
Abstract
Protein 3D structures, determined by their amino acid sequences, are the support of major crucial biological functions. Post-translational modifications (PTMs) play an essential role in regulating these functions by altering the physicochemical properties of proteins. By virtue of their importance, several PTM databases have been developed and released in decades, but very few of these databases incorporate real 3D structural data. Since PTMs influence the function of the protein and their aberrant states are frequently implicated in human diseases, providing structural insights to understand the influence and dynamics of PTMs is crucial for unraveling the underlying processes. This review is dedicated to the current status of databases providing 3D structural data on PTM sites in proteins. Some of these databases are general, covering multiple types of PTMs in different organisms, while others are specific to one particular type of PTM, class of proteins or organism. The importance of these databases is illustrated with two major types of in silico applications: predicting PTM sites in proteins using machine learning approaches and investigating protein structure–function relationships involving PTMs. Finally, these databases suffer from multiple problems and care must be taken when analyzing the PTMs data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proteins are mainly composed of a succession of 20 standard amino acid types. Their 3D structures, determined by their sequences, are the support of major crucial biological functions. But it was found that post-translational modifications (PTMs) influence the structure and regulate the function of proteins. It is speculated that nearly every protein undergoes some form of PTMs (Lodish 2013) which involve the attachment of chemical groups to the amino acid side chains, and in rare cases, to the backbone of proteins (Muller 2018). Although proteins can be modified pre-, co- or post-translationally, all protein modifications are generally referred to as PTMs, because they are typically made post-translationally, after the protein is folded, and they can be reversible or irreversible.
PTMs alter the physicochemical properties of proteins and thereby play a critical role in modulating various biological functions. However different PTMs display different physicochemical properties; thus, the same protein may exhibit different functions upon different modifications (Jungblut et al. 2008; Mann and Jensen 2003). As a result of their high diversity and their reversibility reflecting the dynamic nature of a cell, PTMs have also been reported to play essential roles in many cellular control mechanisms, folding, conformational change, stability, activity, localization, turnover, and molecular interactions with partners (Mann and Jensen 2003; Deribe et al. 2010; Walsh et al. 2005). PTMs influence protein function both in orthosteric and allosteric modes (Berezovsky et al. 2017).
Apart from normal cellular processes, it was also shown that dysregulation of PTMs and mutation of PTM sites are implicated in a number of human diseases (Vidal 2011) such as cancer (Bode and Dong 2004; Dai and Gu 2010; Radivojac et al. 2008), diabetes (Donnelly and Williams 2020; Sidney et al. 2018; Lernmark 2013), cardiovascular disorders (Van Eyk 2011; Aggarwal et al. 2020; Gao et al. 2020) and neurodegenerative disorders (Gong et al. 2005; Nekooki-Machida and Hagiwara 2020; Ajit et al. 2019). It was also shown that proteins modified by multiple types of PTMs are significantly more prone to participate in disease than proteins carrying no known PTM sites (Huang et al. 2014). The formers were found notably implicated in protein complexes with many partners with a preference to act as hubs in protein–protein interaction (PPI) networks.
In the past, PTMs were primarily studied with the aid of low-throughput biological techniques. Nowadays, the current high-throughput MS-based approaches and proteomic studies allow many more novel sites to be identified and produce a wealth of new information regarding PTMs. For instance, the PRoteomics IDEntifications PRIDE database (Perez-Riverol et al. 2019) is the world’s largest data repository of mass spectrometry-based proteomics data and is one of the founding members of the global ProteomeXchange (PX) consortium (http://www.proteomexchange.org) (Deutsch et al. 2017); many of the deposited data are related to PTMs, mainly glycosylation, such as glycoproteome associated with prostate cancer progression (Kawahara et al. 2021). With this increased amount of PTM data, Researchers encountered challenges and difficulties to include all this information in a consistent and structured way by standardizing the annotation of PTM features and adopting a controlled vocabulary associated with every described PTM (Farriol-Mathis et al. 2004), to facilitate easy retrieval and promote understanding by biologist expert users as well as computer programs. Providing a precise number of the different types of PTMs is a highly difficult task as some, such as Phosphorylation and N- and O-linked Glycosylation are ubiquitous, while others are specific to a clade. 682 types of PTMs have been reported in the UniProt database (UniProt 2019) using a controlled vocabulary (2021 3rd release of 02-Jun-2021), spanning all domains of life.
