
Abstract
Metal dyshomeostasis in the brain (BMD) has often been proposed as a possible cause for several neurodegenerative disorders (NDs). Nevertheless, the precise nature of the biochemical mechanisms of metal involvement in NDs is still largely unknown. Mounting evidence suggests that normal aging itself is characterized by, among other features, a significant degree of metal ion dysmetabolism in the brain. This is probably the result of a progressive deterioration of the metal regulatory systems and, at least in some cases, of life-long metal exposure and brain accumulation. Although alterations of metal metabolism do occur to some extent in normal aging, they appear to be highly enhanced under various neuropathological conditions, causing increased oxidative stress and favoring abnormal metal–protein interactions. Intriguingly, despite the fact that most common NDs have a distinct etiological basis, they share striking similarities as they are all characterized by a documented brain metal impairment. This review will primarily focus on the alterations of metal homeostasis that are observed in normal aging and in Alzheimer’s disease. We also present a brief survey on BMD in other NDs (Amyotrophic Lateral Sclerosis, Parkinson’s, and Prion Protein disease) in order to highlight what represents the most reliable evidence supporting a crucial involvement of metals in neurodegeneration. The opportunities for metal-targeted pharmacological strategies in the major NDs are briefly outlined as well.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Neurodegenerative disorders (NDs) include a group of heterogeneous diseases often characterized by the deposition of proteins within neurons or brain parenchyma and/or by oxidative stress exacerbation. These events typically occur because of the failure of several proteins to fold correctly, or to remain correctly folded, giving rise to many different types of biological malfunctions such as misfolding and aggregation and, therefore, to many forms of diseases (Soto and Estrada 2008). The substantial inability to treat NDs most likely arises from the lack of a sufficient understanding of their etiology, which hampers a clear discrimination between the true causes and the consequences of the neurodegenerative processes.
Along with many other etiological factors, metal ions such as copper (Cu), zinc (Zn), iron (Fe), and aluminum (Al) have been all advocated as modulators of the aggregation of some specific proteins that are directly linked to these diseases. In addition, many NDs show, among other features, a common impairment of metal ion brain homeostasis. The above metals, apart from Al, are fundamental for correct brain functioning; however, at the same time they need to be strictly regulated to avoid the triggering of detrimental cell processes; indeed, depletion and accumulation of these metals can lead both to abnormal interactions with proteins or nucleic acids and to consequent cell damage. The brain, therefore, strictly regulates the metal ion fluxes as there is no passive transport of metal ions across the blood–brain barrier (BBB). Thus, for the major NDs, the described metal imbalance is not simply and solely due to an increased exposure to metals but, rather, to a more complicated impairment in relevant homeostatic mechanisms.
Intriguingly, aging is considered as one of the most relevant risk factors for NDs and accumulating evidence has revealed a general age-related increase for the above metals in the brain (see next paragraph). Brain metal accumulation, especially for redox metals such as Cu and Fe, leads to increased oxidative stress (with the production of excess superoxide and hydroxyl radicals), and is associated with severe neuronal damage in physiological aging as well as in NDs (e.g., Alzheimer’s disease) (Butterfield et al. 2007). In a way, metals may provide the link between protein misfolding and aggregation, oxidative stress and the cascade of biochemical alterations, eventually leading to neuronal cell death. Consequently, the transport and the distribution/accumulation processes of metals are of particular interest especially because NDs with a distinct etiology could share some common pathogenetic pathways. The essential role of metals for a variety of general cellular functions is unanimously recognized, as well as the fact that they are required by at least one quarter of all proteins as cofactors (Ferrer et al. 2007).
Brain Metal Dyshomeostasis
Several excellent reviews dealing with metal ions physiology have been published recently (e.g., Garrick and Garrick 2009 for Fe; Nakashima and Dyck 2009 for Zn). The issue is so complicated that it is very difficult to effectively summarize the countless aspects of the Cu, Fe, and Zn metabolic pathways and for this reason we refer to those specific reviews. In turn, the occurrence of metal dysregulation or dyshomeostasis in the brain has been described by a vast literature under a variety of conditions ranging from normal aging to genetic diseases, from reduced metal supplementation to excessive metal exposure and in relation to a variety of NDs (Zatta 2003; Zatta et al. 2009). However, the detailed significance of brain metal dyshomeostasis (BMD) is still a matter of intense debate. It is generally accepted that BMD can be defined in terms of the occurrence of relevant modifications of metal concentrations either in the total mass of the brain or in specific brain areas as a consequence of physio-pathological events. According to this definition, the assessment of metal dyshomeostasis would primarily depend on the obtainment of reliable and unambiguous analytical data on brain tissues, mainly by sophisticated techniques such as atomic absorption spectroscopy or inductively coupled plasma-mass spectrometry measurements. Indeed, metal dyshomeostasis may arise from an abnormal metabolic activity of a specific metal ion due to either misallocation or the lack or insufficiency of specific metal binding proteins. This latter kind of dyshomeostasis is far more difficult to establish given that either a detailed metal speciation or a metal distribution studies are required. Unfortunately, so far, the availability of adequate sets of analytical and/or speciation data for brain metals is disappointingly scarce, in that most studies report only fragmentary and sporadic data which are sometimes contradictory. The collection of reliable analytical data for brain metals is complicated further by the extreme structural, functional, topographical, and architectural complexity of the brain itself. Thus, the assessment of BMD is currently founded on several independent, often unrelated, qualitative indications rather than on conclusive quantitative data. It follows that the new and systematic analytical data on brain metal concentration distribution and speciation are strongly required.
Metal Dysmetabolism in Aging
The aging process, though physiological in nature, has been considered as a critical condition characterized by a progressive deterioration of the overall homeostatic mechanisms. In the human brain, aging implies a variety of morphological alterations which include enlargement of ventricles and progressive decrease of brain weight (Bertoni-Freddari et al. 2008). Moreover, a significant reduction in the number of synapses has been reported for different regions of the central nervous system (CNS) both in animals and in humans, confirming that this is a characteristic feature of the aging brain (Bertoni-Freddari et al. 1996). Thus, a distinction between the neurological alterations occurring in normal aged brains and in NDs is not always easy to identify.
Interestingly, it has been demonstrated in different animal species, including humans, that aging itself is characterized by an alteration in brain metal content and specific topographical distribution. The mechanisms responsible for this imbalance are not clearly understood; therefore, one possible explanation might lie in the age-induced progressive failure of metal controlling systems and ineffective functionality of physiological barriers (e.g., BBB, gastrointestinal tract) due to cumulative errors occurring throughout the course of life. Tarohda et al. (2004) observed in rats a variation with age of manganese (Mn), Fe, Cu, and Zn. The concentrations of each of these metals were quite specific. Cross et al. (2006) showed, in aged rats, reduced bulk transport of Mn from the olfactory bulb to the anterior tract, highlighting a significantly decreased rate of Mn transport when compared to young rats.
We also reported the analysis of Cu, Zn and Mn in the brains of two series of young (8–16 months) and adult (9–12 years) bovines. We found relevant age-dependent differences in the distribution of Cu and Zn, the concentrations of which were markedly higher in older animals. In contrast, Mn seemed to redistribute in the different cerebral areas rather than drastically change with age, a fact that is most evident in basal and central regions of the brain, including the hypothalamus, thalamus, and corpus callosum (Zatta et al. 2008).
Metal distribution in relation to age has also been widely analyzed in humans. Zn concentration in the brain is typically 150 μmol/l (Takeda et al. 2003). However, its distribution is not homogeneous since it is significantly higher in gray than in white matter. Brain regions particularly rich in this metal are the hippocampus, the amygdala, and the cortex (Weiss et al. 2000). Even if during aging Zn distribution in the brain changes in relation to the region considered, many studies have reported little or no decrease in its levels in mice and humans (Del Corso et al. 2000).
Investigation of the Cu content in the aged human brain has not yet allowed a definite conclusion to be reached. However, normal aging seems to increase Cu levels in several tissues, including the brain (Morita et al. 1994). The highest concentrations of Cu were found in substantia nigra (SN) and in other cerebellar regions (Rajan et al. 1997).
The total brain Fe, after the very early stage of life, increases rapidly and remains stable for the rest of the lifespan. According to the recent reports, it increases in the aged brain (Stankiewicz and Brass 2009) particularly in SN and globus pallidus (GP) (Gotz et al. 2004).
Al concentration during the lifespan appears to be biphasic: there is an increase with age up to 40 years, followed by a plateau up to 70 years. A second increase is observed in the course of the eighth and ninth decade of life (Roider and Drasch 1999). Of the different brain areas analyzed, GP, SN, and nucleus ruber (NR) were found to be the richest in Al in the aged population (Speziali and Orvini 2003). It therefore seems that a breakdown of metal regulation could be an inevitable consequence of aging.
Metal Dysmetabolism in NDs
The progressive loss of specific neuronal populations and the extra-intra neuronal accumulation of protein deposits produce dysfunctions which lead to neurodegeneration (Jellinger 2003). It is generally accepted that NDs have a multifactorial origin (e.g., genetic, environmental, and endogenous factors) but that the “primum movens” still remains elusive. Much literature suggests that, of the numerous contributing factors, an altered metal metabolism may accelerate/initiate the neurodegenerative process (Allsop et al. 2008; Lovell 2009). Despite intense study of the role of metals in NDs, it has not yet been convincingly proven whether metal impairment could be an aggravating factor or simply the consequence of neurodegenerative progression. Nevertheless, this alteration is relevant and common to different kinds of NDs, suggesting a high and probably exclusive correlation. It is worth noticing that NDs are also called conformational diseases, in that they are characterized by altered protein conformations, which give rise to different types of cellular dysfunctions (Drago et al. 2008b). Direct interactions between these altered proteins and several metal ions have been widely documented (Ricchelli et al. 2005, 2006, 2007; Kenward et al. 2007; Barnham and Bush 2008); it follows that metals could play a role in the sequence of events which leads to protein misfolding. In addition, aberrant interaction between transition metals (e.g., Cu, Fe) and proteins might be a source of reactive oxygen species (ROS) which contribute to cell death.
