
Abstract
Introduction
A genome-wide association analysis revealed a rheumatoid arthritis (RA)-risk-associated genetic locus on chromosome 9, which contained the tumor necrosis factor receptor-associated factor 1 (TRAF1). However, the detail mechanism by TRAF1 signaled to fibroblast-like synoviocytes (FLSs) apoptosis remains to be fully understood.
Materials and methods
Synovial tissue of 10 RA patients and osteoarthritis patients were obtained during joint replacement surgery. We investigated TRAF1 level and FLSs apoptosis percentage in vivo and elucidated the mechanism involved in the regulation of apoptotic process in vitro.
Results
We proved the significant increase of TRAF1 level in FLSs of RA patients and demonstrated that TRAF1 level correlated positively with DAS28 score and negatively with FLSs apoptosis. Treatment with siTRAF1 was able to decrease MMPs levels and the phosphorylated forms of JNK/NF-κB in vitro. Moreover, JNK inhibitor could attenuate expression of MMPs and increase percentage of apoptosis in RA-FLSs, while siTRAF1 could not promote apoptosis when RA-FLSs were pretreated with JNK activator.
Conclusions
High levels of TRAF1 in RA synovium play an important role in the synovial hyperplasia of RA by suppressing apoptosis through activating JNK/NF-kB-dependent signaling pathways in response to the engagement of CD40.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder characterized by joint involvement and systemic features which affects 0.28% of Chinese people. The lining layer of synovial tissues obtained from RA patients displays an increase in cellularity, which is composed mostly of activated macrophages with an underlying layer of fibroblast-like synoviocytes (FLSs) [1]. FLSs from the intimal lining are considered major effectors of cartilage destruction in RA based on their ability to produce massive amounts of degradative enzymes [2]. The c-Jun N-terminal kinase (JNK)/nuclear factor kappa-B (NF-κB) pathway is one of the important signal transduction pathways related to RA-FLSs apoptosis. Studies have confirmed that the JNK/NF-κB signaling pathway is involved in the occurrence and development of RA synovitis by reducing the apoptosis of FLSs [3,4,5].
In 2007, a genome-wide association analysis revealed an additional RA-risk-associated genetic locus on chromosome 9, which contained the tumor necrosis factor receptor-associated factor 1 and the complement 5 genes (TRAF1-C5) [6]. TRAF1 is a cytoplasmic adaptor protein that has been shown to interact directly or indirectly with tumor necrosis factor receptor (TNFR) family-related molecules [7, 8]. Although the functions of TRAF1 remain to be investigated further, they appear to act as adapter proteins leading to the assembly of larger signaling complexes that consist of effector proteins with enzymatic functions. TRAF1, thereby, regulate the balance between cell survival and death by activating JNK mitogen-activated protein kinases and the transcription factor NF-κB. A number of studies have confirmed that TRAF1 regulates the balance between cell survival and death by regulating JNK mitogen-activated protein kinases and the transcription factor NF-κB; however, the results were controversial. [9,10,11,12,13,14]. Existing studies suggested that the specific role of TRAF1 in the JNK/NF-κB signaling pathway depend on upstream inducing factors. This enlightens us to identify the mechanism involving TRAF1 actions in RA.
In the present study, we investigated the effect of TRAF1 on RA-FLSs apoptosis, and elucidated the mechanism involved in the process. TRAF1 has been shown to decrease apoptosis as it enhances JNK/NF-κB activation in response to the engagement of CD40.
Materials and methods
Study subjects
10 patients of RA diagnosed according the 1987 revised criteria of the American College of Rheumatology (ACR) were enrolled in the study [15]. Of the patients, 5 were female and 5 were male. The median age was 58.50 ± 9.40 years. The following measures were recorded at screening: course of disease, morning stiffness, swollen and tender joint count (both 0–28), patient-assessed global score (0–100), and erythrocyte sedimentation rate (ESR). Disease activity in RA patients was determined according to disease activity score (DAS28). Components of DAS28 score were ESR, swollen and tender joint count and patient-assessed global score. Synovial tissue samples from 10 RA patients and age- and sex-matched osteoarthritis (OA) patients were obtained during joint replacement surgery in the First Affiliated Hospital of Soochow University and The Affiliated Drum Tower Hospital, Medical School of Nanjing University.
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the ethics committee of the First Affiliated Hospital of Soochow University or The Affiliated Drum Tower Hospital, Medical School of Nanjing University. All subjects provided written informed consent before participating in the study.