A decade after the release of the Human Proteome Project (HPP) in 2010 (Legrain et al. 2011), 191,837 PTMs across the 20,379 proteins of the human proteome have been already detected by mass spectrometry in the different cell types that comprise the human body as indicated on the neXtProt platform in its February 2021 release (Adhikari et al. 2020; Zahn-Zabal et al. 2020). All these PTMs, altering protein properties, are in part responsible for the largely unmapped complexity and diversity of the human proteoforms (Aebersold et al. 2018). For instance, the human histone H4 (UniProt accession: P62805) mapped 75 frequent proteoforms overs its length of only 103 residues.
Owing to the importance of PTMs, several databases have been developed and released in decades, but very few of these databases incorporate 3D structural data (i.e., with real 3D coordinates). Since PTMs influence the function of the protein and their aberrant states are frequently implicated in human diseases, providing structural insights to understand the influence and dynamics of PTMs is crucial for unraveling cellular processes. Many web-based protein structure databases exist providing the scientific community access to a wide variety of structural information. The primary repository of 3D structural data on proteins (and other biological macromolecules) is the Protein Data Bank that was founded in 1971 with only seven experimentally determined protein structures at that time (Berman et al. 2000). In 2021, the PDB is celebrating its 50 years anniversary with more than 178,000 entries by June 2021. Despite the PDB being a rich reservoir of structural information for biological macromolecules and having powerful querying interfaces, it turns out that specialized databases, derived from the PDB and cross-annotated with other types of data, are often easier, faster, and more informative for some specific scientific/research questions and goals. These databases have also the added value of being built, maintained, and updated by experts in the field of structural biology.
To illustrate some of the points discussed above, the human Cyclin-Dependent Kinase 2 (CDK2) protein (UniProt ID: P24941) is selected. This kinase is involved in the control of the cell cycle and its modifications are essential to regulate its activity (to cite a few of the published research works Gu et al. 1992; Welburn et al. 2007; Timofeev et al. 2010; Choudhary et al. 2009). Figure 1 represents an overall view of the superimposition of 4 human CDK2 protein structures downloaded from the Protein Data Bank and exhibiting different types of PTMs highlighted in the 4 zoomed views located on the sides: PDB id 2CJM (Welburn et al. 2007), colored in green, highlighting two phosphorylation on residues Tyr15 and Thr160; PDB id 1H01 (Beattie et al. 2003) and PDB id 4RJ3 (Hanan et al. 2014), colored in gold and blue, displaying a carboxylation and acetylation on the same Lys33 residue, respectively; finally PDB id 1GZ8 (Gibson et al. 2002), colored in dark red, showing a sulfino-alanine on residue 177. We can clearly observe the conformational changes induced by the phosphorylation of the two residues (Tyr15 and Thr160) affecting mainly the N-terminal domain and a few loops on the CDK2 structure colored in green.

Overall view of the superimposition of 4 human CDK2 protein structures. The CDK2 fold is rendered in ribbon representation and colored in green, gold, blue, and dark red for the PDB IDs 2CJM, 1H01, 4RJ3 and 1GZ8, respectively. The PTMs present in each of these structures are drawn in ball-and-stick mode with carbon atoms colored similarly to the overall structure while nitrogen, oxygen, phosphorus, and sulfur atoms colored in dark blue, red, orange, and yellow, respectively. RMSD calculations, between all pairs of CDK2 structures, showed values ranging from 0.25 Å (for 1H01 with 4RJ4) to 4.85 Å (for 1GZ8 with 2CJM); The structure colored in green (2CJM) presented most local and global conformational changes, consisting of loop motions and tilt of the smaller N-terminal lobe
The immense majority of currently existing PTM databases predominantly focus on protein sequence information and basic modification site metadata while the 3D structural data related to PTMs have been largely overlooked. In this review, we focus on the current state and development of the limited number of PTM 3D structural databases, i.e., with 3D coordinates. Then we highlight their importance and application in many research studies. Sample cases where these databases have been used to aid computational and modeling studies of PTM structures or to advance our knowledge about biological macromolecules are referenced briefly. Finally, we conclude with the many promising in silico research area that still needs to be further explored for a better understanding of PTM’s impacts on protein structures and functions, and improvement of their predictions.
