ppm for carbon [39], respectively). For the chemical characterization of the hydrazonic ligand, HPCFur was dissolved in DMSO-d6 and the spectra were calibrated through the residual peaks of this solvent at 2.50 (1H) and 39.52 ppm (13C). Additional NMR experiments demonstrated that this hydrazone is stable under the conditions used in the peptide’s oxidation studies. Sample acquisition and processing were performed using the TopSpin 3.5pl7 software (Bruker Biospin). Electrospray ionization mass spectrometry (ESI-MS) analysis on the ternary system constituted by dMKHA, copper(II), and HPCFur was carried out in a Waters ZQ 4000 Single Quadrupole mass spectrometer. Standard configuration parameters for the positive mode were used.
Theoretical values for log P and log S were calculated using the software Osiris Property Explorer: DataWarrior™, freely available for download at http://www.organic-chemistry.org/prog/peo/, accessed on 15/11/2017.
Results and discussion
Synthesis and characterization of HPCFur
Just like HPCIH, HPCFur is a white powder, obtained at 48% yield. Its melting point is 164 ± 1 °C. The structural characterization of the ligand was done through mid-infrared vibrational spectroscopy, NMR spectroscopy, and thermogravimetry, allowing for the confirmation of the product’s identity, as well as its molar mass and purity.
Thermogravimetric analysis
The thermal decomposition of HPCFur occurs in three steps, as shown in the TG curve of Figure S1 (Supplementary Information). The first stage is related to the loss of one molecule of hydration water, around 100 °C (HPCFur·H2O, MM = 233.20 g mol−1). The ligand then decomposes in two steps, presenting no stable residue after 650 °C.
HPCIH, on the other hand, presents two water molecules in its structure, and decomposes at a faster rate, in only one step [21].
Vibrational spectroscopy (FT-IR)
The two ligands inhere described are very closely related to one another. Thus, as expected, their infrared spectra share great similarities (Figure S2). The main bands of both hydrazones and their assignments are displayed in Table 1.
The vibration of the foremost chemical groups present are identified, such as the C=Nazomethine bond, which is formed upon reaction between pyridine-2-carboxaldehyde and the respective aroylhydrazide and constitutes the main band to determine the success of the syntheses. The key difference between the ligands is the presence of the furan ring, which can be observed only in HPCFur’s spectrum, at 1085 cm−1, as a characteristic stretching band involving the carbon–oxygen bond [40]. Both ligands contain aromatic and pyridine-related absorptions, as well as bands arising from the presence of hydration water in the structure, in agreement with the thermogravimetric analyses.
Nuclear magnetic resonance
We have previously reported a complete study of the HPCIH species existing in DMSO-d6 solution [21], in which a rich equilibrium between tautomers and (E)/(Z) isomers was identified. HPCFur, on the other hand, presents only one quantifiable main species in the same conditions—the (E)-amido tautomer. Routine 2D NMR techniques such as COSY, 1H–13C HSQC, and 1H–13C HMBC were also employed to facilitate full assignment (Figures S3–7). Table 2 summarizes the signals and attributions made for the 1H and 13C spectra of HPCFur in comparison to the previously published HPCIH [21].
Since HPCIH and HPCFur are structure-related compounds, the electronic effect of their heterocyclic substituents can be evaluated through 13C NMR, as the chemical shift of the vicinal carbonyl group. For HPCIH, carbonyl resonates at 162.90 ppm, while for HPCFur, this group is more shielded, being registered at 154.35 ppm. The carbonyl’s electronic environment may have direct impact on the intensity of electron donation in the coordination of copper(II), resulting in different metal-binding affinities.
UV–Vis molecular absorption spectroscopy
In HPCIH’s absorption spectrum, using DMSO as solvent, just one asymmetric band, at 301 nm, is observed, probably corresponding to a π → π* transition [21]. As expected, due to their great similarity, HPCFur also presents an asymmetric band (Figure S8A), which can be computationally decomposed into two constituents centered at 268 and 310 nm (green and red dotted lines, respectively).
Although hydrazones may be susceptible to undergo hydrolysis of the C=N bond, HPCFur, as well as the previously reported HPCIH, is very stable in an aqueous-rich environment (1% DMSO/water). A decrease of less than 2% in the absorbance of the hydrazone band (at 310 nm) is observed in a series of spectra taken at regular intervals for 12 h, while the absorption at higher energy increases accordingly, giving rise to an isosbestic point at 272 nm (Figure S8B).