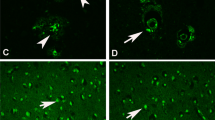
It is also worth noting that several NDs share a common alteration in the brain expression of metallothioneins (MTs), a ubiquitous family of low molecular weight cysteine rich proteins. MTs fulfill different functions which include homeostasis and transport of essential metals (Cu and Zn), metal detoxification and protection against oxidative stress (see Vašák and Meloni 2008). Therefore, as MTs directly correlate with the buffering of metals, their over-expression in NDs, and in aging (Fig. 1), may reflect a protective endogenous response to a sub-chronic state of oxidative stress (Zatta 2008).

MTs in young and old rat brain. a Quantification of total MTs by a silver saturation method (as described in Zatta et al. 2008). MTs level was significantly increased in cerebellum and hemisphere of 3-year-old rat compared to younger animals. b Immunohistochemical studies showed an increased number of reactive astrocytes in the older rat compared to the younger. This was shown with glial fibrillary acidic protein (GFAP) as well as with MT I–II staining. GFAP-reactive cells were present in all the sections of the two experimental series, being the cytoplasm of astrocytes strongly positive especially in the older series. The capillary vessels were only positive because they were associated with astrocytes. Positive MT staining was found in capillary vessels. In young rats we observed a total negativity to MTs as compared with the old rats
Metal Dysmetabolism in Alzheimer’s Disease
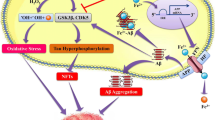
Alzheimer’s disease (AD) is the most frequent cause of dementia in the elderly. It is characterized by the abnormal formation of extracellular senile plaques (SP), composed of, in addition to other features, Aβ, and of intracellular neurofibrillary tangles containing hyperphosphorylated tau protein (Haass and Selkoe 2007; Iqbal et al. 2009). Many studies have implicated biometals in the development and/or progression of AD (Crichton et al. 2008) (Fig. 2). In analytical studies, the hippocampus and amygdala were sites where the concentrations of various elements were most frequently found to be different between control and AD groups (Speziali and Orvini 2003). Convincing results have shown increased metal concentrations within the SP compared with surrounding tissues both in transgenic mice (Rajendran et al. 2009), as well as in humans (Lovell et al. 1998).

Aβ oligomerization and metal ions. Aβ aggregation follows a sequence which includes the formation of soluble, low molecular weight oligomers. The aggregation process is extremely dynamic and oligomers associated rapidly to form higher-order aggregates. Among many other factors, metal ions (e.g., Al, Cu, Fe, and Zn) have been shown to affect the pathways of Aβ folding. The abnormal aggregation of Aβ could stimulate astrocyte activation as well as oxidative stress events, which both result in impairment of neuronal functions
Zinc
The potential role of Zn as a cofactor in the pathogenesis of AD was strengthened when Lovell et al. (1998) found a Zn enrichment in SP and a Zn elevation in the neuropil of AD patients as compared to controls. These results were later confirmed by several other research groups (Stoltenberg et al. 2005; Miller et al. 2006). However, controversial results have been published in an attempt to quantify Zn in AD serum (Rulon et al. 2000; Dong et al. 2008) or cerebrospinal fluid (CSF) (Molina et al. 1998; Gerhardsson et al. 2008).
Despite these controversial reports, studies on the role of Zn in AD have shown that variation of brain Zn levels may contribute to precipitate Aβ, giving rise to protease-resistant unstructured aggregates which synergistically increase Aβ neurotoxicity (Cuajungco and Lees 1997; Bush 2003). In addition, Zn binding to Aβ could also reduce Zn availability at the synaptic cleft leading to deleterious effects in terms of the role of Zn in neuronal signaling and synaptic plasticity (see Frederickson et al. 2005). As a second observation, several studies have supported the hypothesis that vesicular Zn released in the synaptic cleft during neurotransmission may be one contributing factor for the recruitment of Aβ oligomers to synaptic terminals (Deshpande et al. 2009).
Furthermore, when transgenic mice lacking Zn transporter 3 (ZnT3) were crossed with Aβ-producing mice, a marked decrease of the plaque load was observed (Lee et al. 2002), suggesting that synaptic Zn may play a role in enhancing Aβ aggregation and plaque deposition. Moreover, it is worth noting that MTs I and II were histochemically found to be dramatically increased in astrocytes of AD brains compared with those of controls (Zambenedetti et al. 1998). Considering that Zn is an inducer of this family of proteins, a correlation between Zn levels and the pathology should be further investigated.
Behavioral studies on transgenic mice examining the effect of Zn supplementation reported an increased impairment of spatial memory, but with a concomitant unexpected reduction of Aβ deposits (Linkous et al. 2009). In contrast, compounds affecting Zn homeostasis have been shown to decrease Aβ brain deposition (Lee et al. 2004; Adlard et al. 2008).
Cuajungco and Faget (2003) conducted a good review of several controversial findings and conclude in favor of a paradoxical role for Zn in AD. It could indeed be that Zn is released following oxidative/nitrosative stress factors implicated in AD etiology. In turn, this Zn increase can trigger neuronal death giving rise to a vicious cycle.
In any case, several aspects of the potential involvement of Zn in AD pathogenesis remain unclear. Better understanding the correlation between Zn and AD is of key interest given that Zn supplementation has been found to be protective in the treatment of age-related macular degeneration (AREDS 2001) and it has been proposed as having beneficial properties for the elderly (see Mocchegiani et al. 2005). In any case, studies reporting the effect of Zn supplementation in AD patients are still very scarce. Although it is difficult to evaluate the real effect of Zn supplementation, given that several other micronutrients were contextually administered, much caution in designing Zn protocols is required. It is well known that increased dietary Zn causes Cu deficiency and that in turn, given that Cu is involved in Fe transport, it can induce anemia (Salzman et al. 2002). Thus, if any Zn supplementation therapy is proposed then a strict regulation of other nutrients must be implemented.
Copper
The potential involvement of Cu in physiology (Kramer et al. 2003) and pathology (Bush 2003) is even more complicated. There is a general agreement with the hypothesis that the AD brain could be characterized by an excess of Cu in the extracellular space (Crouch et al. 2006), since the metal ion accumulates in large quantities in the SP (Lovell et al. 1998), and at the same time by an intracellular decrease of Cu as compared to healthy control brain. Nevertheless, the analytical data are often controversial; Deibel et al. (1996) reported a general decrease in total Cu brain level of approximately 20% in AD brain compared to controls, even if other groups have failed to confirm this data (Loeffler et al. 1996).
The link between AD and Cu metabolism could be explained by the potential control exerted by Cu on Aβ levels. It has been demonstrated that exposing cells over-expressing amyloid precursor protein (APP) to high Cu levels results in a decrease of secreted Aβ (Borchardt et al. 1999). The same effect was obtained by elevating Cu levels in the brain both by genetic method (Phinney et al. 2003) as well as with dietary supplementation (Bayer et al. 2003) in AD animal models. It has been proposed that an overproduction of APP, and consequently of Aβ, can lead to a Cu efflux from the cell which then causes a concomitant reduction of the protective superoxide dismutase (SOD-1) enzyme (Fig. 3). Thus, Cu supplementation, or better proper delivery into the brain, could be beneficial (Bayer et al. 2006). In this regard, Crouch et al. (2009) demonstrated that increased intracellular Cu availability inhibited the accumulation of Aβ oligomers and τ phosphorylation. Kessler et al. (2008) recently evaluated the effect of oral Cu supplementation on AD CSF biomarkers in a pilot phase 2 clinical trial. They reported a stabilizing effect of the supplementation in terms of contrasting the decrease of Aβ1–42 in the CSF, which is generally reported in AD patients compared to controls (Lewczuk et al. 2004). However, this effect does not correspond to any improvement in cognitive performance and, more generally, as highlighted by Quinn et al. (2009), none of the above studies have demonstrated a Cu deficiency in AD patients.

Potential interaction between APP and Cu. It has been proposed that Aβ overproduction could stimulate Cu efflux from cell cytoplasm, which consequentially causes the reduction of superoxide dismutase 1 activity (SOD-1)
On the other hand, concomitantly with these observations, other studies reported that dietary Cu intake could stimulate a cascade of detrimental effects on neuronal cells. Cu binding to APP is likely to promote APP dimerization and its localization into lipid rafts, where it is processed more rapidly, generating Aβ peptides (Kawarabayashi et al. 2004). In addition, Sparks and Schreurs (2003) showed that the addition of Cu (0.12 ppm) to drinking water (which is largely below the maximum contaminant level for Cu, as fixed by the Environmental Protection Agency at 1.3 ppm) exacerbated Aβ accumulation and learning deficit in a cholesterol-fed rabbit model of AD, thus suggesting that Cu may negatively influence Aβ clearance from the brain. The detrimental effect of combining trace amounts of Cu and a cholesterol rich diet was also confirmed in mice apparently caused, at least in part, by the activation of the apoptotic pathway (Lu et al. 2006). More recently, epidemiological studies hinted that high Cu dietary intake, associated with a high saturated and trans fat diet, accelerated cognitive decline (Morris et al. 2006). Cu was found to exert neurotoxic effects even if there was no correlation with elevated cholesterol diet. Indeed, Kitazawa et al. (2009) reported alteration of APP processing, enhanced Aβ production and τ phosphorylation in young triple transgenic (3×Tg-AD) mice after chronic Cu exposure. In conclusion, it is still a matter of debate as to whether Cu intake (both dietary as well as excess) can alter APP and Aβ metabolism and with what results.
In relation to this matter, recent studies on AD patients have evaluated a specific fraction of Cu in the serum, namely the levels of non-ceruloplasmin bound Cu (called “free”), which despite being extremely low, can cross the BBB. It has been demonstrated that in AD patient serum “free” Cu increases, and this correlates with the main cognitive deficits of the disease (Squitti et al. 2005, 2006; Babiloni et al. 2007). It has been proposed that about 3% of the serum “free” Cu can enter the brain, potentially stimulating Aβ aggregation (Squitti et al. 2006). An interesting case report compared serum Cu in a pair of elderly monozygotic twins discordant for AD. The AD twin had a 44% increment of Cu and peroxide concentrations compared to the non-AD twin (Squitti et al. 2004).
Overall, despite the large number of studies, the mechanism which links the potential Cu dysfunction to AD is still currently unclear.