PTM structural databases
The focus of this review is to discuss the current status of structural databases providing three-dimensional data that are experimentally confirmed and/or predicted on PTM sites in proteins. These databases can be general, covering multiple types of PTMs in different organisms, or specific to one particular type of PTM, class of proteins or organism. The emphasis is on repositories offering open-access data through a web user interface. Some of the significant historical projects, no longer maintained, will be also mentioned in this review. The information discussed below are summarized in Table 1. For each database, its name, the year of first and last publications, a brief description of the various features, the data coverage and their citation references are provided.
The most famous database giving access to PTM sites in protein is dbPTM (http://dbptm.mbc.nctu.edu.tw), firstly published in 2006 (Lee et al. 2006), and often updated (Lu et al. 2013; Huang et al. 2016, 2019; Lee et al. 2009). The first release of this database includes all the experimentally validated PTM sites from three external biological databases related to protein PTM information [SwissProt (Wu et al. 2003), PhosphoELM (Diella et al. 2004) and O-GLYCBASE (Gupta et al. 1999)] and authors have developed computational tools to systematically identify and predict three major types of protein PTM (phosphorylation, glycosylation and sulfation) sites against the SwissProt proteins. Protein structural properties and functional information, such as the solvent accessibility of residues, protein isoforms, non-synonymous single nucleotide polymorphism (SNP), protein tertiary structures and protein functional domains, are provided for researchers who are investigating the protein PTM mechanisms by integrating the following external data sources: Ensembl (Hubbard et al. 2005), InterPro (Mulder et al. 2002) and PDB (Deshpande et al. 2005). Solvent accessibility and secondary structure of residues, when experimental 3D structures are not available, are computationally predicted and are mapped to the PTM sites. To help access the database content, a web query interface and graphical visualization were designed and implemented.
In the second version of dbPTM (Lee et al. 2009), the database was enhanced to comprise a variety of new features and collected literature related to PTM, protein conservations and the specificity of substrate site. Furthermore, a variety of prediction tools have been developed for more than ten PTM types (Zhou et al. 2006), such as phosphorylation, glycosylation, acetylation, methylation, sulfation and sumoylation. The interface was also redesigned and enhanced.
In 2013, the dbPTM database in its third version (Lu et al. 2013) was updated to integrate experimental PTMs obtained from public resources as well as manually curated MS/MS peptides associated with PTMs from research articles. The aim is to become an informative resource for investigating the substrate specificity of PTM sites and functional association of PTMs between substrates and their interacting proteins. Additionally, the information of structural topologies on transmembrane (TM) proteins is integrated into dbPTM to delineate the structural correlation between the reported PTM sites and TM topologies. To facilitate the investigation of PTMs on TM proteins, the PTM substrate sites and the structural topology are graphically represented. Also, literature information related to PTMs, orthologous conservations and substrate motifs of PTMs are also provided in the resource. Finally, this version features an improved web interface to facilitate convenient access to the resource.
In 2014, the authors have developed topPTM (http://topptm.cse.yzu.edu.tw) (Su et al. 2014), a new dbPTM module that provides a public resource for identifying the functional PTM sites on transmembrane (TM) proteins with structural topology giving the crucial roles of TM proteins in various cellular processes and the importance of PTMs in their functioning. Experimentally verified TM topology data were integrated from TMPad (Lo et al. 2011), TOPDB (Tusnady et al. 2008), PDBTM (Kozma et al. 2013) and OPM (Lomize et al. 2012). In addition to the PTMs obtained from dbPTM, experimentally verified PTM sites were manually extracted from research articles by text mining. The PTM sites on the tertiary structures of TM proteins can be visualized using a Jmol plugin.