In vitro assays with the M112A PrP103–112 mutant fragment dMKHA
dMKHA was chosen from the series of mutants beforehand studied [15], since there is no significant difference in the oxidation process in comparison to the one of the wild-type (WT) fragment. The employed mutant contains a unique methionine residue instead of two, which allows for a simplified assessment of compounds’ effects, since, in the WT fragment, oxidation could occur in either one of the methionine sites, or even in both. In terms of coordination chemistry, the use of dMKHA is also very suitable since this mutant maintains all the donor atoms involved in the coordination sphere of copper(II) observed in the human WT 103–112 fragment. Furthermore, in a very recent publication by Sánchez-López et al. [41], the preference of copper(II) for the H111 residue instead of the H96 one was demonstrated, and the fact was associated with the presence of M109 in the vicinity of the former site. This constitutes an additional support for the use of the short dMKHA fragment, which displays the M112A mutation but keeps the auxiliary anchoring methionine residue at the 109 position.
NMR characterization of dMKHA and its interaction with copper(II)
Copper(II) coordination chemistry of full-length PrPC and many of its mutants and fragments have been systematically investigated in the past years [15, 42,43,44], including under a theoretical perspective [45]. In spite of this, some aspects still deserve more attention to, for example, confirm by complementary means the involvement of the methionine thioether sulfur atom in the metal anchoring system constituted by the H111 side chain. In the present work, 1D and 2D NMR spectroscopy were employed to pursue this goal.
The intrinsic absorptions of the HEPES buffer prevented the observation of the peptide’s signals between 4.0 and 2.7 ppm. However, the most important resonances regarding the present study are located outside of this region. The aromatic hydrogens of the histidine residue are observed as a pair of doublets (4J = 1.1 Hz) at 7.729 and 6.934 ppm. Cross peaks in the 1H–1H TOCSY contour plot confirm the assignments. The most shielded of those aromatic nuclei also couples with a resonance at around 3 ppm, which should correspond to the histidine β-methylene hydrogen atoms. Thus, the signal at 6.934 ppm was assigned to the δ2 imidazole hydrogen. On the other hand, the ε methyl hydrogens from the methionine residue resonate at 2.022 ppm and those from the N-terminal acetyl group, at 1.992 ppm. Amide hydrogens from the side chain of asparagine (N108) are observed as two broad signals at 7.526 and 6.827 ppm. Figure 1A shows the main assignments concerning the Hα (in a 1H–13C HSQC contour plot) and some “key” hydrogen signals belonging to potentially coordinating side chains in the peptide (highlighted in the 1H spectrum of dMKHA). The 1H–1H TOCSY spectrum obtained from a 5.0 mM solution of the peptide at 5 °C is given as Figure S9.

NMR study of the coordination of copper(II) by dMKHA. 1H–13C HSQC contour plots and 1H NMR spectra of a dMKHA apo-form, b dMKHA + 0.1 eq copper(II) and c dMKHA + 1.0 eq copper(II). pH 7.4 at 25 °C
During the last 20 years, NMR has become a widely employed tool for the study of metal interactions with unstructured soluble protein domains [46]. In the present work, the interaction between dMKHA and copper(II) ions at pH 7.4 and 25 °C was characterized by 1D and 2D NMR spectroscopy. Usually, the loss of NMR signals due to the paramagnetic effects related to this cation occurs within 9–10 Å, affecting the peptide’s nuclei in a relatively local manner. The addition of 0.1 eq copper(II) disturbed immediately the Hα/Cα H111 cross peak in the 1H–13C HSQC map, as well as the signals from histidine’s δ2 and ε1 aromatic hydrogens in the 1H NMR spectrum of dMKHA, which were broadened almost beyond detection (Fig. 1b). This confirms the high affinity of histidine for copper(II) as well as its role as an anchoring site for this metal, and is in accordance to data previously published for the PrP106–113 fragment [47]. Additionally, a significant intensity decrease is observed in the ε methyl signal from methionine, which could indicate a weak interaction with the sulfur atom of this residue as a part of the coordination process. Increasing the copper concentration until reaching 1.0 eq (Fig. 1c), we observed that only three out of ten Hα/Cα cross peaks seem to be not directly affected by the metal. They are, as