FLSs culture and stimulation
Synovial tissue specimens were obtained from RA patients and OA patients by synovectomy or joint replacement surgery. Carefully minced tissues were digested with 1 mg/ml collagenase I (Sigma-Aldrich, St. Louis, MO) in serum-free Dulbecco’s modified Eagle’s medium (DMEM) (Gibco BRL, Grand Island, NY) for 4–6 h at 37 C in a standard cell culture chamber. The tissue digest was filtered through a 70 μm cell strainer (BD, Durham, NC) to enrich for cells. The cell suspension was thoroughly washed with serum-free DMEM and finally cultured in DMEM supplemented with 10% fetal bovine serum (Gibco BRL, Grand Island, NY), 100 U penicillin, and 100 µg/ml streptomycin overnight in a cell culture chamber containing 5% CO2. After removal of non-adherent cells after 1 day, adherent fibroblast-like synoviocytes were cultured until near confluence (90%) and then were split using a 1/3 ratio for serial passage. A relatively homogeneous population of cells was obtained after passage 3 by visual inspection of cell morphology by light microscopy. Cells from passage 3–5 were used for subsequent experiments.
The FLSs were treated with either 30 μM SP600125, a specific JNK inhibitor (Selleck, Houston, TX, USA), or 5 μM Anisomycin, a TRAF1 activator (Selleck, Houston, TX, USA). In some experiments, 10 μg/ml CD40 blocking antibody (Functional Grade, eBioscience) and 1 ng/ml CD154 (CD40 Ligand) (Functional Grade, eBioscience) were added to the cocultures for 48 h. Cells treated with DMSO alone were used as controls.
Western blotting
Rabbit antibodies to SAPK/JNK (9252), phosphor-SAPK/JNK (4668), NF-κB P65 (8242), phosphor-NF-κB P65 (3033) (Cell Signaling Technology), MMP-1 (10371-1-AP), MMP-3 (17873-1-AP), MMP-13 (18165-1-AP) (Proteintech), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology) and Goat anti-RabbitIgG (H + L) (SA00001-2) (Proteintech) were used for western blotting. FLS were washed twice with PBS and lysed on ice for 30 min with 1× RIPA buffer (Cell Signaling Technology) containing 1% 100× protease/phosphatase inhibitor Cocktail (Cell Signaling Technology). Lysates were centrifuged at 12,000 g at 4 C for 20 min, and the supernatants were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Proteins were then transferred to polyvinylidene fluoride membranes (Millipore), blocked for 1 h in 5% nonfat milk (in 10 mM Tris pH 7, 150 mM NaCl, 0.1% Tween 20, TBST), and then immunoblotted with indicated primary antibodies and appropriate horseradish peroxidase-conjugated secondary antibodies. The bands were visualized in a luminol-based detection system with piodophenol enhancement.
Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total cellular RNA was extracted using Trizol reagent (Invitrogen) and 1 μg RNA was used in RT reactions. cDNA was synthesized by Primer Script RT reagent Kit (TaKaRa). Sequences of the forward and reverse primers were: TRAF1 5ʹ-CGGCGCCGAGATGGAG-3ʹ and 5ʹ-GTGTGGTTCAACGTCACAGC-3ʹ; MMP-1 5ʹ-CTGGCCACAACTGCCAAATG-3ʹ and 5ʹ-CTGTCCCTGAACAGCCCAGTACTTA-3ʹ; MMP-3 5ʹ-ATTCCATGGAGCCAGGCTTTC-3ʹ and 5ʹ-CATTT GGGTCAAACTCCAACTGTG-3ʹ; MMP-13 5ʹ-TCCCAGGAATTGGTGATAAA GTAGA-3ʹ and 5ʹ-CTGGCATGACGCGAACAATA-3ʹ; GAPDH 5ʹ-TGGCCTTCCGTGTTCCTAC-3ʹ and 5ʹ-GAGTTGCTGTTGAAGTCGCA-3ʹ. For real-time PCR experiments, reactions containing SYBR Premix EX Taq (Takara), ROX Reference Dye (Takara), cDNA, and gene primers were run on a Step One Plus real-time PCR system and analyzed using Step One Software, version 2.1 (Applied Biosystems). Relative gene quantification was calculated by the 2ΔCt method and then normalized to the level of GAPDH.
Cell counting kit-8 (CCK-8)
FLSs were incubated in a 96-well plate. Pre-incubation of the plate was in a humidified incubator 37℃, 5% CO2, followed by addition of 10 μl of the CCK-8 solution to each well of the plate. Incubation of the plate was for 2 h in the incubator. Measured absorbance at 450 nm was done using a micro-plate reader. A calibration curve was prepared using the data obtained from the wells that contain known numbers of viable cells.