Iron
Much of the literature reports increased Fe levels and Fe-binding proteins in the AD brain (Lovell et al. 1998; Connor and Lee 2006; Cahill et al. 2009) and several hypotheses have been proposed which attempt to clarify this involvement (see Altamura and Muckenthaler 2009). Smith et al. (2007) stated this could be a secondary effect caused by, for example, increased heme oxygenase activity in response to oxidative stress (Schipper, 2004). This could be further strengthened by the fact that, despite being found to interact with Aβ in vitro (Hu et al. 2006), Fe does not co-purify with Aβ extracted from plaques (Opazo et al. 2002). Most recently, other pathways have been explored; in particular, it has been assumed that Fe could directly influence Aβ production through the modulation of furin, a ubiquitous enzyme, whose proteolytic activity is required for many cellular processes, including α- and γ-secretase processing. According to Silvestri and Camaschella (2008) high cellular Fe levels lower furin activity, which in turn reduces α-secretase activity favoring β- and γ-secretase activity with the consequent enhancement of Aβ production. In accordance, the furin mRNA level was reduced in AD brain patients (Hwang et al. 2006).
A further correlation between Fe and AD is based on the observation that oxidative stress markers are highly expressed in AD-affected brain regions (e.g., Zambenedetti et al. 1998) and this matches with the redox-active nature of Fe. Fe-dependent ROS production is indeed able to increase Fe cellular uptake (Pantopoulos and Hentze 1998) which, in turn, could increase oxidative damage giving rise to a vicious cycle.
Another aspect highlighted by Rogers et al. (2002a, b) was the presence of a functional Fe-regulatory element in the 5′-URT mRNA encoding the APP. Intracellular Fe levels were shown to modulate APP synthesis in neuroblastoma cells, while the addition of a Fe-chelator reduced APP levels.
In summary, although the mechanism for Fe accumulation in AD is still unknown, it is necessary to consider Fe as an important cofactor. It is also undeniable that diseases directly related to increased levels of this metal (e.g., haemochromatosis) are not characterized by enhanced deposition of SP, proving that it could be one of many other contributing factors.
Aluminum
Since the 1970’s it has been hypothesized that exposure to Al may enhance the pathogenesis of AD, mainly in genetically predisposed subjects (Campbell 2006). Significantly raised levels of Al were indeed reported in the parietal cortex of the AD brain as compared with controls (Srivastava and Jain 2002; Yumoto et al. 2009). Moreover, early studies using Laser Microprobe Mass Analysis (LAMMA) showed high Al concentrations within the AD neurofibrillary tangles (Good and Perl 1993; Bouras et al. 1997). Up until now, no physiological role has been established for this element (Bala Gupta et al. 2005). Some studies have summarized the effects of occupational exposure to Al suggesting that it induces relevant neurotoxic effects following acute or subacute exposure (see Krewski et al. 2007).
Alfrey et al. (1976) described for the first time a neurological condition resembling AD dementia which was called dialysis encephalopathy (DE). DE consists of an abnormal accumulation of Al in the brain of uremic patients with renal failure undergoing chronic dialysis, which occurs when tap water, without any further purification, was used in the dialysis process (Zatta et al. 2004; Zatta 2006). The effects of Al on cognitive functions were reversible since DE patients greatly improved following treatment with desferrioxamine (DFO) (Yokel 1994). Once Al was removed from the “dialysis bath” the DE practically disappeared. These findings have given rise to widespread speculation as to whether AD and Al could be linked, but no conclusive results were established (Reusche 2003). Indeed, the epidemiological results which addressed the problem of Al in drinking water in connection with the incidence of AD were controversial (Reusche 2003). In addition, many nephrologists currently use Al salts to decrease the hyperphosphatemia in uremic subjects with no major incidence of AD among these patients with respect to general population. Thus, Al itself cannot be a sufficient trigger of AD and there must be another reason for the potential AD–Al connection.
In this complex scenario, we have recently proposed that the binding of Al to Aβ can promote a conformational change which stabilizes the peptide in its oligomeric form (Drago et al. 2008a; Zatta et al. 2009), considered by the recent literature as the most toxic specie (Glabe 2005). Such Aβ–Al metal complex showed a dramatic reduction in the sequestration in the brain microcapillaries and an increased high permeability across BBB, a phenomenon that led to intra-cerebral accumulation of Aβ–Al (Banks et al. 2006). Al favors the exposure of peptide hydrophobic clusters which results in a peculiar aggregation pattern compared to the other metals tested (Cu, Fe, Zn). Furthermore, we have demonstrated that in neuronal cell cultures exposed to different Aβ–metal complexes (Aβ–Cu, Aβ–Zn, Aβ–Fe, and Aβ–Al), only the Aβ–Al complex was able to alter glutamate-driven intracellular calcium (Ca) rises and to inhibit the oxidative respiration in isolated rat brain mitochondria (Drago et al. 2008c). Finally, Al dyshomeostasis was also recently found in a triple transgenic AD mouse, the 3×Tg-AD: experiments employing mass spectrometry indicate that, when compared with the distribution of other AD-relevant metals (Zn, Cu, and Fe), Al is the only metal ion that increased significantly in the cortex of 14-month-old 3×Tg-AD mice (Drago et al. 2008c).
In summary, the potential involvement of Al in AD still remains of great interest yet controversial along with many other hypotheses on AD etiology (www.alzforum.com).
Finally, several other arguments pro and con the possible role of Al in AD are represented in the literature; however, we pinpoint two aspects that necessarily have to be considered before approaching this issue. Firstly, Al has a complex hydrolysis pH-dependent chemistry in biological systems which can account for many inconsistencies reported in the literature on the effects of Al on animal or cellular models. As an example, when Al inorganic salts such as chloride, sulfate, hydroxide or perchlorate are dissolved in water at a calculated concentration of 10 mM, the analytical Al concentration in solution is about 50 μM. The use of Al-lactate or Al-aspartate, however, increases the soluble Al concentration to 50–330 μM (Zatta 2002). Hence, the examination of the metal bioavailability under physiological conditions has to be taken into account while designing Al studies. Secondly, a distinction has to be made between the concepts of neurotoxicity and neurodegeneration. Al has been aptly described as a neurotoxic element (Zatta 2002) if it cannot be physiologically excreted or it is in direct contact with the brain. Besides the neurotoxicity of Al at high concentration, the role of this metal ion in affecting pathways related to neurodegenerative mechanisms should be further investigated.
Metal Dysmetabolism in Parkinson’s Disease (PD)
Parkinson’s disease (PD) is a fatal disease characterized by a progressive and slow degeneration of neurons in SN, a small area of dopaminergic cells located in the mid-brain (Kaur and Andersen 2002). Dopamine depletion produces several typical symptoms of PD such as bradykinesia, resting tremor and postural dysfunction. Remaining neurons display cytosolic inclusions called Lewy bodies. The major component of Lewy bodies is α-synuclein which is a soluble, acidic and natively unfolded protein (for a recent review see Olivares et al. 2009). The cause of PD remains unknown but mitochondrial dysfunction, oxidative damage, environmental factors and genetic predisposition have all been proposed as etiological factors (Sayre et al. 2005; Olivares et al. 2009).
Several early epidemiological studies have also shown that exposure to metals such as Cu, Zn, Mn, Fe, and Al can be associated with increased incidence of PD (Powers et al. 2003). Furthermore, post-mortem analysis of PD brain tissues revealed an increased concentration of Fe in nigral neurons (Jellinger 1999) and of Al and Fe in Lewy bodies (Hirsch et al. 1991) using various quantitative methods and histochemical techniques (Zecca et al. 2004). Consistent with these findings, Dexter et al. (1991) also found increased total brain Fe levels as well as decreased ferritin content. Fe may contribute to the progression of the pathology by catalyzing oxidative reaction (Andersen 2004). In dopaminergic neurons Fe2+ is likely to react with hydrogen peroxide produced during deamination of dopamine, thus producing reactive radicals which are detrimental to cells (Andersen 2004). In contrast, serum Fe was found to be in reduced levels in PD as compared to control subjects (Alimonti et al. 2007). Although the explanation for this imbalance is unknown it has been proposed that it has a role in enhancing free radical production (Faucheux et al. 2003). As a matter of fact, the oxidation of methionine residues represents a crucial factor in the formation of soluble α-synuclein oligomers (Leong et al. 2009). Moreover, Fe and free radical generators (e.g., hydrogen peroxide and dopamine) have been found to stimulate the production of intracellular aggregates containing α-synuclein (Osterova-Golts et al. 2000). Hence, a recent study by Bayir et al. (2009) proposed that α-synuclein may have a dual function: preventing apoptosis through the oligomerization of cytochrome c but that this is, at the same time, associated with the formation of the peroxidase complex representing a source of oxidative stress (Zhou et al. 2008).
Indeed, increased oxidative stress and reduction in antioxidative defense have been shown in SN of PD patients (Sofic et al. 2006). It follows that oxidative stress has been linked to both the initiation and the progression of PD, but several findings suggest that the neurodegenerative process starts with neurons being affected by some etiological factor, resulting in a cascade of events which include formation of ROS (see Zhou et al. 2008 for a recent review).
The study of metals/α-synuclein interaction, besides being associated with oxidative stress, has also been stimulated by the findings that metals are able to induce structural change in α-synuclein conformation. Many studies have reported that enhanced α-synuclein aggregation in the presence of Cu (Binolfi et al. 2008) but also Al, Fe, and Mn (Paik et al. 1997; Uversky et al. 2001) can effectively cause acceleration in the rate of α-synuclein fibril formation. The clinical significance of this interaction, as well as the involvement of oxidative stress in the etiology of the disease, deserve further investigation, despite both being recognized as important pathophysiological features.
Metal Dysmetabolism in Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is characterized by the selective loss of upper and lower motor neurons resulting in muscular atrophy, including respiratory muscles, complete paralysis and death (Eisen 2009). The exact etiology of the disease is unknown but approximately 10% of ALS cases are caused by inherited mutations in gene encoding Cu/Zn–superoxide dismutase 1 (SOD-1), a cytosolic enzyme with a Zn and Cu binding site, which catalyzes the conversion of superoxide radicals to hydrogen peroxide (Rosen et al. 1993). The remaining cases are sporadic, probably multifactorial, with environmental and genetic components contributing to disease susceptibility (Eisen 2009).