expected, those from S103, K104, and P105, which confirm the idea that N-terminal residues are not involved in the interaction with copper(II) ions due to the presence of proline, a well-known break point for metal coordination. At this 1:1 stoichiometry, also the ε methyl signal of methionine is severely affected. This strongly supports the propositions regarding a methionine contribution to coordination. However, other signals considered in the present study, as those related to the –NH2 hydrogens from the amide side chain of asparagine, remain quite unchanged, which allows to rule out the involvement of this residue in the dMKHA–metal interaction. From the protonation equilibria studies published by Csire et al. [15], we assume that, at pH 7.4, an almost equimolar mixture of the [Cu(H−2dMKHA)]3+ and [Cu(H−3dMKHA)]2+ species occurs. The conditional stability constants for these complexes are, respectively, − 7.95 and − 15.43. Therefore, we can conclude that the most likely donor atoms in the coordination sphere of copper(II) at this pH value are an imidazole nitrogen from histidine and two (in H−2dMKHA, where dMKHA represents the peptide protonated at the three lysine residues) or three (in the case of H−3dMKHA) deprotonated amide nitrogens from the main-chain peptide bonds involving H111, K110 and, for the species [Cu(H−3dMKHA)]2+, also M109. It is well known that copper(II) is able to deprotonate and bind amide nitrogen atoms when anchored to imidazole groups from histidine residues [48], a process which, for this system, happens above pH 5 in a cooperative manner [15]. The thioether sulfur atom from methionine should be axially coordinated. Figure 2 shows the proposed structures for the metal–peptide adducts at pH 7.4. Special attention should be paid to the labile equatorial site displaying a coordinated water molecule in [Cu(H−2dMKHA)]3+, since it constitutes a putative spot for the interaction and subsequent oxidation of H2O2 to the O2− radical, giving rise to a reduced copper(I) center which, in turn, could be able to trigger dMKHA oxidation through Fenton-type reactions [49, 50]. Finally, a contact with the hydroxyl oxygen atom from the T107 side chain, alternating with the apical water molecule in a supposed multipart equilibrium, cannot be completely discarded at this point, as the cross peak related to threonine Hβ/Cβ coupling is also very affected by the presence of copper.

Proposed structures for the main species present in equilibrium at pH 7.4 in the dMKHA–copper(II) system. The peptide’s main chain is highlighted in bold gray, while the main amino acid residues involved in coordination are labeled
Oxidation of the peptide dMKHA catalyzed by copper(II)
The oxidation of dMKHA in the presence of copper(II) and hydrogen peroxide has been previously investigated by RP-HPLC and mass spectrometry [15]. Figure 3a presents a series of representative chromatograms involving the studied systems after 30 min of oxidation. The ratio of the main products at the end of this period is presented in Fig. 3b. For the system containing only the peptide and copper(II), oxidation does not occur at significant extent under our experimental conditions. In the presence of H2O2, however, the product dM(O)KHA, observed at a retention time of 13.2 min, is generated, corresponding to the oxidation of the methionine residue to methionine sulfoxide. The number of oxidation sites present, as well as their precise identification, was determined by MS/MS measurements. The fragment ions are indicated by b if the charge is retained on the N terminus and by y if the charge is maintained on the C terminus, as proposed by Roepstorff and Fohlman [51]. The subscript refers to the number of amino acid residues in the fragment. The fragmentation of the singly oxidized product at the methionine residue is displayed in Fig. 4. The m/z value at 599.822 (calculated value: m/z 599.822) corresponds to the doubly charged singly oxidized molecular ion of dMKHA (i.e., [y10 + O]2+). The most convincing proof of methionine oxidation is the elimination of a CH3SOH molecule (63.998 Da) from the [y10 + O]2+ and [y8 + O]2+ ions, as it is considered a diagnostic evidence for the formation and presence of methionine sulfoxide [52]. Furthermore, the formation of [y4 + O]+ supports that the oxygen atom is attached to one of the four amino acids at the C terminus. Since the [y2]+ ion was observed in the MS/MS spectrum, the oxidation of the histidine residue can be ruled out.