Overexpression and knockdown of TRAF1
To create FLS cells that overexpressed TRAF1, the open reading frame of the human TRAF1 gene was cloned into pcDNA3.1 vector (RiboBio, Guangzhou, China) to construct TRAF1 expressing plasmid. To silence TRAF1 expression, FLS were transfected with control siRNA (siNC) or TRAF1 small interfering RNA (siTRAF1, TGTGGAAGATCACCAATGT) (RiboBio, Guangzhou, China) using Lipofectamine 2000 (Invitrogen).
Enzyme-linked immunosorbent assay (ELISA)
MMP-1 level was measured by ELISA kit (Fcmacs, China) according to the manufacturer’s protocol. MMP-3 and MMP-13 level were measured using human total MMP-3 and MMP-13 ELISA Kits (R&D system, USA), respectively, according to the manufacturers’ instructions.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
TUNEL assay (In Situ Cell Death Detection Kit; Roche, Basel, Switzerland) was used on tissue sections. Sections were paraformaldehyde-fixed and hydrated. We randomly selected three representative slides from each group. The TUNEL assay was then performed according to the manufacturer’s protocol. Slides were mounted with ProLong Antifade (Invitrogen–Molecular Probes, Carlsbad, CA) containing 4’, 6-diamidino-2-phenylindole (DAPI). The slides were analyzed using a fluorescent microscope (BZ-X710; Keyence, Osaka, Japan). Dead cells were quantified by counting TUNEL-positive nuclei in 10 random microscopic fields (20×).
Flow cytometry analysis
Cells were harvested and washed with pre-cooled PBS three times, followed by being resuspended using binding buffer to a concentration of 1 × 106/ml. The rates of apoptosis for FLS were detected by staining with Annexin V and 7AAD (BD Pharmingen) according to the manufacturer’s instruction. Data were collected with a fluorescence-activated cell sorting (FACS) Calibur flow cytometer (BD Biosciences) and analyzed by FlowJo software (Tree Star).
Statistical analysis
The data were expressed as mean ± standard error of mean and analyzed with Prism 5 (GraphPad Software). Student’s t test was used to analyze significance between the two groups, and comparisons among more than two groups were analyzed using one-way ANOVA. The value of p < 0.05 was considered statistically significant.
Results
TRAF1 expression in RA synovial tissue correlates negatively with apoptosis
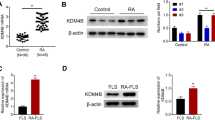
The mRNA expression of TRAF1 was determined in FLSs from RA and OA patients by RT-PCR analysis. As shown in Fig. 1A B, TRAF1 mRNA and protein level were significantly higher in RA patients than in control patients. We used the DAS28 to explore the relationship between disease activity and the TRAF1 mRNA level. Positive correlation of the TRAF1 level and the DAS28 score was observed with r = 0.806, p = 0.005 (Fig. 1C). In addition, FLSs apoptosis percentage was calculated by TUNEL, which was negatively correlated with TRAF1 mRNA level (Fig. 1D, E). These results indicate that TRAF1 mediates FLSs apoptosis in RA patients.

TRAF1 expression correlates negatively with apoptosis in FLSs of RA patients. A TRAF1 mRNA level in FLSs of RA and OA patients. B TRAF1 protein level in FLSs of RA and OA patients. C Correlation of TRAF1 level and the DAS28 score (r = 0.806, p = 0.005). D FLS apoptosis percentage by TUNEL in synovial tissue. E Correlation of TRAF1 level and FLSs apoptosis percentage (r = − 0.860, p = 0.002). n = 10/group. **p < 0.01; ***p < 0.001
TRAF1 decreases RA-FLSs apoptosis through JNK/NF-κB and increases MMP expressions in vitro
We postulated that a potential mechanism of TRAF1 could decrease FLSs apoptosis through suppressing JNK/NF-κB pathway in RA. To test this, we used FLSs isolated from RA patients to examine the influence of TRAF1 on synoviocytes in vitro. FLSs were co-cultured with TRAF1siRNA at different concentrations (30 nM or 60 nM) for 48 h. The results showed that the TRAF1siRNA could not only decreased the levels of TRAF1 (Supplementary information, Fig. S1), but also significantly decreased the levels of MMP-1, MMP-3 and MMP-13 in a dose-dependent manner, consistent with the secreted protein level in the cell culture supernatant detected by ELISA (Fig. 2A–D). However, the percentage of FLSs apoptosis was increased while the proliferation rate was decreased in TRAF1siRNA group (Fig. 2E, F). In addition, we found TRAF1siRNA significantly decreased levels of the phosphorylated forms of JNK and NF-κB/p65 proteins in FLSs (Fig. 2G). These data suggest that TRAF1 contributes to the RA-FLSs apoptosis by activating JNK/NF-κB pathway and increase MMP-1, MMP-3 and MMP-13 expression.