In its most recent publication, dbPTM integrates more than 30 different PTM databases leading to 908,917 experimentally verified PTM sites (571,032 experimentally verified phosphorylation sites, 137,442 acetylation and 118,495 ubiquitination, …) and 347,984 predicted putative sites. It described more than 130 PTM types (Huang et al. 2019).
dbPTM strength is not only limited to the fact that the database has been well maintained for over 10 years and that it integrates many experimentally validated PTMs from available databases and through manual curation of literature but also it provides PTM-disease associations based on non-synonymous single nucleotide polymorphisms (nsSNPs). Some 3D structures are shown with some information of secondary structures; this last is predicted when no 3D structures are available. A JSmol applet allows the visualization of the molecules (Huang et al. 2016).
PTMcode, constructed by Minguez et al., is another general database that integrates 3D structural data, co-evolution and literature curation (http://ptmcode.embl.de) (Minguez et al. 2013, 2015). The PTM residues were searched in the Protein Data Bank (Berman et al. 2000) and specific works have been performed to analyze distance between two PTMs in the same proteins; their conformation could have been visualized using the Jmol plugin. However, the complete list of PTMs, within and between interacting proteins, can be downloaded under the Data” tab in a tab-separated flat files. Based on Flash technology, that is now obsolete, it is complex to browse the database. The second version of PTMcode was released in 2014 and it includes a new strategy to propagate PTMs from validated modified sites through orthologous proteins. This second release covers 19 eukaryotic species from which more than 300,000 experimentally verified PTMs were collected of 69 types (Minguez et al. 2015).
Another publicly available 3D structure database for a wide range of PTMs, named PRISMOID [PRoteIn Structure MOdIfication Database (http://prismoid.erc.monash.edu)] was recently developed (Li et al. 2020). The focus is the 3D structural context of PTMs sites and mutations that occur on PTMs and neighboring PTM sites with functional impact. PRISMOID provides the users with a variety of interactive and customizable search options and data browsing functions to access the data for the target of interest via keywords, PDB/UniProt ID. For each entry in the database, a comprehensive page includes a detailed PTM annotation on the 3D structure and biological information in terms of mutations affecting PTMs, secondary structure assignment, solvent accessibility features of PTM sites and predicted disordered regions. In addition, visualization tools are employed to underline the position of the PTM. However, it is not possible to highlight it in a specific and direct way; the user must do it with its own tool. The DSSP assignment is not provided as classically done by the succession of 3- or 8-states but with 3D coordinates of the Cα residues and backbone angles, e.g., phi, psi and alpha angles.
Few years back, we have built and continue to maintain the Post Translational Modification Structural Database (PTM-SD, http://dsimb.inserm.fr/dsimb_tools/PTM-SD), a curated database that provides access to proteins for which PTMs are both experimentally annotated and structurally resolved (Craveur et al. 2014). It combines different PTM information and annotation gathered from other databases; it crosses information from the PDB, UniProt, PTMCuration and dbPTM.
While most databases and web servers concerning PTMs are dedicated to their compilation and prediction, PTM-SD is probably the only database that focuses on the experimentally resolved amino acid modifications in view of the proteins 3D structures as retrieved from the PDB.
PTM-SD can be browsed using different criteria and users can compute statistics and conduct some analyses on the selected subset of data. PTM-SD gives valuable information on observed PTMs in protein 3D structures facilitating sequence–structure–function analyses in light of PTMs and could provide insights for comparative modeling and PTM prediction protocols.
We can also notice the existence of novPTMenzy (http://202.54.249.142/~shradha/PTM/master.html), a database cataloging information on the sequence, structure, active site and genomic neighborhood of experimentally characterized enzymes involved in five novel PTMs, namely AMPylation, Eliminylation, Sulfation, Hydroxylation and Deamidation (Khater and Mohanty 2015). Based on a comprehensive analysis of the sequence and structural features of these known PTM catalyzing enzymes, an interesting feature of novPTMenzy is the availability of Hidden Markov Model profiles for the identification of similar PTM catalyzing enzymatic domains in genomic sequences.