The toxicity of SOD-1 mutants seems not to be due to the loss of enzymatic activity, but rather to a toxic gain of function (Boillée et al. 2006) (Fig. 4). Several studies demonstrated that mutations in SOD-1 altered its metal affinity or coordination (Goto et al. 2000) and in particular decreased the affinity of SOD-1 for Zn up to 50-fold compared to the wild-type form (Crow et al. 1997) and increased the affinity for Cu (Lyons et al. 1996). SOD-1-decreased affinity for Zn led to an increment in nitrotyrosine formation and induced apoptosis in cultured motor neurons (Estevez et al. 1999) while Cu-increased affinity enhanced Cu-mediated oxidative stress which may lead to neuronal death (Said Ahmed et al. 2000). Moreover, recent evidence has shown the presence of enhanced amyloid-like aggregate formation (Oztug Durer et al. 2009) as well as increased hydrophobicity (Tiwari et al. 2009) upon removal of metals from SOD-1.

CuZnSOD and ALS. There are several different mutations associated with familial ALS, but about 20% of these cases have a defect in the gene encoding CuZnSOD. a The mutant enzyme catalyzes H2O2-dependent peroxidase-type reactions and forms nitrating species from peroxynitrite. b Shape and dimension of CuZnSOD active site channel (from Yim et al. 2003)
In addition to these in vitro findings, high Fe levels have been reported in spinal neurons of ALS patients (Kasarskis et al. 1995). Fe accumulation has been shown in a mouse model of ALS both in neurons and glia but it has been proposed that different molecular mechanisms may contribute to Fe accumulation in these two cell types (Jeong et al. 2009). Also serum ferritin levels were significantly increased in ALS patients as compared to controls which may reflect a general increase in stored Fe (Goodall et al. 2008). On the contrary, no differences were found in the level of Fe in the serum (Goodall et al. 2008). Subramaniam et al. (2002) observed no alteration in total Cu content in mutant SOD-1 transgenic mice, despite the fact that Cu, Zn, and Ca chelators have been shown to be protective (Petri et al. 2007).
All these findings suggest that the involvement of metal ion imbalance in ALS cannot be excluded even if further investigation is clearly necessary to establish its contribution in ALS development.
Metal Dysmetabolism in Prion Protein Disease (PrPD)
Prion protein disease (PrPD) is a group of fatal and rare NDs which include Gerstmann-Straussler-Scheinker (GSS) disease, kuru, fatal familial insomnia, sporadic and variant form of Creutzfeldt-Jacob disease (CJD) (Wadsworth and Collinge 2007). The common feature shared by these disorders is the accumulation in the CNS of an abnormally folded isoform (PrPSc) of the cellular prion protein (PrPC) (Fig. 5). PrPC forms soluble monomers, while PrPSc forms insoluble aggregates with increased β-sheet secondary structure (Prusiner 1998). The etiology of the disease is still elusive but recent investigations have hypothesized that PrPC might provide neuroprotection (Nicolas et al. 2009) which is suppressed with PrPSc (Harris and True 2006). In addition, PrPC may be involved in neurotransmitter release (Re et al. 2006).

PrP protein. a Schematic representation of the human PrP primary sequence showing the main Cu binding site. b PrPC is an extracellular protein which is anchored to the plasma membrane. Its detrimental conversion to PrPSc results in the formation of fibrils which accumulate in the extracellular space. c Proposed mechanism of interaction between PrP and Aβ oligomers as hypothesized by Laurén et al. (2009)
The fact that PrP is able to bind Cu2+ with moderate affinity (Rachidi et al. 2003) suggests that it could be involved in Cu metabolism (Sassoon and Brown 2003). An increasing body of evidence suggests that the function of PrPC is that of an antioxidant and/or a Cu-sequestering protein, active in the prevention of Cu toxicity in neurons (Sassoon and Brown 2003). Thus, although PrPC undeniably binds Cu, it is unclear whether PrPSc acts in the same way. Owing to this connection to Cu, many efforts have been made to characterize the metal binding process and to investigate more thoroughly the role of metal ions in general in the etiology of the disease.
Miura et al. (1996) reported an increase in PrP α-helix content in the presence of Cu. These kinds of studies gave rise to many subsequent investigations. Nevertheless, despite the huge number of published papers, no clear conclusions on the relation between Cu and PrP have been drawn and conflicting results have been reported.
It is generally accepted that PrPC expression can alter Cu uptake into cells and enhance Cu incorporation into superoxide dismutase (SOD) (Brown et al. 1999). Wells et al. (2006) proposed that the physiological binding of Cu to normal PrPC in the brain occurs only when the concentration of Cu increases locally as, for instance, during depolarization. Furthermore, Cu itself has recently been shown to induce expression of cellular PrP in primary cell cultures, upregulating the expression of this protein both at the cell surface, as well as within intracellular compartments (Varela-Nallar et al. 2006). PrPC also has the capacity to bind other divalent metal such as Mn (Brown et al. 2000). These findings stimulated the investigations on metal ion alteration in PrPD human brains. Changes in the levels of Cu and Mn bound to PrPSc were found in sporadic CJD compared to PrPC in normal subjects (Wong et al. 2001). Moreover, it was also observed that there was a decrease of up to 50% of Cu and a tenfold increase of Mn in brain tissues of CJD compared to controls (Wong et al. 2001). In addition, a group reported evidence for accumulation of large amounts of Mn in the brain of both animals and patients affected by the disease (Hesketh et al. 2007, 2008). Similarly, studies with recombinant PrP demonstrated that binding of Mn produced a change in protein conformation (Tsenkova et al. 2004). Overall these studies indicate that the binding of metal ions to PrP could be involved in the pathology (Thackray et al. 2002; Brown 2009). However, Waggoner et al. (2000) reported that brain Cu levels and cuproenzyme activity in mice overexpressing PrP levels were unaffected when compared to controls. According to Pushie et al. (2009) this could be explained by considering that Cu is not required for the propagation of PrPSc, but only for its initial formation, and therefore, Cu is not expected to be enriched in prion plaques. Accordingly, the Cu chelator D-penicillamine seems to delay, and not to cure, the onset of the disease (Sigurdsson et al. 2003). These contrasting results create uncertainty on the putative role of Cu in PrPD (Zatta and Frank 2007) and warrant further work to explore the connection existing between PrP and metal ion metabolism.
The “Domino Effect”
When approaching ND it seems reasonable not to consider metal dyshomeostasis as the only causative factor for disease etiology. However, in the context of the impairment of the homeostatic mechanisms, largely reported in the aged brain, this imbalance may play a relevant role in the progression of these pathologies. It also follows that the imbalance of only one metal cannot be the exclusive triggering factor, as for example, Cu excess as well as deficiency have been well characterized in Wilson’s disease and Menke’s disease, respectively. These genetic disorders are described by a specific neurological scenario which is different from the kind of impairment seen in, for example, AD. Nevertheless, the above-mentioned biometals can all interact with a key protein enhancing their neurotoxicity (e.g., Aβ) or activating detrimental processing pathways.
The understanding of the real role played by metal dyshomeostasis is greatly hindered by the scarce knowledge of the mechanisms underlying these diseases. In any case, there is no clear consensus about the real metabolic state of the different metals in the diseases (e.g., Cu in AD). Thus, for the main biometals supplementation, lowering as well as redistribution strategies have all been proposed. We think it must be highlighted that focusing only on a single metal as the culprit of the disease could be misleading or reductive. The change of a single metal can indeed upset the whole metal pool or part of it (e.g., the Cu deficiency following Zn deficiency). This effect produced by modifying the uptake or the metabolism of one single element as the cause for the alteration in the physiological distribution, concentration, and excretion of several other elements is called “domino effect” and it must be taken into account when proposing metal-modulating strategies as a therapeutic approach. Thus, if a single-metal modulating strategy must be undertaken, it will also be important to monitor the distribution of the other metals.
Opportunities for Metal-Targeted Treatments of ND
The extreme complexity and the multiplicity of the biochemical processes leading to neurodegeneration make identification of any useful therapeutic approach very difficult. As stated above, although metal imbalance is only one aspect of the complex array of alterations observed in ND, the use of chelating compounds aimed at restoring correct metal homeostasis certainly deserves greater attention as it is considered of potential benefit to patients for slowing down the course of the disease.
Among recent possible treatments for ND, chelation therapy, or more conceptually correctly “metal-targeted therapy”, are rapidly emerging as a promising and effective therapeutic option (see review Bolognin et al. 2009; Hegde et al. 2009; Kim et al. 2009). The design of an effective metal-targeted therapy should necessarily take into account that metal dyshomeostasis associated with ND often does not correspond to a relevant overload of a specific metal but rather it is characterized by focal marked alterations of one or more metals in specific brain areas. Sometimes such alterations just consist of an abnormal metal distribution inside the cell, with no evidence of changes in total metal concentration. It should be highlighted that an effective metal ligand has to efficiently remove the metal/s from target proteins or tissues and destroy anomalous and deleterious metal/protein interactions, but must not scavenge metals from metalloenzymes and metalloproteins that physiologically require them for their correct functioning. If this happens, it is predicted that treatment-limiting toxicity occurs.
Hence, future research should aim to develop increasingly sophisticated approaches directed at attenuating abnormal protein/metal interactions without causing any systemic metal depletion. Targeting, for instance, the metal ligand to a specific and restricted brain area or to selected brain tissues might be of great benefit and this goal might be afforded by linking the chelator to appropriate nanostructures showing a precise tropism for defined CNS regions.
Moreover, chelators (or metal ligands) should show an acceptable degree of selectivity for the metal of interest compared to the other biometals. Some significant selectivity may be imparted by molecular design criteria inspired by classical coordination chemistry and by “hard and soft acids and bases” (HSAB) principles. Thus, several chelators are now available which are capable of distinguishing between Cu or Zn, on one hand, and Fe or Al, on the other. In contrast, achieving a sufficient discrimination between Al and Fe or between Cu and Zn is far more problematic, owing to the close chemical similarity of these pairs of metal ions.