TRAF1 decreases RA-FLSs apoptosis through JNK/NF-κB pathway and increases MMPs expressions in vitro. FLSs from RA patients were cultured alone or with siTRAF1 at different concentrations (30 nM and 60 nM) for 48 h. MMP-1, MMP-3 and MMP-13 level of FLSs were quantitated by qPCR (A). The levels of MMP-1, MMP-3 and MMP-13 in cell supernatants were measured by ELISA (B–D). The apoptosis percentage was determined by flow cytometry in siNC and siTRAF1 (60 nM) treated RA-FLSs (E). The proliferation rate was measured by CCK-8 assay in siNC and siTRAF1 (60 nM) treated RA-FLSs (F). Western blotting analysis of the protein levels of JNK, p-JNK, NF-κB and p-NF-κB in siNC and siTRAF1 (60 nM) treated RA-FLSs. GAPDH was used as a protein loading control (G). n = 5/group. All experimental data were verified in at least two independent experiments. *p < 0.05; **p < 0.01
JNK is required for TRAF1-mediated apoptosis of FLSs in RA patients
Next, we examined whether JNK is sufficient and necessary for TRAF1 to decrease the apoptosis of FLSs in RA patients. We found that pretreatment of RA-FLSs with the JNK inhibitor-SP600125 decreased MMPs expression and increased percentage of apoptosis cells (Fig. 3A, B). Conversely, pretreatment of RA-FLSs with a selective activator of JNK, Anisomycin, inhibited the apoptosis induced by TRAF1siRNA (Fig. 3C, D). Collectively, these data reveal that JNK is a key mediator whereby TRAF1 decreases apoptosis of FLSs in RA patients.

JNK is a mediator of TRAF1 decreasing apoptosis of FLSs in RA patients. The protein expression of JNK, p-JNK, MMP-1, MMP-3, MMP-13 and the apoptosis percentage in JNK inhibitor-SP600125 and DMSO-treated RA-FLSs for 48 h by Western blotting and flow cytometry (A, B). RA-FLSs and siTRAF1 were cultured alone or together in the presence or absence of Anisomycin. The expression of JNK, p-JNK, MMP-1, MMP-3 and MMP-13 were quantified by Western blotting (C). The apoptosis percentage was determined by flow cytometry (D). GAPDH was used as a protein loading control. n = 3/group. All experimental data were verified in at least two independent experiments. *p < 0.05; **p < 0.01
TRAF1 elevates MMPs expression and secretion via CD40
Next, we examined whether the receptor CD40 existing on FLSs membrane responsible for TRAF1 induced signal transduction. We demonstrated TRAF1 overexpression plasmid alone increased TRAF1 level (Supplementary information, Figure S2). Co-culture of CD40 antibody-CD40L and TRAF1 with RA-FLSs for 48 h showed decreased MMP-1, MMP-3, MMP-13 expression compared with TRAF1 overexpression plasmid alone (Fig. 4A). In addition, the presence of MMP-1, MMP-3, and MMP-13 in the supernatant from FLSs cultures showed similar results (Fig. 4B–D). Collectively, these data suggest that TRAF1 increases MMPs expression via CD40 in RA-FLSs.

TRAF1 increases MMPs expression via CD40 in RA-FLSs. RA-FLSs and TRAF1 were cultured alone or together in the presence or absence of CD40 antibody. The mRNA expression of MMP-1, MMP-3 and MMP-13 were quantified by qPCR (A). The MMP-1, MMP-3 and MMP-13 levels in cell supernatants were measured by ELISA (B–D). n = 3/group. All experimental data were verified in at least two independent experiments. *p < 0.05
Discussion
Synovial hyperplasia is a major pathophysiologic feature of RA, ultimately causing bone invasion to lose its integrity. Various genome-wide association studies have identified single-nucleotide polymorphisms in the TRAF1-C5 locus on chromosome 9 as risk factors for RA patients since 2007 [6, 16, 17]. Evidence for TRAF1 showed both positive and negative regulator of immune signaling in various diseases [18]. However, limited studies addressed the role of TRAF1 in FLSs of RA patients. In 2002, Youn et al. showed that the expression of TRAF1 was the most dramatically enhanced in RA synoviocytes after TNF-α stimulation [19]. Nishimoto T et al. reported a single-nucleotide polymorphism of TRAF1 predicted the clinical response to anti-TNF treatment in Japanese patients with RA in 2009 [20]. Here, we proved the significant increase of TRAF1 level in FLSs of RA patients and demonstrated the TRAF1 level correlated positively with DAS28 score. These results suggest that the disorder of TRAF1 in FLSs is an important factor in the pathogenesis of RA.