Other databases are specific to a particular PTM type. ADPriboDB (http://adpribodb.leunglab.org) is a database dealing with ADP-ribosylation; it was firstly developed in 2016 by Vivelo et al. to facilitate studies in uncovering insights into the mechanisms and biological significance of ADP-ribosylation (Vivelo et al. 2017). This protein modification refers to the addition of one or more ADP-ribose units onto proteins and is responsible for many biological processes such as DNA repair, RNA regulation, cell cycle and biomolecular condensate formation. Its dysregulation is implicated in cancer, inflammatory diseases, and neurological disorders. This database was updated in 2020 by Ayyappan et al. (2021). ADPriboDB 2.0 comprises 48 346 entries and 9097 ADP-ribosylated unique proteins, of which 35,946 and 6708 were newly identified, respectively, since the original database release, showing an acceleration of ADP-ribosylation related research. In addition, the authors have created a new interactive tool to visualize the local context of ADP-ribosylation, such as structural and functional features as well as other post-translational modifications.
Another interesting, specialized database is dbSNO 2.0 (http://dbSNO.mbc.nctu.edu.tw) that firstly released in 2012 (Lee et al. 2012; Chen et al. 2015); it focuses only on S-nitrosylation (SNO). This reversible PTM involves the covalent attachment of nitric oxide (NO) to the thiol group of cysteine (Cys) residues, regulating protein activity, localization and stability. SNO is associated with a large panel of pathologies like cancers (Bignon et al. 2018). 298 3D S-nitrosylation are included in the database and are presented with a Java applet, in addition to secondary structure assignment and surface solvent accessibility calculation using DSSP (Kabsch and Sander 1983), modified residues positions, link to PubMed and experiments if available. This website does not allow multiple queries, but only individually.
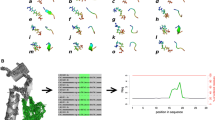
Many databases are dedicated to phosphorylation, one of the most abundant PTM in proteins. Phospho.ELM (Dinkel et al. 2011) (http://phospho.elm.eu.org) is an anciently established database, manually curated, dedicated to eukaryotic phosphorylation sites in proteins. The data is extracted from the literature and phospho-proteomics analyses. Its last version comprises more than 42,500 non-redundant phosphorylation sites in more than 11,000 different protein sequences. The user can query the database by keyword or sequence identifier (UniProt or Ensembl) to get the information about single proteins/substrates, or by kinase name to retrieve all the phosphorylated substrates of a particular kinase. It is also possible to restrict the query to different taxonomy groups. Figure 2 illustrates the usage of Phospho.ELM with the similarity search feature. At first, a sequence is provided in the Phospho.ELM Blast search (see Fig. 2a); different hits can be found by the search engine (see Fig. 2b); the selection of one of these hits (see Fig. 2c) provides many information such as the potential interactions with other proteins. In this case, a PDB file is also available, and the user can follow the link to the PDBe (Gutmanas et al. 2014; Velankar et al. 2010) website (see Fig. 2d).

Presentation of Phospho.ELM with the example of BLAST Search feature. a The sequence is provided, b a list of results is returned, c by selecting one of them multiple information such as SMART and MINT interactions are provided, but also sequences, PMID, disorder prediction and link to the structure that is d link to PDBe
Phospho3D (http://phospho3d.org), a database of three-dimensional structures of phosphorylation sites (P-sites), is derived from Phospho.ELM database previously discussed. It collects information on the P-site instance, its flanking sequence (10 residues) and the P-site 3D zone (the set of residues in a 12 Å radius surrounding the P-site in space). The database uses the latter to conduct large-scale structural comparison to identify structurally similar sites in other proteins (Zanzoni et al. 2011, 2007). It was also enriched with structural annotation at the residue level, including secondary structure and solvent accessibility as defined by DSSP (Kabsch and Sander 1983) and residue conservation as from the Consurf-HSSP database (Glaser et al. 2005). In the same field, we must note the defunct mtcPTM database (mitocheck.org/cgi-bin/mtcPTM/) that stored a large number of structural models of phosphorylatable structures (Jimenez et al. 2007).