DFO and deferiprone (Fig. 6a, d, respectively) are ligands which manifest a high selectivity for Fe that should be primarily considered for iron removal in the case of documented brain Fe overload. Deferiprone appears to be the most appropriate ligand for brain Fe scavenging due to its favorable physicochemical properties; indeed, it is currently considered for treating those NDs where severe Fe overload has been conclusively demonstrated (see for instance neuroferritinopathy and aceruloplasminemia). It seems obvious to extend this type of Fe chelation strategy to the more important NDs showing less severe Fe accumulation provided that the drug administration schemes are finely adjusted and that much attention is paid to avoid systemic Fe depletion.

Chelator molecules. Molecules proposed to chelate metals for the treatment of neurodegenerative diseases. a DFO; b CQ; c D-penicillamine; d Deferiprone; and e Trientine
On the other hand, clioquinol (CQ), trientine, and D-penicillamine (Fig. 6b, c, e, respectively) are typical ligands that show some selectivity for Cu and Zn. CQ, in particular, embodies a paradigmatic story in the field of metal targeted therapies of ND. Indeed, quite encouraging results have been reported a few years ago in vivo upon CQ treatment, with evidence of a significant slow down or even a reversal of the cognitive decline in AD (Ritchie et al. 2003). Several studies were thus devoted to uncover the mechanism of action of this apparently successful drug (White et al. 2006). Remarkably in contrast to the initial idea that CQ might work as a Cu chelator, it was demonstrated that CQ may act as a metal protein attenuation compound (MPAC) and even as an ionophore increasing Cu cellular uptake (Cherny et al. 2001; White et al. 2006). However, the scenario of the biochemical action of CQ and derivatives appears to be more controversial and certainly deserves further investigation (Bolognin et al. 2008).
Many encouraging results have been recently reached both with the design of new molecules and with the improvement of drug delivery systems. Remarkably, several attempts have been made to design new ways to deliver well-known molecules to specific brain areas. As an example, Liu et al. (2005) proposed the use of nanoparticles linked with metal chelators as a tool to overcome limitations due to scarce lipophilicity of the molecule itself. However, a greater understanding of the role of metals in the etiology of these diseases is still required before metal ligand therapies can be effectively exploited as an effective treatment option.
Conclusions
Metal ions such as Fe, Cu, Zn, and Al may play an important role in the etiology of NDs either as triggers or as modulators of pathology progression. Despite the wide diversity in their etiopathogenesis, similar findings for different NDs indicate overlapping mechanisms of homeostatic failure for metal ions. The hypothesis of metal involvement has generally gained considerable acceptance but an unanimous agreement regarding the quantitative/qualitative entity of this imbalance has not been reached. Contrasting reports on the actual concentrations of the mentioned metals in the brain (both in aging as well as in ND) have been published and the goal of recognizing and characterizing unambiguously their distribution patterns has not been accomplished yet. Reasons for the several analytical discrepancies may be numerous. Bearing in mind that the neurodegenerative process is mainly the result of collective and cumulative metabolic mistakes occurring over decades of life (Liu et al. 2006), it is conceivable that environmental and genetic factors might also account for these inconsistencies and for the reported high variability. Furthermore, the use of different experimental and methodological approaches and the improvement of the technologies compared to early studies constitute other sources of variability. Clarification of the role played by metal dyshomeostasis will certainly add to the basic understanding of the neurodegenerative process, thus verifying whether addressing metal dyshomeostasis may represent a valuable therapeutic option.
References
Adlard, P. A., Cherny, R. A., Finkelstein, D. I., Gautier, E., Robb, E., Cortes, M., et al. (2008). Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron, 59, 43–55.
Age-Related Eye Disease Study Research Group. (2001). A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Archives of Ophthalmology, 119, 1417–1436.
Alfrey, A. C., LeGendre, G. R., & Kaehny, W. D. (1976). The dialysis encephalopathy syndrome. Possible aluminum intoxication. New England Journal of Medicine, 294, 184–188.
Alimonti, A., Ristori, G., Giubilei, F., Stazi, M. A., Pino, A., Visconti, A., et al. (2007). Serum chemical elements and oxidative status in Alzheimer’s disease, Parkinson disease and multiple sclerosis. Neurotoxicology, 28, 450–456.
Allsop, D., Mayes, J., Moore, S., Masad, A., & Tabner, B. J. (2008). Metal-dependent generation of reactive oxygen species from amyloid proteins implicated in neurodegenerative disease. Biochemical Society Transactions, 36, 1293–1298.
Altamura, S., & Muckenthaler, M. U. (2009). Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. Journal of Alzheimers Diseases, 16, 879–895.
Andersen, J. K. (2004). Iron dysregulation and Parkinson’s disease. Journal of Alzheimers Diseases, 6, 47–52.
Babiloni, C., Squitti, R., Del Percio, C., Cassetta, E., Ventriglia, M. C., Ferreri, F., et al. (2007). Free copper and restino temporal EEG rhythms correlate across healthy, mild cognitive impairment, and Alzheimer’s disease subjects. Clinical Neurophysiology, 118, 1244–1260.
Bala Gupta, V., Anitha, S., Hedge, M. L., Zecca, L., Garruto, M. R., Ravid, R., et al. (2005). Aluminium in Alzheimer’s disease: Are we still at a crossroad? Cellular and Molecular Life Sciences, 62, 143–158.
Banks, W. A., Niehoff, M. L., Drago, D., & Zatta, P. (2006). Aluminum complexing enhances amyloid beta protein penetration of blood-brain barrier. Brain Research, 1116, 215–221.
Barnham, K. J., & Bush, A. I. (2008). Metals in Alzheimer’s and Parkinson’s diseases. Current Opinion in Chemical Biology, 12, 222–228.
Bayer, T. A., Schäfer, S., Breyhan, H., Wirths, O., Treiber, C., & Multhaup, G. (2006). A vicious circle: Role of oxidative stress, intraneuronal Abeta and Cu in Alzheimer’s disease. Clinical Neuropathology, 25, 163–171.
Bayer, T. A., Schäfer, S., Simons, A., Kemmling, A., Kamer, T., Tepest, R., et al. (2003). Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proceedings of the National Academy of Sciences of the United States of America, 100, 14187–14192.
Bayir, H., Kapralov, A. A., Jiang, J., Huang, Z., Tyurina, Y. Y., Tyurin, V. A., et al. (2009). Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome c: Protection agains apoptosis versus delayed oxidative stress in Parkinson’s disease. Journal of Biological Chemistry, 284, 15951–15961.
Bertoni-Freddari, C., Fattoretti, P., Casoli, T., Di Stefano, G., Giorgetti, B., & Balietti, M. (2008). Brain aging: The zinc connection. Experimental Gerontology, 43, 389–393.
Bertoni-Freddari, C., Fattoretti, P., Paoloni, R., Caselli, U., Galeazzi, L., & Meier-Ruge, W. (1996). Synaptic structural dynamics and aging. Gerontology, 42, 170–180.
Binolfi, A., Lamberto, G. R., Duran, R., Quintanar, L., Bertoncini, C. W., Souza, J. M., et al. (2008). Site-specific interactions of Cu(II) with α and β-synuclein: Bridging the molecular gap between metal binding and aggregation. Journal of the American Chemical Society, 130, 11801–11812.
Boillée, S., Vande Velde, C., & Cleveland, D. W. (2006). ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron, 52, 39–59.
Bolognin, S., Drago, D., Messori, L., & Zatta, P. (2009). Chelation therapy for neurodegenerative diseases. Medicinal Research Reviews, 29, 547–570.
Bolognin, S., Zatta, P., Drago, D., Parnigotto, P. P., Tognon, G., & Ricchelli, F. (2008). Mutual stimulation of beta-amyloid fibrillogenesis by clioquinol and divalent metals. Neuromolecular Medicine, 10, 322–332.
Borchardt, T., Camakaris, J., Cappai, R., Master, C. L., Beyreuther, K., & Multhaup, G. (1999). Copper inhibits beta-amyloid production and stimulates the non-amyloidogenic pathway of amyloid-precursor protein secretion. Biochemical Journal, 344, 461–467.
Bouras, C., Giannakopoulos, P., Good, P. F., Hsu, A., Hof, P. R., & Perl, D. P. (1997). A laser microprobe mass analysis of brain aluminum and iron in dementia pugilistica: Comparison with Alzheimer’s disease. European Neurology, 38, 53–58.
Brown, D. R. (2009). Brain proteins that mind metals: A neurodegenerative perspective. Dalton Transactions, 21, 4069–4076.
Brown, D. R., Hafiz, F., Glassmith, L. L., Wong, B. S., Jones, I. M., Clive, C., et al. (2000). Consequences of manganese replacement of copper for prion protein function and proteinase resistance. EMBO Journal, 19, 1180–1186.
Brown, D. R., Wong, B. S., Hafiz, F., Clive, C., Haswell, S. J., & Jones, I. M. (1999). Normal prion protein has an activity like that of superoxide dismutase. Biochemical Journal, 344, 1–5.
Bush, A. I. (2003). The metallobiology of Alzheimer’s disease. Trends in Neurosciences, 26, 207–214.
Butterfield, D. A., Reed, T., Newman, S. F., & Sultan, R. (2007). Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radical Biology and Medicine, 43, 658–677.
Cahill, C. M., Lahiri, D. K., Huang, X., & Rogers, J. T. (2009). Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochimica et Biophysica Acta, 1790, 615–628.
Campbell, A. (2006). The role of aluminum and copper on neuroinflammation and Alzheimer’s disease. Journal of Alzheimers Diseases, 10, 165–172.
Cherny, R. A., Atwood, C. S., Xilinas, M. E., Gray, D. N., Jones, W. D., Mclean, C. A., et al. (2001). Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron, 30, 665–676.
Connor, J. R., & Lee, S. Y. (2006). HFE mutations and Alzheimer’s disease. Journal of Alzheimers Diseases, 10, 267–276.
Crichton, R. R., Dexter, D. T., & Roberta, J. W. (2008). Metal based neurodegenerative diseases—from molecular mechanisms to therapeutic strategies. Coordination Chemistry Reviews, 251, 1189–1199.
Cross, D. J., Flexman, J. A., Anzai, Y., Morrow, T. J., Maravilla, K. R., & Minoshima, S. (2006). In vivo imaging of functional disruption, recovery and alteration in rat olfactory circuitry after lesion. Neuroimage, 32, 1265–1272.