The imbalance between FLSs apoptosis inhibitors and apoptosis-promoting factors leads to prolonged synovial cell survive and increases the secretion of MMPs, which promotes articular cartilage and bone tissue damage. Insufficient synovial cell apoptosis is considered to be the pathogenesis of RA [2]. Extensive studies have demonstrated that TRAF1 was involved in multiple signaling pathways and thus influenced apoptotic responses. TRAF1 has been widely reported as an anti-apoptotic gene, including protecting staurosporine-induced macrophages apoptosis [21] and resistant to CD30-mediated apoptosis in classical Hodgkin lymphoma [22]. It should be noted that the anti-apoptotic effect of TRAF1 is not universal; some exceptions have been reported in hepatocyte and cerebral cell apoptosis via ASK1 [23, 24]. These data suggested that TRAF1 differentially affects receptor-transduced signals and controls critical proliferative and anti-apoptotic functions in a cell type-dependent manner. Herein, we demonstrate that TRAF1 level correlated negatively with FLSs apoptosis in vivo and treatment with siTRAF1 was able to increase percentage of FLSs apoptosis in vitro. These results suggest that the reduction of apoptosis in FLSs of RA patients is related with TRAF1 and this might be associated with consequent hyper-proliferation of FLSs.
TRAF1 belongs to a group of NF-κB-dependent gene products that function cooperatively at the earliest check-point to suppress TNFα-mediated apoptosis [25, 26]. Interestingly, not only TRAF1 can cause NF-κB activation but also the expression is up-regulated by NF-κB, suggesting that they could form part of a positive feedback loop sustaining the NF-κB-activated state of TNFα-stimulated cells [27]. Previous reports showed that binding of NF-κB to three of five putative binding sites within the human TRAF1 promoter was found in electrophoretic mobility shift assay studies. Moreover, triggering of TNF-R1, CD40, and the interleukin-1 receptor resulted in transcription of the TRAF1 gene [28]. Researchers have shown that a deficiency of TRAF1 in cultured hepatocytes led to the inhibition of NF-κB-mediated inflammatory responses and the suppression of the JNK pro-death pathway [24]. JNK activation in several cell types, including FLSs, contributed to pathology in part through cellular production of inflammatory cytokines and MMPs [29]. However, the specific regulation of TRAF1 on the JNK/NF-κB signaling pathway was controversial [9,10,11,12,13,14]. Our previous research provided evidence for the increased expression levels of CD40 and TRAF1, as well as total NF-κB p65, phospho-NF-κB and NF-κB-related gene expression in synovial tissue of CIA mice model [30]. In this study, we confirmed the role of TRAF1 and JNK/NF-κB signaling pathways in FLSs of RA patients. We demonstrated that treatment with siTRAF1 was able to decrease MMPs levels and the phosphorylated forms of JNK/NF-κB in vitro. Moreover, JNK inhibitor could attenuate expression of MMPs and increase percentage of apoptosis cells in RA-FLSs, while siTRAF1 could not promote apoptosis when RA-FLSs were pretreated with JNK activator. Taken together, these data suggest that JNK is a mediator for TRAF1 reduced apoptosis of FLSs in RA patients. The mechanism of TRAF1 that controls the JNK/NF-κB pathway was not confirmed in this study and required further investigation. Related researches showed that TRAF1 expression enhances the ubiquitination of ERK5 on lysine 184, which is necessary for its kinase activity and AP-1 family members (c-Fos/c-Jun) activation in UVR-induced skin carcinogenesis [31]. Other studies showed that TRAF1 promotes myocardial injury and hepatic steatosis through regulating ASK1 (apoptosis signal-regulating kinase 1)-mediated JNK/p38 cascades [32, 33].
CD40-CD40L interaction results in the activation of a variety of signaling cascades, which ultimately determine the diverse physiologic effects. In 2007, Eeva et al. showed the CD40-induced protection against CD95-mediated apoptosis is associated with a rapid upregulation of anti-apoptotic c-FLIP [34]. Other studies showed that CD40-CD40L inhibits apoptosis and stimulates proliferation of B cells by upregulating bcl-xL expression and blocking oxidant accumulation [35, 36]. However, some functional studies have produced conflicting results on its apoptotic function. For example, CD40 induced Fas-dependent apoptosis in human intrahepatic biliary epithelial cells and endothelial cells [37, 38]. In addition, some researches revealed that CD40 antibody had no effect on apoptosis [39, 40]. These results raise a possibility that the phenotypic consequences of CD40 signaling on apoptosis, therefore, appear to be dependent on the cell types and proinflammatory microenvironment. TRAF1 interacted directly with a subset of TNFR family members. As one of the upstream inducers, CD40 mediated its specific role in the TRAF1 signaling pathway in RA-FLSs remains unclear. In this study, we also showed that co-culture of CD40 monoclonal antibody and TRAF1 with RA-FLSs significantly reduced MMPs expression compared with TRAF1 expression plasmid alone. These data suggest that TRAF1 increases MMPs expression in response to the engagement of CD40 in RA-FLSs.