PhosphoSitePlus (PSP) (http://phosphosite.org) (Hornbeck et al. 2015) is an open and continuously curated database for studying experimentally observed PTMs in the regulation of biological processes. It was reengineered from the PhosphoSite (Hornbeck et al. 2004) resource that was solely dedicated to phosphorylation in proteins. PSP now covers other commonly studied PTMs including acetylation, methylation, ubiquitination, and O-glycosylation. The interface provides the users with multiple features to browse the database. For each specific modification sites, PSP provides structural and functional information, and many powerful tools for interpreting this data in different contexts: diseases, tissues, subcellular localization, protein domains, sequences, motifs, etc. When available, a list of PDB ids for the protein in question is provided with the possibility of downloading a PyMOL or Chimera script to visualize the location of the different modified residues on the protein structure. It is important to note that not all the PDB structures have the PTM experimentally resolved.
Another database providing structural data on phosphosites is Scop3P (http://iomics.ugent.be/scop3p) (Ramasamy et al. 2020) developed by Ramasamy et al. Scop3P integrates sequences (UniProtKB/Swiss-Prot), structures (PDB), and uniformly reprocessed PRIDE (Perez-Riverol et al. 2019) phospho-proteomics data to annotate all known human phosphosites. Furthermore, these sites are put into biophysical context by annotating each phosphoprotein with per-residue structural propensity, solvent accessibility, disordered probability, and early folding information. The web interface presents a 3D plugin for visualization and analysis of phosphosites, and for the understanding of phosphosite structure–function relationships.
Finally, carbohydrates constitute a specific research area of PTMs by themselves due not only to the impressive diversity of saccharides, links, and dispersion in every clade in addition to their biotechnology applications and implications in multiple diseases. The number of databases is impressive with the large majority focusing mainly on the carbohydrates without their target macromolecules i.e. Carbohydrate Structure Database (http://csdb.glycoscience.ru) (Egorova et al. 2015), while others take into account both the proteins with their glycosylation. One particularly interesting web portal is Glycosciences.DB (http://glycosciences.de/database/) (Bohm et al. 2019) that provides databases and tools to support glycobiology and glycomics research. Its focuses on 3D structures, including 3D structural models as well as references to PDB entries that feature carbohydrates. Another website is Glyco3D (http://glyco3d.cermav.cnrs.fr) (Pérez et al. 2015) that have a large number of information on free carbohydrates and some on linked ones, such as the recent GAG database (http://gagdb.glycopedia.eu) (Perez et al. 2020) that contains the 3D structure of glycosaminoglycan (GAG) binding proteins that have been crystallized with their ligands.
We will not delve into the structural databases on glycosylation in this manuscript as Scherbinina and Toukach have dedicated a recent review to approaches of chemo- and glyco-informatics towards 3D structural data generation, deposition and processing in regard to carbohydrates and their derivatives (Scherbinina and Toukach 2020). They focus on the important aspects of carbohydrate 3D structure availability to researchers including structural repositories, glycoinformatics tools and workflows, carbohydrate 3D structure presentation and visualization methods.
In silico applications
Many of the above-mentioned PTM structural databases have been employed in a variety of in silico applications and computational studies. In this section, we will be briefly reporting two types of applications. The chosen examples were selected based on their remarkability.
Predictions
One of the main applications is the computer-aided prediction of PTM sites which is essential for the functional annotation of uncharacterized proteins (Eisenhaber and Eisenhaber 2010). During the last decades, machine learning has become a valuable approach for understanding the large amount of biological data being generated and made accessible to the scientific community; bibliographic databases are witnessing an exponential growth of ML publications. Many methodologies based on machine learning and deep learning approaches have been developed to predict the modification sites for certain specific types of PTM and the PTM databases, highlighted above, constitute benchmark datasets for training the predictive tools and measuring their performance.