HPLC study of the oxidation of dMKHA in the presence of copper(II) and H2O2. a Representative chromatograms of the different studied systems after 30 min of reaction. b Ratio of products at the end of this time point

MS/MS spectrum of the methionine oxidized dM(O)KHA species
When an excess of ascorbic acid is present, more products are formed: besides dM(O)KHA (tR = 13.1 min), oxidation of the histidine residue also takes place, giving rise to the doubly oxidized product, [dMKHA + 2O], and the singly histidine oxidized product dMKH(O)A. These species elute at 13.4 min and 16.6 min, respectively. The m/z value of the product in which the histidine residue is oxidized is equal to the previous one (m/z 599.822), but it is detected at a different retention time: 16.6 instead of 13.1 min. The oxidation of histidine is also proved in the doubly oxidized product ([y10 + 2O]2+, with m/z 607.814, calculated value: m/z 607.819), as can be seen in Figure S10. Additionally, the [y3 + O]+ ion, with the amino acid sequence –KHA–, is only singly oxidized, being the oxygen atom consequently attached to the histidine residue. From the results discussed above, one can conclude that the presence of ascorbic acid slightly hinders the oxidation of methionine, promoting that of histidine (Fig. 3b). This effect is not favorable since the methionine oxidation can be easily reversed by the methionine sulfoxide reductase systems (methionine sulfoxide reductase A or B), but one of histidine is irreversible.
A similar effect on the oxidation of dMKHA is observed when an equimolar amount (with respect to the peptide) of the aroylhydrazone HPCIH is added. However, the compound promotes the oxidation of histidine to a lesser extent than ascorbic acid. Only a small amount of the doubly oxidized product is generated (around 3%), while the single oxidized product dMKH(O)A is negligible. Even if the amount of this compound is decreased to one-tenth, its effect is still significant.
The protecting role of HPCFur is even more expressive. When this compound is present at the same concentration as dMKHA, almost 85% of the peptide remains unchanged, being only 15% of it oxidized, but exclusively on the methionine residue. In the presence of ascorbic acid, the protective effect of HPCFur remains, although to a lesser degree. Analyzing the effect of this ligand on the oxidation profile of the system dMKHA:Cu(II):H2O2:ascorbic acid, we observe almost 10% more of unoxidized peptide, due to the reduction of histidine-associated oxidation products, since the amount of methionine-oxidized peptide remains almost the same.
To further characterize the oxidation of the peptide, NMR spectroscopy was employed. As stated above, both methionine and histidine residues of dMKHA are sensitive to oxidation in the presence of hydrogen peroxide, a reaction catalyzed by copper(II) ions. Methionine is an amino acid highly susceptible to oxidation, even under mild conditions, and the methionine sulfoxide-containing derivative constitutes the main product identified by RP-HPLC and mass spectrometry experiments. Therefore, the oxidation of this residue of the peptide at 25 °C was followed by 1H NMR, in a reaction mixture containing 0.1 eq Cu2+ and excess (4.0 eq) of H2O2.
Throughout the oxidation process of dMKHA, the decrease in intensity of the ε methyl hydrogens’ signal from the methionine residue at 2.02 ppm was accompanied by the rise of an asymmetric absorption at 2.65 ppm, which was assigned to the ε methyl hydrogens from the methionine sulfoxide of the oxidized product (Fig. 5a). These outcomes agree with the observations made on the single amino acid which, after oxidation to methionine sulfoxide, displays two very close singlets for the methyl group, which are shifted by around 0.60 ppm downfield when compared to the signal related to the methyl group in unoxidized methionine [53]. Weihong Du et al. also found a similar NMR spectral behavior when studying the methionine oxidation in peptides by peroxovanadium complexes [54]. A qualitative analysis of the results obtained was performed by measuring the peak intensities related to radiofrequency absorptions, since the strong signal overlapping prevented accurate integrations to be carried out. After 45 min of reaction, intensities corresponding to the oxidized and unoxidized forms of dMKHA seem to reach a plateau (Fig. 5a, inset). At this point, approximately 87% of the peptide units contain methionine sulfoxide. However, if the reaction is additionally followed until 120 min, a second oxidation phase, probably related to the formation of the doubly oxidized product, occurs (Fig. 5b, black line). During this second step, another 6% of unoxidized dMKHA are lost. Besides those related to the methyl group of the methionine residue, other minor changes were observed in the spectra as well, probably related to the generation of a new stereogenic center in the sulfur atom introduced by the oxidation of methionine [53]. A series of magnetic anisotropy effects related to the presence of the sulfoxide group can be identified too, being also responsible for these small alterations.