In conclusion, our ex vivo and in vitro experiments demonstrated the first time that CD40/TRAF1 decreases RA-FLSs apoptosis through the JNK/NF-κB pathway in RA patients. These findings not only extend our knowledge of the immunoregulatory function of TRAF1 in RA, but also offer TRAF1 as a potential biomarker or therapeutic target to further explore in the future.
References
Nygaard G, Firestein GS (2020) Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol 16:316–333. https://doi.org/10.1038/s41584-020-0413-5
Korb-Pap A, Bertrand J, Sherwood J, Pap T (2016) Stable activation of fibroblasts in rheumatic arthritis-causes and consequences. Rheumatology (Oxford) 55:64–67. https://doi.org/10.1093/rheumatology/kew347
Yang Y, Ye Y, Qiu Q, Xiao Y, Huang M, Shi M, Liang L, Yang X, Xu H (2016) Triptolide inhibits the migration and invasion of rheumatoid fibroblast-like synoviocytes by blocking the activation of the JNK MAPK pathway. Int Immuno pharmacol 41:16. https://doi.org/10.1016/j.intimp
Galliqan CL, Siminovitch KA, Keystone EC, Bykerk V, Perez OD, Fish EN (2010) Fibrocyte activation in rheumatoid arthritis. Rheumatology 49:640–651. https://doi.org/10.1093/rheumatology/kep265
Xia ZB, Meng FR, Fang YX, Wu X, Zhang CW, Liu Y, Liu D, Li GQ, Feng FB, Qiu HY (2018) Inhibition of NF-κB signaling pathway induces apoptosis and suppresses proliferation and angiogenesis of human fibroblast-like synovial cells in rheumatoid arthritis. Medicine 97:e10920. https://doi.org/10.1097/MD.0000000000010920
Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF et al (2007) TRAF1-C5 as a risk locus for rheumatoid arthritis-a genome wide study. N Engl J Med 357:1199. https://doi.org/10.1056/NEJMoa073491
Xie P, Hostager BS, Munroe ME, Moore CR, Bishop GA (2006) Cooperation between TNF receptor-associated factors 1 and 2 in CD40 Signaling. Bishop J Immunol 176:5388–5400. https://doi.org/10.4049/jimmunol.176.9.5388
Dunn IF, Sannikova TY, Geha RS, Tsitsikov EN (2000) Identification and characterization of two CD40-inducible enhancers in the mouse TRAF1 gene locus. Mol Immunol 37:961–973. https://doi.org/10.1016/s0161-5890(01)00015-3
Cabal-Hierro L, Rodríguez M, Artime N, Lglesias J, Ugarte L, Prado MA, Lazo PS (2014) TRAF-mediated modulation of NF-kB AND JNK activation by TNFR2. Cell Signal 26:2658–2666. https://doi.org/10.1016/j.cellsig.2014.08.011
McPherson AJ, Snell LM, Mak TW, Watts TH (2012) Opposing roles for TRAF1 in the alternative versus classical NF-κB pathway in T cells. J Biol Chem 29:23010–23019. https://doi.org/10.1074/jbc.M112.350538
Choudhary S, Kalita M, Fang L, Patel KV, Tian B, Zhao Y, Edeh CB, Brasier AR (2013) Inducible TNF receptor associated factor-1 expression couples the canonical to the non-canonical NF-κB Pathway in TNF stimulation. J Biol Chem 288:14612–14623. https://doi.org/10.1074/jbc.M113.464081
Oussa NA, Soumounou Y, Sabbagh L (2013) TRAF1 phosphorylation on Serine 139 modulates NF-κB activity downstream of 4-1BB in T cells. Biochem Biophys Res Commun 432:129–134. https://doi.org/10.1016/j.bbrc.2013.01.073
Bin W, Ming X, Wen-Xia C (2019) TRAF1 meditates lipopolysaccharide-induced acute lung injury by up regulating JNK activation. Biochem Biophys Res Commun 511:49–56. https://doi.org/10.1016/j.bbrc.2019.01.041
Greenfeld H, Takasaki K, Walsh MJ, Ersing I, Bernhardt K, Ma Y, Fu B, Ashbaugh CW, Cabo J, Mollo SB, Zhou H, Li S, Gewurz BE (2015) TRAF1 coordinates polyubiquitin signaling to enhance Epstein-Barr Virus LMP1-mediated growth and survival pathway activation. PLos Pathog 11:e1004890. https://doi.org/10.1371/journal.ppat.1004890