We can note the historical work of Wilkins et al. who developed a tool based on MS data, FindMod (http://web.expasy.org/findmod/), to predict 22 PTM types, including acetylation, phosphorylation, and less classical ones (Wilkins et al. 1999a, 1999b). As the main purpose of this manuscript is not to review PTM prediction methods, we decided to only list a few recent works: Wang R. et al. have employed Support Vector Machine (SVM) and Random Forest (RF) machine learning methods to identify lysine crotonylation sites in both plant and mammalian (Wang et al. 2020c). Zhang et al. have developed a succinylation site prediction tool based on protein sequences (Zhang et al. 2020). The training data were collected from dbPTM. Another study by Wang H. et al. have applied an improved word-embedding scheme based on the transfer learning strategy incorporated with the multilayer convolutional neural network (CNN) for identifying protein ubiquitylation sites in plant (Wang et al. 2020b). Finally, Wang D. et al. have developed MusiteDeep (http://musite.net) (Wang et al. 2020a); it combines deep learning approaches with evolutionary information to predict 13 different PTM types (including N6-acetyllysine, methylarginine, methyllysine and pyrrolidone-carboxylic-acid), with excellent results. It integrates an interesting feature, supported by the NGL viewer (Rose and Hildebrand 2015), to visualize the predicted PTM sites in the 3D context of homologous proteins that have known 3D structures. It is important to mention that no structural information is used in the prediction by itself.
However, since these methods operate through a learning process with positive and negative observations, it is essential to construct clean datasets for training purposes. Ideally, the positive set should only consist of protein sites where experimental proof of their modification has been found. On the other hand, creating a negative dataset is a difficult task, because experimental negative results are rarely described in scientific papers and the protein to be included in the negative set must be located in the same cellular compartment as the modification enzyme, to make sure that the sequence motif is not recognized by the enzyme. Readers are advised to consider these various ML studies with caution and check whether all the good practices of ML (including data collection and splitting, features engineering and selection, model training and optimization with parameters and hyperparameters tuning, model performance and generalization on unseen data with the appropriate evaluation metrics) were applied properly. Other issues to consider are the comparison of the performance of a certain ML predictor with other similar tools and the reproducibility of the results. This type of comparison is likely to be biased because these models were not trained on the same datasets or using the same evaluation metrics. As for reproducibility, it is often impossible due to the unavailability of the source code and the used dataset, and the lack of details in the original publications.
In a study published in 2012, Schwartz discussed the metrics and procedures used to assess predictive tools and surveyed 11 online computational tools aimed at the prediction of the four most widely studied lysine post-translational modifications (acetylation, methylation, SUMOylation and ubiquitination) (Schwartz 2012). His findings suggested that nine of the 11 tools performed no better than random or have false-positive rates which make them unusable by the experimental biologist when assessed using unbiased testing datasets. Another similar study was recently published in which proline hydroxylation was considered as a case study to compare the performance of seven predictors on two newly constructed independent datasets (Piovesan et al. 2020). The self-reported performance is found to widely overestimate the real accuracy measured on independent datasets indicating overfitting and lack of generalization to detect new sites.
To counter these above-mentioned phenomena, recommendations for machine-learning-based analyses applied to biological studies have recently been proposed for non-experts in the field to help improve machine learning assessment and reproducibility focusing on four aspects related to data, optimization, model and evaluation (DOME) (Walsh et al. 2021). Finally, some PTM types are limited by the size of the training data. A close collaboration between data scientists and experimentalists could help generate appropriate experimental datasets for model training and the experimental validation of these ML methods.
Impact of PTMs on protein structure
Appending PTMs repositories with 3D structural data opens the way for the computational modeling of PTMs structures at atomic resolution. Such studies allow to examine the association of PTMs with the structural rearrangements of their target proteins and to provide critical insights into the mechanics behind the dynamic regulation of protein function.
Recently, we have investigated the local and global impacts of PTMs on the backbone conformation of the modified proteins (Craveur et al. 2019). We have considered two main PTM types (N-glycosylation and phosphorylation) in non-redundant datasets extracted from PTM-SD, and four examples of proteins were selected to illustrate our findings and compare the backbone flexibility in the presence and absence of PTMs. We used a structural alphabet to analyze the structural local protein conformations, namely the Protein Blocks, able to approximate in a very fine way the structural architecture (Etchebest et al. 2005). We observed that PTMs could either stabilize or destabilize the backbone structure, at a local and global scale, and that the impact of multiple PTMs is not additive on protein structure flexibility and lastly that these effects depend on the PTM types. A similar study was conducted by Xin and Radivojac (Xin and Radivojac 2012). Their results provide evidence that PTMs induce conformational changes at both local and global level. However, the proportion of large changes is unexpectedly small.