NMR study of the oxidation of dMKHA in the presence of copper(II) and H2O2. a A series of 1H NMR acquired at regular intervals for 45 min, at 25 °C and pH 7.4. Inset: peak intensities of the ε methyl hydrogens’ signal from methionine at 2.02 ppm (black) and the oxidized product formed (methionine sulfoxide) at 2.65 ppm (red). b Normalized intensity of the ε methyl hydrogens’ signal from methionine at 2.02 ppm (black), in the presence of 0.1 eq (red) and 0.5 eq (blue) HPCFur. Inset: magnification of the methionine ε methyl signal intensity versus reaction time curves
As the compound HPCFur showed the most promising prospect regarding its protecting role toward dMKHA oxidation, the effect of 0.1 and 0.5 eq of this ligand was also evaluated through NMR in the system constituted by 1:0.1:4 dMKHA (5.0 mM):Cu2+:H2O2. As shown in Fig. 5b (red and blue lines), this aroylhydrazone is able to prevent methionine oxidation to some extent, as assessed by the peak intensity of the signal at 2.02 ppm. After 120 min of reaction, around 12% of the peptide remains unoxidized for both concentrations (0.5 and 2.5 mM) of HPCFur, which represents a protection of around 70% (0.12 against 0.07) compared to dMKHA oxidation in the absence of the compound. It is worth noting that these values are not directly comparable to those obtained by RP-HPLC, since solvent system and reactant concentrations are not the same. However, the protective effect of HPCFur is nonetheless confirmed by the NMR results. Probably more relevant is the corroboration of the compound inhibition effect in histidine oxidation, which seems to occur in a concentration-dependent manner. The inset of Fig. 5b shows a magnification of the methionine ε methyl signal intensity versus reaction time curve, in which one can observe that, upon addition of 0.5 eq HPCFur (blue line), the second oxidation step detected for dMKHA (black line) is completely abolished. This is again in agreement with the data obtained from RP-HPLC and mass spectrometry experiments.
Impact of the ligand HPCFur on the dMKHA interaction with copper(II)
The observed HPCFur protective effect toward dMKHA oxidation certainly deserves some deeper considerations regarding this ligand’s interactions with the peptide–copper(II) system. First of all, 1H NMR spectra showed that HPCFur does not interact directly with dMKHA under the experimental conditions employed in the present study (data not shown). Upon addition of 0.1 eq of Cu2+ to a mixture constituted by dMKHA (5.0 mM) and 0.5 eq HPCFur, all the NMR signals related to the hydrogen atoms in the ligand, altogether with the imidazole signals of the peptide, are broadened beyond detection, indicating the interaction with the paramagnetic ion. The ε methyl group from M109 is also affected, although to a lesser extent. This is in accordance with 1H NMR titration experiments performed for the dMKHA (5 mM):Cu2+ (0.1 eq):HPCFur (0.1–0.5 eq) system (Fig. 6a). However, in the absence of HPCFur, the H111 imidazole signals, while broadened and slightly shifted, are not completely abolished (Fig. 6a, inset). This seems to indicate that the presence of the ligand at the [HPCFur]:[Cu2+] = 1:1 ratio strengthens the interaction of dMKHA with copper instead of preventing it, probably through the formation of a ternary complex whose proposed structure can be observed in Fig. 6b. Coordination of copper to dMKHA is, in this sense, modulated by HPCFur. This is reflected in the large amount of signal loss detected after the first ligand addition. Increasing amounts of HPCFur (up to a ligand-to-metal stoichiometry of 5:1, limited by solubility issues) are able to rescue the signals’ intensities only to the level registered after the addition of copper(II). Recovery of M109 methyl signal, on the other hand, is more significant and, at the end of the titration, the observed signal was around 65% higher than that measured after Cu2+ addition (Fig. 6a, inset).

Suggested interactions in the ternary system dMKHA–copper(II)–HPCFur. a1H NMR titration performed for the dMKHA (5 mM): Cu2+ (0.5 mM): HPCFur (0.5–2.5 mM) system, at 5 °C and pH 7.4. Insets: zooms of the Hε1 signal from H111 (left) and Hε from M109 (right). b Proposed schematic structure for the ternary complex [Cu(H−1dMKHA)(HPCFur)]4+
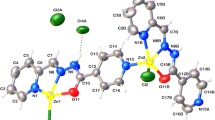
It is worth noting that the ternary coordination compound was identified via ESI–MS(+) measurements, through the peak at m/z 499.32 (Figure S11), assigned to the [Cu(H−1dMKHA)(HPCFur)Cl]3+ species (calculated: m/z 499.54). This may represent a different mechanism of action from the one usually associated with “classical” MPACs, in which competition with the peptide for the binding to the metal ion is replaced by an active modulation of such interactions. Our proposition is that steric- or kinetic-related factors may impair the production of a coordinated hydroxyl radical, proposed to be the actual ROS involved in amino acid oxidation catalyzed by copper in the presence of hydrogen peroxide [49]. In fact, in a very recent publication by the groups of Luigi Casella and Bilha Fischer, the potential of an aminomethylene-phosphonate derivative to inhibit the 4-methylcatechol oxidation induced by the PrP84–114–copper(II) system was associated to the formation of a ternary, redox inactive, PrP84–114–copper(II)–ligand complex [55].