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS (1988) The American rheumatism association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31:315–324. https://doi.org/10.1002/art.1780310302
Zhang X, Li W, Zhang X, Zhang X, Jiang L, Guo Y, Wang X (2014) Association between polymorphism in TRAF1/C5 gene and risk of rheumatoid arthritis: a meta-analysis. Mol Biol Rep 41:317–324. https://doi.org/10.1007/s11033-013-2864-0
Van Steenbergen HW, Rodríguez-Rodríguez L, Berglin E, Zhernakova A, Knevel R, Ivorra-Cortés J, Huizinga TW, Fernández-Gutiérrez B, Gregersen PK, Rantapää-Dahlqvist S, van der Helm-van Mil AHM (2015) A genetic study on C5-TRAF1 and progression of joint damage in rheumatoid arthritis. Arthritis Res Ther 17:1. https://doi.org/10.1186/s13075-014-0514-0
Edilova MI, Abdul-Sater AA, Watts TH (2018) TRAF1 signaling in human health and disease. Front Immunol 9:2969. https://doi.org/10.3389/fimmu.2018.02969
Youn J, Kim HY, Park JH, Hwang SH, Lee SY, Cho CS, Lee SK (2002) Regulation of TNF-a-mediated hyperplasia through TNF receptors, TRAFs, and NF-κB in synoviocytes obtained from patients with rheumatoid arthritis. Immunol Lett 83:85–93. https://doi.org/10.1016/s0165-2478(02)00079-2
Nishimoto T, Seta N, Anan R, Yamamoto T, Kaneko Y, Takeuchi T, Kuwana M (2014) A single nucleotide polymorphism of TRAF1 predicts the clinical response to anti-TNF treatment in Japanese patients with rheumatoid arthritis. Clin Exp Rheumatol 32:211–217
Okuda J, Arikawa Y, Takeuchi Y, Mahmoud MM, Suzaki E, Kataoka K, Suzuki T, Okinaka Y, Nakai T (2006) Intracellular replication of Edwardsiella tarda in murine macrophage is dependent on the type III secretion system and induces an up-regulation of anti-apoptotic NF-kappaB target genes protecting the macrophage from staurosporine-induced apoptosis. Microb Pathog 41:226–240. https://doi.org/10.1016/j.micpath.2006.08.002
Dürkop H, Hirsch B, Hahn C, Foss HD, Stein H (2005) Differential expression and function of A20 and TRAF1 in Hodgkin lymphoma and anaplastic large cell lymphoma and their induction by CD30 stimulation. Anticancer Res 25:2367–2379. https://doi.org/10.1002/path.1351
Lu Y-Y, Li Z-Z, Jiang D-S, Wang L, Zhang Y, Chen K, Zhang X-F, Liu Y, Fan G-C, Chen Y, Yang Q, Zhou Y, Zhang X-D, Liu D-P, Li H (2013) TRAF1 is a critical regulator of cerebral ischaemia-reperfusion injury and neuronal death. Nat Commun 4:2852. https://doi.org/10.1038/ncomms3852
Zhang XF, Zhang R, Huang L, Wang PX, Zhang Y, Jiang DS, Zhu LH, Tian S, Zhang XD, Li H (2014) TRAF1 is a key mediator for hepatic ischemia/reperfusion injury. Cell Death Dis 5:e1467. https://doi.org/10.1038/cddis.2014.411
Dunn IF, Geha RS, Tsitsikov EN (1999) Structure of the murine TRAF1 gene. Mol Immunol 36:611–617
Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr (1988) NF-κB antiapoptosis: Induction of TRAF-1 and TRAF-2 and cIAP2 to suppress caspase-8 activation. Science 281:1680–1683
Duckett CS, Gedrich RW, Gilfillan MC, Thompson CB (1997) Induction of NF-kB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol Cell Biol 17:1535–1542. https://doi.org/10.1128/MCB.17.3.1535
Schwenzer R, Siemienski K, Liptay S, Schuber G, Peters N, Scheurich P, Schmid RM, Wajant H (1999) The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-kB and c-Jun N-terminal kinase. J Biol Chem Actions Search 274:19368–19374. https://doi.org/10.1074/jbc.274.27.19368
Guma M, Firestein GS (2012) c-Jun N-terminal kinase in inflammation and rheumatic diseases. Open Rheumatol J 6:220–231. https://doi.org/10.2174/1874312901206010220