Crouch, P. J., Barnham, K. J., Bush, A. I., & White, A. R. (2006). Therapeutic treatments for Alzheimer’s disease based on metal bioavailability. Drug News Perspective, 19, 469–474.
Crouch, P. J., Hung, L. W., Adlard, P. A., Cortes, M., Lal, V., Filiz, G., et al. (2009). Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proceedings of the National Academy of Sciences of the United States of America, 106, 381–386.
Crow, P., Sampson, J. B., Zhuang, Y., Thompson, J. A., & Beckman, J. S. (1997). Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. Journal of Neurochemistry, 69, 1936–1944.
Cuajungco, M. P., & Faget, K. Y. (2003). Zinc takes the center stage: Its paradoxical role in Alzheimer’s disease. Brain Research Reviews, 41, 44–56.
Cuajungco, M. P., & Lees, G. J. (1997). Zinc and Alzheimer’s disease: Is there a direct link? Brain Research Reviews, 23, 219–236.
Deibel, M. A., Ehmann, W. D., & Markesbery, W. R. (1996). Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. Journal of the Neurological Sciences, 143, 137–142.
Del Corso, L., Pastine, F., Protti, M. A., Romanelli, A. M., Moruzzo, D., Rocco, L., et al. (2000). Blood zinc, copper and magnesium in ageing. A study in healthy home-living elderly. Panminerva Medica, 42, 273–277.
Deshpande, A., Kawai, H., Metherate, R., Glabe, C. G., & Busciglio, J. (2009). A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. Journal of Neuroscience, 29, 4004–4015.
Dexter, D. T., Carayon, A., Javoy-Agid, F., Agid, Y., Wells, F. R., Daniel, S. E., et al. (1991). Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain, 114, 1953–1975.
Dong, J., Robertson, J. D., Merkesbery, W. R., & Lovell, M. A. (2008). Serum zinc in the progression of Alzheimer’s Disease. Journal of Alzheimers Diseases, 15, 443–450.
Drago, D., Bettella, M., Bolognin, S., Cendron, L., Scancar, J., Milacic, R., et al. (2008a). Potential pathogenic role of beta-amyloid(1–42)-aluminum complex in Alzheimer’s disease. International Journal of Biochemistry and Cell Biology, 40, 731–746.
Drago, D., Bolognin, S., & Zatta, P. (2008b). Role of metal ions in the abeta oligomerization in Alzheimer’s disease and in other neurological disorders. Current Alzheimer Research, 5, 5007–5507.
Drago, D., Cavaliere, A., Mascetra, N., Ciavardelli, D., di Ilio, C., Zatta, P., et al. (2008c). Aluminum modulates effects of beta amyloid(1–42) on neuronal calcium homeostasis and mitochondria functioning and is altered in a triple transgenic mouse model of Alzheimer’s disease. Rejuvenation Resarch, 11, 861–871.
Eisen, A. (2009). Amyotrophic lateral sclerosis: A 40-year personal perspective. Journal of Clinical Neuroscience, 16, 505–512.
Estevez, A. G., Crow, J. P., Sampson, J. B., Reiter, C., Zhuang, Y., Richardson, G. J., et al. (1999). Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science, 286, 2498–2500.
Faucheux, B. A., Martin, M. E., Beaumont, C., Hauw, J. J., Agid, Y., & Hirsch, E. C. (2003). Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson’s disease. Journal of Neurochemistry, 86, 1142–1148.
Ferrer, M., Golyshina, O. V., Beloqui, A., Golyshin, P. N., & Timmis, K. N. (2007). The cellular machinery of Ferroplasma acidiphilum is iron-protein-dominated. Nature, 445, 91–94.
Frederickson, C. J., Koh, J. Y., & Bush, A. I. (2005). The neurobiology of zinc in health and disease. Nature Reviews Neuroscience, 6, 449–462.
Garrick, M. D., & Garrick, L. M. (2009). Cellular iron transport. Biochimica et Biophysica Acta, 1790, 309–325.
Gerhardsson, L., Lundh, T., Minthon, L., & Londos, E. (2008). Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders, 25, 508–515.
Glabe, C. C. (2005). Amyloid accumulation and pathogensis of Alzheimer’s disease: Significance of monomeric, oligomeric and fibrillar Abeta. SubCellular Biochemistry, 38, 167–177.
Good, P. F., & Perl, D. P. (1993). Aluminium in Alzheimer’s? Nature, 362, 418.
Goodall, E. F., Haque, M. S., & Morrison, K. E. (2008). Increased serum ferritin levels in amyotrophic lateral sclerosis (ALS) patients. Journal of Neurology, 255, 1652–1656.
Goto, J. J., Zhu, H., Sanchez, R. J., Nersissian, A., Gralla, E. B., Valentie, J. S., et al. (2000). Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis. Journal of Biological Chemistry, 275, 1007–1014.
Gotz, M. E., Double, K., Gerlach, M., Youdim, M. B., & Riederer, P. (2004). The relevance of iron in the pathogenesis of Parkinson’s disease. Annals of the New York Academy of Sciences, 1012, 193–208.
Haass, C., & Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nature Reviews Molecular Cell Biology, 8, 101–112.
Harris, D. A., & True, H. L. (2006). New insights into prion structure and toxicity. Neuron, 40, 547–586.
Hegde, M. L., Bharathi, P., Suram, A., Venugopal, C., Jagannathan, R., Poddar, P., et al. (2009). Challenges associated with metal chelation therapy in Alzheimer’s disease. Journal of Alzheimers Diseases (in press).
Hesketh, S., Sassoon, J., Knight, R., & Brown, D. R. J. (2008). Elevated manganese levels in blood and CNS in human prion disease. Molecular and Cellular Neurosciences, 37, 590–598.
Hesketh, S., Sassoon, J., Knight, R., Hopkins, J., & Brown, D. R. J. (2007). Elevated manganese levels in blood and central nervous system occur before onset of clinical signs of scrapie and bovine spongiform encephalopathy. Journal of Animal Science, 85, 1596–1609.
Hirsch, E. C., Brandel, J. P., Galle, P., Javoy-Agid, F., & Agid, Y. (1991). Iron and aluminum in the substantia nigra of patients with Parkinson’s disease: An X-ray microanalysis. Journal of Neurochemistry, 56, 446–451.
Hu, W. P., Chang, G. L., Chen, S. J., & Kuo, Y. M. (2006). Kinetic analysis of beta-amyloid peptide aggregation induced by metal ions based on surface plasmon resonance biosensing. Journal of Neuroscience Methods, 154, 190–197.
Hwang, E. M., Kim, S. K., Sohn, J. H., Lee, J. Y., Kim, Y., Kim, Y. S., et al. (2006). Furin is an endogenous regulator of alpha-secretase associated APP processing. Biochemical and Biophysical Research Communications, 349, 654–659.
Iqbal, K., Liu, F., Gong, C. X., Alonso Adel, C., & Grundke-Iqbal, I. (2009). Mechanisms of tau-induced neurodegeneration. Acta Neuropathologica, 118, 53–69.
Jellinger, K. A. (1999). The role of iron in neurodegeneration: Prospects for pharmacotherapy of Parkinson’s disease. Drugs and Aging, 14, 115–140.
Jellinger, K. A. (2003). General aspects of neurodegeneration. Journal of Neural Transmission (Supplementum), 65, 101–144.
Jeong, S. Y., Rathore, K. I., Schulz, K., Ponka, P., Arosio, P., & David, S. (2009). Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. Journal of Neuroscience, 29, 610–619.
Kasarskis, E. J., Tandon, L., Lovell, M. A., & Ehmann, W. D. (1995). Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: A preliminary study. Journal of the Neurological Sciences, 130, 203–208.
Kaur, D., & Andersen, J. K. (2002). Ironing out Parkinson’s disease: Is therapeutic treatment with iron chelators a real possibility? Aging Cell, 1, 17–21.
Kawarabayashi, T., Shoji, M., Younkin, L. H., Wen-Lang, L., Dickson, D. W., Murakami, T., et al. (2004). Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. Journal of Neuroscience, 24, 3801–3809.
Kenward, A. G., Bartolotti, L. J., & Burns, C. S. (2007). Copper and zinc promote interactions between membrane-anchored peptides of the metal binding domain of the prion protein. Biochemistry, 46, 4261–4271.
Kessler, H., Bayer, T. A., Bach, D., Schneider-Axmann, T., Supprian, T., Herrmann, W., et al. (2008). Intake of copper has no effect on cognition in patients with mild Alzheimer’s disease: A pilot phase 2 clinical trial. Journal of Neural Transmission, 115, 1181–1187.
Kim, Y., Lee, J. H., Ryu, J., & Kim, D. J. (2009). Multivalent & multifunctional ligands to beta-amyloid. Current Pharmaceutical Design, 15, 637–658.
Kitazawa, M., Cheng, D., & LaFerla, F. M. (2009). Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. Journal of Neurochemistry, 108, 1550–1560.
Konoho, K., Sadakane, Y., & Kawahara, M. (2006). Zinc neurotoxicity and its role in neurodegenerative diseases. Journal of Health Sciences, 52, 1–8.
Kramer, D. R., Llanos, R. M., & Mercer, J. F. B. (2003). Molecular basis of copper transport: Cellular and physiological functions of Menkes and Wilson disease proteins. In Metal ions and neurodegenerative disorders (pp. 183–206). Singapore, London: World Scientific.
Krewski, D., Yokel, R. A., Nieboer, E., Borchelt, D., Cohen, J., Harry, J., et al. (2007). Human health risk assessment for aluminium, aluminium oxide, and aluminium hydroxide. Journal of Toxicology and Environmental Health B: Critical Review, 10, 1–269.
Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., & Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature, 457, 1128–1132.
Lee, J. Y., Cole, T. B., Palmiter, R. D., Sush, S. W., & Koh, J. Y. (2002). Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proceedings of the National Academy of Sciences of the United States of America, 99, 7705–7710.
Lee, J. Y., Friedman, J. E., Angel, I., Kozak, A., & Koh, J. Y. (2004). The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human beta-amyloid precursor protein transgenic mice. Neurobiology of Aging, 25, 1315–1321.