Final remarks
Concerning HPCFur’s potential as a CNS-targeting drug, it is relevant to mention that its structure is in accordance to the Lipinski’s rule of five [56]. The calculated values for the studied hydrazone are − 2.632 and 1.443 for log S and log P, respectively. Bernhardt et al. [30] determined the experimental log P as being equal to 2.47 for this hydrazone. In spite of the significant differences between these values, both of them are in the optimal range for blood–brain barrier crossing, which is between 0 and 3 [57]. In this sense, and concerning the more than acceptable calculated solubility of around 2 mM, as well as the hydrolysis resistance profile observed for this compound, HPCFur presents suitable properties that allow it to be delivered in the brain at potential effective concentrations.
Moreover, as aroylhydrazones can readily be obtained from the reaction between an aldehyde or ketone and an aroylhydrazide, being this synthetic simplicity the key for an extraordinary chemical diversity, we consider that there are many still unexplored possibilities for the structural improvement of this compound. Therefore, new protective agents, even more potent against undesired oxidative damage, can be expected in the coming years.
Conclusions
The effect of two pyridine-2-carboxaldehyde-derived aroylhydrazones, namely, HPCIH and HPCFur, on the copper-catalyzed oxidation of the M112A PrP103–112 short mutant fragment dMKHA was studied by RP-HPLC, mass spectrometry, and NMR spectroscopy. As done previously for HPCIH, HPCFur was fully characterized by FT-IR and NMR spectroscopies, as well as by thermogravimetric analysis. Usually hydrazones present a rich equilibrium of different species in DMSO-d6 solution, which is the case of HPCIH. HPCFur, on the other hand, presented only one species, which was proposed to be the amido tautomer. This ligand also shows an enhanced stability than HPCIH against hydrolysis in a 1% DMSO/water (v/v) solution.
Copper(II) interacts with dMKHA at pH 7.4 primarily through the anchoring site constituted by the side chain of histidine, in a coordination pattern completed by two or three deprotonated amide nitrogens from the main-chain peptide bonds involving H111, K110 and, possibly, M109. The thioether sulfur atom from methionine should be axially coordinated. Additionally, a contact with the oxygen atom from the T107 side chain cannot be completely ruled out, especially if considering the putative formation of linkage isomers in this kind of systems. This redox-active metal is able to catalyze the oxidation of the peptide by hydrogen peroxide through the generation of ROS. When only the peptide, copper(II) and H2O2 are present, one major product is formed, corresponding to the oxidation of the methionine residue to methionine sulfoxide. On the other hand, the presence of compounds such as ascorbic acid hinders the oxidation of methionine, promoting the oxidation of histidine. NMR spectroscopy showed that the consumption of methionine upon reaction with hydrogen peroxide seems to occur in two well-defined steps, with the first one, mainly related to the production of methionine sulfoxide, finishing at around 45 min. The second phase of methionine depletion appears to involve a more intricate process, probably with the side production of the doubly oxidized species, although to a very small extent.
Concerning the impact of the synthesized aroylhydrazones in the oxidation of dMKHA, one could state that HPCIH possesses a similar effect to that of ascorbic acid, protecting methionine from oxidation and, as an undesirable side effect, promoting, although to a lesser extent, histidine oxidation. Only a small amount of doubly oxidized product is generated, while the single oxidized product in which the additional oxygen atom is on the histidine residues is negligible. The compound HPCFur, on the other hand, not only prevented the irreversible oxidation of histidine, but it also protected 85% of the peptide from undergoing methionine oxidation. In the presence of ascorbic acid, the protective effect of HPCFur is still observed. NMR experiments confirmed the potential protective role of HPCFur toward methionine oxidation and, even more important, the elimination of the second step of methionine depletion, which argues favorably to the protection of the histidine residue. Although aroylhydrazones are able to compete for the binding of copper with some amyloidogenic proteins, experimental evidences seem to point to a different mechanism in the present case, through the formation of a dMKHA–Cu(II)–HPCFur ternary complex. Our proposition is that steric- or kinetic-related factors associated with this species may impair the production of a coordinated hydroxyl radical, thus partially protecting the peptide from oxidative damage.