Cheng T, Wang M, Chen L, Guo Y, Chen Z, Wu J (2018) Increased expression of CD40/TRAF1 and activation of nuclear factor-κB-dependent proinflammatory gene expression in collagen-induced arthritis. Scand J Rheumatol 47:455–460. https://doi.org/10.1080/03009742.2018.1432684
Yamamoto H, Ryu J, Min E, Oi N, Bai R, Zykova TA, Yu DH, Moriyama K, Bode AM, Dong Z (2017) TRAF1 Is Critical for DMBA/Solar UVR-Induced Skin Carcinogenesis. J Invest Dermatol 137:1322–1332. https://doi.org/10.1016/j.jid.2016.12.026
Xu W, Zhang L, Zhang Y, Zhang K, Wu Y, Jin D (2019) TRAF1 Exacerbates Myocardial Ischemia Reperfusion Injury via ASK1-JNK/p38 Signaling. J Am Heart Assoc 8:e012575. https://doi.org/10.1161/JAHA.119.012575
Xiang M, Wang PX, Wang AB, Zhang XJ, Zhang Y, Zhang P, Mei FH, Chen MH, Li H (2016) Targeting hepatic TRAF1-ASK1 signaling to improve inflammation, insulin resistance, and hepatic steatosis. J Hepatol 64:1365–1377. https://doi.org/10.1016/j.jhep.2016.02.002
Eeva J, Ropponen A, Nuutinen U, Eeva ST, Mättö M, Eray M, Pelkonen J (2007) The CD40-induced protection against CD95-mediated apoptosis is associated with a rapid upregulation of anti-apoptotic c-FLIP. Mol Immunol 44:1230–1237. https://doi.org/10.1016/j.molimm.2006.06.005
Wang D, Freeman GJ, Levine H, Ritz J, Robertson MJ (1997) Role of the CD40 and CD95 (APO-1/Fas) antigens in the apoptosis of human B-cell malignancies. Br J Haematol 97:409–417. https://doi.org/10.1046/j.1365-2141.1997.422688.x
Fang W, Nath KA, Mackey MF, Noelle RJ, Mueller DL, Behrens TW (1997) CD40 inhibits B cell apoptosis by upregulating bcl-xL expression and blocking oxidant accumulation. Am J Physio 272:C950-956. https://doi.org/10.1152/ajpcell.1997.272.3.C950
Afford SC, Ahmed-Choudhury J, Randhawa S, Russell C, Youster J, Crosby HA, Eliopoulos A, Hubscher SG, Young LS, Adams DH (2001) CD40 activation-induced, Fas-dependent apoptosis and NF-kappaB/AP-1 signaling in human intrahepatic biliary epithelial cells. FASEB J 15:2345–2354. https://doi.org/10.1096/fj.01-0088com
Longo CR, Arvelo MB, Patel VI, Daniel S, Mahiou J, Grey ST, Ferran C (2003) A20 protects from CD40-CD40 ligand-mediated endothelial cell activation and apoptosis. Circulation 108:1113–1118. https://doi.org/10.1161/01.CIR.0000083718.76889.D0
Villarroel Dorrego M, Whawell SA, Speight PM, Barrett AW (2006) Transfection and ligation of CD40 in human oral keratinocytes affect proliferation, adhesion and migration but not apoptosis in vitro. Clin Exp Dermatol 31:266–271. https://doi.org/10.1111/j.1365-2230.2005.02018.x
Kim L-H, Eow G-I, Peh SC, Poppema S (2003) The role of CD30, CD40 and CD95 in the regulation of proliferation and apoptosis in classical Hodgkin’s lymphoma. Pathology 35:428–435. https://doi.org/10.1080/00313020310001602567
Funding
This work was supported by Suzhou Minsheng science and technology project (SYS2020108, SYS2019043).
Author information
Authors and Affiliations
Contributions
TC and JW participated in study design, data collection, data analysis, data interpretation, and drafting the paper. MW, YX, CL and HZ participated in patient recruitment, experiments, and data collection. TC and MW supervised the whole research, designed the study, interpreted the data, and wrote the paper. All the authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Consent for publication
All the authors agree to publish.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
About this article

Cite this article
Cheng, T., Wu, J., Xu, Y. et al. CD40/TRAF1 decreases synovial cell apoptosis in patients with rheumatoid arthritis through JNK/NF-κB pathway. J Bone Miner Metab 40, 819–828 (2022). https://doi.org/10.1007/s00774-022-01350-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-022-01350-6