Leong, S. L., Pham, C. L., Galatis, D., Fodero-Tavoletti, M. T., Perez, K., Hill, A. F., et al. (2009). Formation of dopamine-mediated alpha-synuclein-soluble oligomers requires methionine oxidation. Free Radical Biology and Medicine, 46, 1328–1337.
Lewczuk, P., Esselmann, H., Otto, M., Maler, J. M., Henkel, A. W., Henkel, M. K., et al. (2004). Neurochemical diagnosis of Alzheimer’s dementia by CSF Abeta42, Abeta42/Abeta40 ratio and total tau. Neurobiology of Aging, 25, 273–281.
Linkous, D. H., Adlard, P. A., Wanschura, P. B., Conko, K. M., & Flinn, J. M. (2009). The effects of enhanced Zinc on spatial memory and plaque formation in transgenic mice. Journal of Alzheimers Diseases (in press).
Liu, G., Garrett, M. R., Men, P., Zhu, X., Perry, G., & Smith, M. A. (2005). Nanoparticle and other metal chelation therapeutics in Alzheimer disease. Biochimica et Biophysica Acta, 1741, 246–252.
Liu, G., Men, P., Harris, P. L., Rolston, R. K., Perry, G., & Smith, M. A. (2006). Nanoparticle iron chelators: A new therapeutic approach in Alzheimer disease and other neurologic disorders associated with trace metal imbalance. Neuroscience Letters, 406, 189–193.
Loeffler, D. A., LeWitt, P. A., Juneau, P. L., Sima, A. A., Nguyen, H. U., DeMaggio, A. J., et al. (1996). Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders. Brain Research, 738, 265–274.
Lovell, M. A. (2009). A potential role for alterations of zinc and zinc transport proteins in the progression of Alzheimer’s disease. Journal of Alzheimers Diseases, 16, 471–483.
Lovell, M. A., Robertson, J. D., Teesdale, W. J., Campbell, J. L., & Markesbery, W. R. (1998). Copper, iron and zinc in Alzheimer’s disease senile plaques. Journal of the Neurological Sciences, 158, 47–52.
Lu, J., Zheng, Y. L., Wu, D. M., Sun, D. X., Shan, Q., & Fan, S. H. (2006). Trace amount of copper induce neurotoxicity in the cholesterol-fed mice through apoptosis. FEBS Letters, 580, 6730–6740.
Lyons, T. J., Liu, H., Goto, J. J., Nersissian, A., Roe, J. A., Graden, J. A., et al. (1996). Mutations in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proceedings of the National Academy of Sciences of the United States of America, 93, 12240–12244.
Miller, L. M., Wang, Q., Telivala, T. P., Smith, R. J., Lanzirotti, A., & Miklossy, J. (2006). Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. Journal of Structural Biology, 155, 30–37.
Miura, T., Hori-i, A., & Takeuchi, H. (1996). Metal-dependent a-helix formation promoted by the glycine-rich octapeptide region of prion protein. FEBS Letters, 396, 248–252.
Mocchegiani, E., Bertoni-Freddari, C., Marcellini, F., & Malavolta, M. (2005). Brain, aging, and neurodegeneration: Role of zinc availability. Progress in Neurobiology, 75, 367–390.
Molina, J. A., Jiménez-Jiménez, F. J., Aguilar, M. V., Meseguer, I., Mateos-Vega, C. J., González-Muñoz, M. J., et al. (1998). Cerebrospinal fluid levels of transition metals in patients with Alzheimer’s disease. Journal of Neural Transmission, 105, 479–488.
Morita, A., Kimura, M., & Itokawa, Y. (1994). The effect of aging on the mineral status of female mice. Biological Trace Element Research, 42, 165–177.
Morris, M. C., Evans, D. A., Tangney, C. C., Bienias, J. L., Schneider, J. A., Wilson, R. S., et al. (2006). Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Archives of Neurology, 63, 1085–1088.
Nakashima, A. S., & Dyck, R. H. (2009). Zinc and cortical plasticity. Brain Research Reviews, 59, 347–373.
Nicolas, O., Gavín, R., & Del Río, J. A. (2009). New insights into cellular prion protein (PrP(c)) functions: The “ying and yang” of a relevant protein. Brain Research Reviews (in press).
Olivares, D., Huang, X., Branden, L., Greig, N. H., & Rogers, J. T. (2009). Physiological and pathological role of alpha-synuclein in Parkinson’s disease through iron mediated oxidative stress; the role of a putative iron-responsive element. International of Journal of Molecular Science, 10, 1226–1260.
Opazo, C., Huang, X., Cherny, R. A., Moir, R. D., Roher, A. E., White, A. R., et al. (2002). Metalloenzyme-like activity of Alzheimer’s disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). Journal of Biological Chemistry, 277, 40302–40308.
Osterova-Golts, N., Petrucelli, L., Hardy, J., Lee, J. M., Farer, M., & Wolozin, B. (2000). The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. Journal of Neuroscience, 20, 6048–6054.
Oztug Durer, Z. A., Cohlberg, J. A., Dinh, P., Padua, S., Ehrenclou, K., Downes, S., et al. (2009). Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PLoS ONE, 4, e5004.
Paik, S. R., Lee, J. H., Kim, D. H., Chang, C. S., & Kim, J. (1997). Aluminum-induced structural alterations of the precursor of the non-A beta component of Alzheimer’s disease amyloid. Archives of Biochemistry and Biophysics, 344, 325–334.
Pantopoulos, K., & Hentze, M. W. (1998). Activation of iron regulatory protein-1 by oxidative stress in vitro. Proceedings of the National Academy of Sciences of the United States of America, 95, 10559–10563.
Petri, S., Calingasan, N. Y., Alsaied, O. A., Wille, E., Kiaei, M., Friedman, J. E., et al. (2007). The lipophilic metal chelators DP-109 and DP-460 are neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis. Journal of Neurochemistry, 102, 991–1000.
Phinney, A. L., Drisaldi, B., Schmidt, S. D., Lugowski, S., Coronado, V., Liang, Y., et al. (2003). In vivo reduction of amyloid-beta by a mutant copper transporter. Proceedings of the National Academy of Sciences of the United States of America, 100, 14193–14198.
Powers, K. M., Smith-Weller, T., Franklin, G. M., Longstreth, W. T., Jr, Swanson, P. D., & Checkoway, H. (2003). Parkinson’s disease risks associated with dietary iron, manganese, and other nutrient intakes. Neurology, 60, 1761–1766.
Prusiner, S. B. (1998). Prions. Proceedings of the National Academy of Sciences of the United States of America, 95, 13363–13383.
Pushie, M. J., Rauk, A., Jirik, F. R., & Vogel, H. J. (2009). Can copper binding to the prion protein generate a misfolded form of the protein. BioMetals, 22, 159–179.
Quinn, J. F., Crane, S., Harris, C., & Wadsworth, T. L. (2009). Copper in Alzheimer’s disease: Too much or too little? Expert Review of Neurotherapeutics, 9, 631–637.
Rachidi, W., Vilette, D., Guiraud, P., Arlotto, M., Riondel, J., Laude, H., et al. (2003). Expression of prion protein increases cellular copper binding and antioxidant enzyme activities but not copper delivery. Journal of Biological Chemistry, 278, 9064–9072.
Rajan, M. T., Jagannatha Rao, K. S., Mamatha, B. M., Rao, R. V., Shanmugavelu, P., Menon, R. B., et al. (1997). Quantification of trace elements in normal human brain by inductively coupled plasma atomic emission spectrometry. Journal of the Neurological Sciences, 146, 153–166.
Rajendran, R., Minqin, R., Ynsa, M. D., Casadesus, G., Smith, M. A., Perry, G., et al. (2009). A novel approach to the identification and quantitative elemental analysis of amyloid deposits insights into the pathology of Alzheimer’s disease. Biochemical and Biophysical Research Communications, 382, 91–95.
Re, L., Rossini, F., Re, F., Bordicchia, M., Mercanti, A., Fernandez, O. S., et al. (2006). Prion protein potentiates acetylcholine release at the neuromuscular junction. Pharmacological Research, 53, 62–68.
Reusche, E. (2003). Aluminium and central nervous system morphology in hemodialysis. In Metal ions and neurodegenerative disorders (pp. 117–138). Singapore, London: World Scientific.
Ricchelli, F., Buggio, R., Drago, D., Forloni, G., Negro, A., Tognon, G., et al. (2006). Aggregation/Fibrillogenesis of recombinant human prion protein and Gestmann-Straussler-Scheinker disease peptides in the presence of metal ions. Biochemistry, 45, 6724–6732.
Ricchelli, F., Drago, D., Filippi, B., Tognon, G., & Zatta, P. (2005). Aluminum-triggered structural modifications and aggregation of beta-amyloids. Cellular and Molecular Life Sciences, 62, 1724–1733.
Ricchelli, F., Fusi, P., Tortora, P., Valtorta, M., Riva, M., Tognon, G., et al. (2007). Destabilization of non-pathological variants of ataxin-3 by metal ions results in aggregation/fibrillogenesis. International Journal of Biochemistry and Cell Biology, 39, 966–977.
Ritchie, C. W., Bush, A. I., Mackinnon, A., Macfarlane, S., Mastwyk, M., MacGregor, L., et al. (2003). Metal-protein attenuation with clioquinol targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Archives of Neurology, 60, 1685–1691.
Rogers, J. T., Randall, J. D., Cahill, C. M., Eder, P. S., Huang, X., Gunshin, H., et al. (2002a). An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. Journal of Biological Chemistry, 277, 45518–45528.
Rogers, J. T., Randall, J. D., Eder, P. S., Huang, X., Bush, A. I., Tanzi, R. E., et al. (2002b). Alzheimer’s disease drug discovery targeted to the APP mRNA 5′ untranslated region. Journal of Molecular Neuroscience, 19, 77–82.
Roider, G., & Drasch, G. (1999). Concentration of Al in human tissues. Investigations on an occupationally non-exposed population in Southern Bavaria (Germany). Trace Elements Electrolytes, 16, 77–86.
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Snapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 362, 59–62.
Rulon, L. L., Robertson, J. D., Lovell, M. A., Deibel, M. A., Ehmann, W. D., & Markesber, W. R. (2000). Serum zinc levels and Alzheimer’s disease. Biological Trace Element Research, 75, 79–85.