Oxidation of the methionine side chain is a reversible physiologically relevant phenomenon, proposed to act as a molecular mechanism for cellular regulation, since a variety of crucial signaling proteins are functionally altered by this amino acid’s cycling between its oxidized and reduced forms [58]. However, in pathology, oxidation of methionine has been implicated in protein misfolding and consequent aggregation. The compounds studied in the present work, especially HPCFur, show improved solubility and stability in aqueous solution compared to our patented lead hydrazone INHHQ, and display a very interesting protective effect regarding the oxidation of methionine and histidine residues from a model peptide and, as a logical extrapolation, biologically relevant proteins, a process related to both physiological and pathological aging.
Abbreviations
- dMKHA:
-
Ac-SKPKTNMKHA-NH2
- HPCIH:
-
Pyridine-2-carboxaldehyde isonicotinoyl hydrazone
- HPCFur:
-
Pyridine-2-carboxaldehyde 2-furoyl hydrazone
- PrPC :
-
Cellular prion protein
- PrPSc :
-
Scrapie prion protein
- ROS:
-
Reactive oxygen species
- RP-HPLC:
-
Reverse-phase high performance liquid chromatography
- TFA:
-
Trifluoroacetic acid
References
Prusiner SB (1982) Science 216:136–144
Geschwind MD (2015) Continuum (Minneap Minn) 21:1612–1638
Riesner D (2003) Brit Med Bull 66:21–33
Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, Dyson HJ (1999) Proc Natl Acad Sci USA 96:2042–2047
Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K (1996) Nature 382:180–182
Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR (2008) Physiol Rev 88:673–728
Brown DR (2001) Trends Neurosci 24:85–90
Wulf MA, Senatore A, Aguzzi A (2017) BMC Biol 15:34
Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE (1993) Proc Natl Acad Sci USA 90:10962–10966
Prusiner SB (1998) Proc Natl Acad Sci USA 95:13363–13383
Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F (1993) Nature 362:543–546
Salmona M, Forloni G, Diomede L, Algeri M, De Gioia L, Angeretti N, Giaccone G, Tagliavini F, Bugiani O (1997) Neurobiol Dis 4:47–57
Chiesa R, Harris DA (2001) Neurobiol Dis 8:743–763
Requena JR, Groth D, Legname G, Stadtman ER, Prusiner SB, Levine RL (2001) Proc Natl Acad Sci 98:7170
Csire G, Nagy L, Várnagy K, Kállay C (2017) J Inorg Biochem 170:195–201
Elmallah MIY, Borgmeyer U, Betzel C, Redecke L (2013) Prion 7:404–411
Wang Z, Feng B, Xiao G, Zhou Z (2016) Biochim Biophys Acta 1864:346–358
Swaim MW, Pizzo SV (1988) J Leukoc Biol 43:365–379
Hauser-Davis RA, de Freitas LV, Cukierman DS, Cruz WS, Miotto MC, Landeira-Fernandez J, Valiente-Gabioud AA, Fernández CO, Rey NA (2015) Metallomics 7:743–747
Cukierman DS, Pinheiro AB, Castiñeiras-Filho SL, da Silva AS, Miotto MC, De Falco A, de Ribeiro TP, Maisonette S, da Cunha AL, Hauser-Davis RA, Landeira-Fernandez J, Aucélio RQ, Outeiro TF, Pereira MD, Fernández CO, Rey NA (2017) J Inorg Biochem 170:160–168
Cukierman DS, Accardo E, Gomes RG, De Falco A, Miotto MC, Freitas MCR, Lanznaster M, Fernández CO, Rey NA (2018) J Biol Inorg Chem 23:1227–1241
Kozlowski H, Luczkowski M, Remelli M, Valensin D (2012) Coordin Chem Rev 256:2129–2141
Gaeta A, Hider RC (2005) Br J Pharmacol 146:1041–1059
Bolognin S, Messori L, Zatta P (2009) Neuromol Med 11:223–238
Hane F, Leonenko Z (2014) Biomolecules 4:101–116
Hane F, Tran G, Attwood SJ, Leonenko Z (2013) PLoS One 8:e59005
Paik SR, Shin HJ, Lee JH, Chang CS, Kim J (1999) Biochem J 340(Pt 3):821–828
Tabner BJ, El-Agnaf OM, German MJ, Fullwood NJ, Allsop D (2005) Biochem Soc Trans 33:1082–1086
Jomova K, Vondrakova D, Lawson M, Valko M (2010) Mol Cell Biochem 345:91–104
Bernhardt PV, Chin P, Sharpe PC, Richardson DR (2007) Dalton Trans 2007:3232–3244
Banerjee R, Hks K, Banerjee M (2012) Int J Rev Life Sci 2(1):7–16
Gholivand K, Farshadfar K, Mark Roe S, Hosseinia M, Gholamia A (2016) Cryst Eng Comm 18:7104–7115
Xu C, Chen P, Mao H, Shen X, Zhang H, Zhu Y (2005) Synth React Inorg M 35(10):773–778
Zamani HA, Ganjali MR, Norouzi P, Adib M, Aceedy M (2006) Anal Sci 22:943–948
Richardson DR, Becker E, Bernhardt PV (1999) Acta Crystallogr C 55(Pt 12):2102–2105
Richardson D, Bernhardt PV, BeckerEM (2006) US6989397 B1
Ferreira C, Pinto I, Soares E, Soares H (2015) RSC Adv 5:30989–31003
Hoffman RE (2006) Magn Reson Chem 44:606–616
Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD (1995) J Biomol NMR 6:135–140
Kwiatkowski JS, Leszczyński J, Teca I (1997) J Mol Struct 436–437:451–480
Sánchez-López C, Rivillas-Acevedo L, Cruz-Vásquez O, Quintanar L (2018) Inorg Chim Acta 481:87–97
Viles JH (2012) Coord Chem Rev 256:2271–2284
Arena G, La Mendola D, Pappalardo G, Sóvágó I, Rizzarelli E (2012) Coord Chem Rev 256:2202–2218
Grande-Aztatzi R, Rivillas-Acevedo L, Quintanar L, Vela A (2013) J Phys Chem B 117:789–799
Quintanar L, Rivillas-Acevedo L, Grande-Aztatzi R, Gómez-Castro CZ, Arcos-López T, Vela A (2013) Coord Chem Rev 257:429–444
De Ricco R, Potocki S, Kozlowski H, Valensin D (2014) Coord Chem Rev 269:1–12
Belosi B, Gaggelli E, Guerrini R, Kozłowski H, Łuczkowski M, Mancini FM, Remelli M, Valensin D, Valensin G (2004) ChemBioChem 5:349–359
Kozłowski H, Kowalik-Jankowska T, Jeżowska-Bojczuk M (2005) Coord Chem Rev 249:2323–2334
Zhao F, Ghezzo-Schöneich E, Aced GI, Hong J, Milby T, Schöneich C (1997) J Biol Chem 272(14):9019–9029
Schöneich C (2000) J Pharmaceut Biomed 21:1093–1097
Roepstorff P, Fohlman J (1984) Biomed Mass Spectrom 11:601
Bridgewater JD, Vachet RW (2005) Anal Biochem 341:122–130
Concetti A, Gariboldi P (1990) Biol Met 3:125–126
He L, Wang X, Zhu D, Zhao C, Du W (2015) Metallomics 7:1562–1572
Cohen NP, Presti EL, Dell’Acqua S, Jantz T, Shimon LJW, Levy N, Nassir M, Elbaz L, Casella L, Fischer B (2019) Inorg Chem (in press)
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Adv Drug Deliv Rev 23:3–25
Pajouhesh H, Lenz GR (2005) NeuroRx 2(4):541–553
Hoshi T, Heinemann SH (2001) J Physiol 531:1–11
Acknowledgements
NAR, DSC, and BNE are grateful for the scientific Brazilian funding agencies FAPERJ (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro, Brazil), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil), and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil) for the research fellowships and scholarships awarded. The financial support from the Hungarian Scientific Research Fund (NKFI-115480 and NKFI-128783) is appreciated. The research was supported by the EU and co-financed by the European Regional Development Fund under the project GINOP-2.3.2-15-2016-00008. The Hungarian co-authors also thank the UNKP-18-4 New National Excellence Program of the Ministry of Human Capacities. This research was also financed by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. The authors wish to thank Prof. Dr. Christian Griesinger, director of the NMR-based Structural Biology Department, for his support and fruitful discussions, and Kerstin Overkamp, from the same institution, for her experimental support in the ESI-MS measurements.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cukierman, D.S., Bodnár, N., Evangelista, B.N. et al. Impact of pyridine-2-carboxaldehyde-derived aroylhydrazones on the copper-catalyzed oxidation of the M112A PrP103–112 mutant fragment. J Biol Inorg Chem 24, 1231–1244 (2019). https://doi.org/10.1007/s00775-019-01700-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-019-01700-2