Said Ahmed, M., Hung, W. Y., Zu, J. S., Hockberger, P., & Siddique, T. (2000). Increased reactive oxygen species in familial amyotrophic lateral sclerosis with mutations in SOD1. Journal of the Neurological Sciences, 176, 88–94.
Salzman, M. B., Smith, E. M., & Koo, C. (2002). Excessive oral zinc supplementation. Journal of Pediatric Hematology/oncology, 24, 582–584.
Sassoon, J., & Brown, D. R. (2003). Copper and prion disease. In Metal ions and neurodegenerative disorders (pp. 279–306). Singapore, London: World Scientific.
Sayre, L. M., Moreira, P. A., Smith, M. A., & Perry, G. (2005). Metal ions and oxidative protein modification in neurological disease. Annals of Ist Super Sanità, 41, 143–164.
Schipper, H. M. (2004). Heme oxygenase expression in human central nervous system disorders. Free Radical Biology and Medicine, 37, 1995–2011.
Sigurdsson, E. M., Brown, D. R., Alim, M. A., Scholtzova, H., Carp, R., Meeker, H. C., et al. (2003). Copper chelation delays the onset o prion disease. Journal of Biological Chemistry, 278, 46199–46202.
Silvestri, L., & Camaschella, C. (2008). A potential pathogenetic role of iron in Alzheimer’s disease. Journal of Cellular and Molecular Medicine, 12, 1548–1550.
Smith, D. G., Cappai, R., & Barnham, K. J. (2007). The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochimica et Biophysica Acta, 1768, 1976–1990.
Sofic, E., Sapcanin, A., Tahirovic, I., Gavrankapetanovic, I., Jellinger, K., Reynolds, G. P., et al. (2006). Antioxidant capacity in post-mortem brain tissues of Parkinson’s and Alzheimer’s diseases. Journal of Neural Transmission (Supplementum), 71, 39–43.
Soto, C., & Estrada, L. D. (2008). Protein misfolding and neurodegeneration. Archives of Neurology, 65, 184–189.
Sparks, D. L., & Schreurs, B. G. (2003). Trace amount of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America, 100, 11065–11069.
Speziali, M., & Orvini, E. (2003). Metals distribution and regionalization in the brain. In Metal ions and neurodegenerative disorders (pp. 15–65). Singapore, London: World Scientific.
Squitti, R., Barbati, G., Rossi, L., Ventriglia, M., Dal Forno, G., Cesaretti, S., et al. (2006). Excess of nonceruloplasmin serum copper in AD correlates with MMSE, CSF [beta]-amyloid, and h-tau. Neurology, 67, 76–82.
Squitti, R., Cassetta, E., Dal Forno, G., Lupoi, D., Lippolis, G., Pauri, F., et al. (2004). Copper perturbation in 2 monozygotic twins discordant for degree of cognitive impairment. Archives of Neurology, 61, 738–743.
Squitti, R., Pasqualetti, P., Dal Forno, G., Moffa, F., Cassetta, E., Lupoi, D., et al. (2005). Excess of serum copper not related to ceruloplasmin in Alzheimer disease. Neurology, 64, 1040–1046.
Srivastava, R. A., & Jain, J. C. (2002). Scavenger receptor class B type I expression and elemental analysis in cerebellum and parietal cortex regions of the Alzheimer’s disease brain. Journal of the Neurological Sciences, 196, 45–52.
Stankiewicz, J. M., & Brass, S. D. (2009). Role of iron in neurotoxicity: A cause for concern in the elderly? Current Opinion in Clinical Nutrition and Metabolic Care, 12, 22–29.
Stoltenberg, M., Bruhn, M., Sondergaard, C., Doering, P., West, M. J., Larsen, A., et al. (2005). Immersion autometallographic tracing of zinc ions in Alzheimer beta-amyloid plaques. Histochemistry and Cell Biology, 123, 605–611.
Subramaniam, J. R., Lyons, W. E., Liu, J., Bartnikas, T. B., Rothstein, J., Price, D. L., et al. (2002). Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nature Neuroscience, 5, 301–307.
Takeda, A., Hirate, M., Tamano, H., & Oku, N. (2003). Release of glutamate and GABA in the hippocampus under zinc deficiency. Journal of Neuroscience Research, 72, 537–542.
Tarohda, T., Yamamoto, M., & Amamo, R. (2004). Regional distribution of manganese, iron, copper, and zinc in the rat brain during development. Analytical and Bioanalytical Chemistry, 380, 240–246.
Thackray, A. M., Knight, R., Haswell, S. J., Bujdoso, R., & Brown, D. R. (2002). Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochemical Journal, 362, 253–258.
Tiwari, A., Liba, A., Sohn, S. H., Seetharaman, S. V., Bilsel, O., Matthews, C. R., et al. (2009). Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. Journal of Biological Chemistry (in press).
Tsenkova, R. N., Iordanova, I. K., Toyoda, K., & Brown, D. R. (2004). Prion protein fate governed by metal binding. Biochemical and Biophysical Research Communications, 325, 1005–1012.
Uversky, V. N., Li, J., & Fink, A. L. (2001). Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. Journal of Biological Chemistry, 276, 44284–44296.
Varela-Nallar, L., Toledo, E. M., Larrondo, F. L., Cabral, A. L., Martins, V. R., & Inestrosa, N. C. (2006). Induction of cellular prion protein gene expression by copper in neurons. American Journal of Physiology and Cell Physiology, 290, 271–281.
Vašák, M., & Meloni, G. (2008). Metallothionein structure and reactivity. In: Metallothioneins in biochemistry and pathology (pp. 3–27). Singapore, London: World Scientific.
Wadsworth, J. D. F., & Collinge, J. (2007). Update on human prion disease. Biochemical and Biophysical Acta, 1772, 598–609.
Waggoner, D. J., Drisaldi, B., Bartnikas, T. B., Casareno, R. L., Prohaska, J. R., Gitlin, J. D., et al. (2000). Brain copper content and cuproenzyme activity do not vary with prion protein expression. Journal of Biological Chemistry, 275, 7455–7458.
Weiss, J. H., Sensi, S. L., & Koh, J. Y. (2000). Zn(2+): A novel ionic mediator of neural injury in brain disease. Trends in Pharmacological Sciences, 21, 395–401.
Wells, M. A., Jackson, G. S., Jones, S., Hosszu, L. L., Craven, C. J., Clarke, A. R., et al. (2006). A reassessment of copper(II) binding in the full length prion protein. Biochemical Journal, 399, 435–444.
White, A. R., Du, T., Laughton, K. M., Volitakis, I., Sharples, R. A., Xilinas, M. E., et al. (2006). Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. Journal of Biological Chemistry, 281, 17670–17680.
Wong, B. S., Brown, D. R., Pan, T., Whiteman, M., Liu, T., Bu, X., et al. (2001). Oxidative impairment in scrapie-infected mice is associated with brain metals perturbations and altered antioxidant activities. Journal of Neurochemistry, 79, 689–698.
Yim, M. B., Chock, P. B., & Stadtman, E. R. (2003). Copper-zinc superoxide dismutase and familial amyotrophic lateral sclerosis. In Metal ions and neurodegenerative disorders (pp. 263–278). Singapore, London: World Scientific.
Yokel, R. A. (1994). Aluminum chelation: Chemistry, clinical, and experimental studies and the search for alternatives to desferrioxamine. Journal of Toxicology and Environmental Health, 41, 131–174.
Yumoto, S., Kakimi, S., Ohsaki, A., & Ishikawa, A. (2009). Demonstration of aluminum in amyloid fibers in the cores of senile plaques in the brains of patients with Alzheimer’s disease. Journal of Inorganic Biochemistry (in press).
Zambenedetti, P., Giordano, R., & Zatta, P. (1998). Metallothioneins are highly expressed in astrocytes and microcapillaries in Alzheimer’s disease. Journal of Chemical Neuroanatomy, 15, 21–26.
Zatta, P. (Ed.). (2002). Recent topics in aluminium chemistry. Coordination Chemistry Reviews, 228, 91–396.
Zatta, P. (Ed.). (2003). Metal ions and neurodegenerative disorders (pp. 1–508). Singapore, London: World Scientific.
Zatta, P. (2006). Aluminum and Alzheimer’s disease: A Vexata Questio between uncertain data and a lot of imagination. Journal of Alzheimers Diseases, 10, 33–37.
Zatta, P. (Ed.). (2008). Metallothioneins in biochemistry and pathology (pp. 1–320). Singapore, London: World Scientific.
Zatta, P., Drago, D., Bolognin, S., & Sensi, S. L. (2009). Alzheimer’s disease, metal ions and metabolic homeostatic therapy. Trends in Pharmacological Sciences, 30, 346–355.
Zatta, P., Drago, D., Zambenedetti, P., Bolognin, S., Nogara, E., Peruffo, A., et al. (2008). Accumulation of copper and other metal ions, and metallothionein I/II expression in the bovine brain as a function of aging. Journal of Chemical Neuroanatomy, 36, 1–5.
Zatta, P., & Frank, A. (2007). Copper deficiency and neurological disorders in man and animals. Brain Research Reviews, 54, 19–33.
Zatta, P., Zambenedetti, P., Reusche, E., Stellmacher, F., Cester, A., Albanese, P., et al. (2004). A fatal case of aluminium encephalopathy in a patient with severe chronic renal failure not on dialysis. Nephrology, Dialysis, Transplantation, 19, 2929–2931.
Zecca, L., Youdim, M. B., Riederer, P., Connor, J. R., & Crichton, R. R. (2004). Iron, brain ageing and neurodegenerative disorders. Nature Reviews Neuroscience, 5, 863–873.
Zhou, C., Huang, Y., & Przedborski, S. (2008). Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Annals of the New York Academy of Sciences, 1147, 93–104.
Acknowledgments
This work was supported by Grants from CNR/MIUR (FIRB No. RBNE03PX83) and PRIN 2007.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bolognin, S., Messori, L. & Zatta, P. Metal Ion Physiopathology in Neurodegenerative Disorders. Neuromol Med 11, 223–238 (2009). https://doi.org/10.1007/s12017-009-8102-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-009-8102-1