It had also been broadly discussed that many PTM sites are found in intrinsically disordered regions (IDRs) (Tompa et al. 2014; Bah and Forman-Kay 2016). Some studies have investigated the correlation between protein disorder and PTMs by integrating data from different databases (UniProt/Swiss-Prot and 3D structures solved by NMR from Protein Data Bank) (Gao and Xu 2012). These studies shed light on the significant preference of PTMs to occur in disordered regions (phosphorylation, hydroxylation, …) or ordered regions (S-nitrosocysteine, most of ADP-ribosylation, …), while acetyllysine does not show any significant preference. Further analysis of NMR structures suggested disorder-to-order transitions might be introduced by some type of modifications. Intrinsically Disorder Proteins (IDPs) are found in sequence databases, the most famous being DisProt (Hatos et al. 2020) and MobiDB (Piovesan et al. 2021). However, the number of resolved cases of IDRs with PTMs remains limited in the PDB. To work with IDP, structural models are often considered (structural models must be handled cautiously as they are theoretical and not experimental) and must be complemented with experimental data; it is one of the most complex art of PTM research using 3D structures.
Lastly, molecular dynamics (MD) simulation is an interesting computational method that is being increasingly used by many research groups in the last few years to investigate the impact of PTM on the dynamics of the modified proteins. Just to cite couple of these studies: Yalinca et al. (2019) have used MD simulations to study the effects of phosphorylation and acetylation as well as cross-talk between these modifications on the energy landscape of huntingtin N-terminus. Their findings provide insights to understand the structural basis underlying the effect of PTMs in the aggregation and cellular properties of huntingtin protein and its implications in Huntington disease. In a more recent study, Rao et al. has investigated the effects of changes in glycan composition on glycoprotein dynamics by considering the example of N-glycans on insulin receptor (Rao et al. 2021). However, it is important to note that one of the challenges in MD simulation is the selection of appropriate force field parameters to correctly simulate the dynamics of the biological systems involving PTMs. Many tools were developed to explore non-standard amino acids and protein modifications using MD simulations such as Privateer (Bagdonas et al. 2020), CHARMM-GUI (Jo et al. 2008) and Vienna-PTM (Margreitter et al. 2013); but for unconventional and rare PTMs, their parameters are not present in the existing force fields and therefore must be calculated using quantum mechanics approaches; the latter can be cumbersome and computationally expensive.
Conclusion and prospects
In this review, we have discussed the existing PTMs structural databases and highlighted their importance in providing the scientific community with the data needed to advance PTM-related research and more specifically to assist structure–function relationship studies.
We first noticed that PTM structure databases are in limited numbers and this observation was confirmed by the 2021 Nucleic Acid Research Database issue (Rigden and Fernandez 2021) that contains 189 papers with only one database on glycan structures (GlycoPOST). Secondly, these databases also suffer from classical database issues such as availability and sustainability. A recent study, screening the availability of thousands of bioinformatics web services published from 2010 to 2020, has shed the light on the factors affecting their lifetime (Kern et al. 2020). Some of the databases we have tested and reviewed became unreachable/unavailable during the writing of the manuscript.
The analysis and correct identification of the modified residues encounters many difficulties that are intrinsic to the properties of the PTM in question. These modifications are dynamic and change over time. A single protein (like the Human CDK2 example discussed in the introduction) can also have several PTM sites that can be modified in different combinations. The analysis of the peptides by the mass spectrometer can also produce doubtful results because it is not possible to identify the exact modified residue and its location especially when the peptide contains several possible modification sites. Some studies have revealed the extent of differences in PTM patterns for the same protein between different species [i.e., Myelin basic protein (MBP) between mammals and lower vertebrates (Zhang et al. 2012)], making the automatic annotation of PTM inferred by similarity and predictions prone to errors. Finally, the major issue severely limiting the structural studies of PTMs are the deficiency of structural data in general and the absence of PTM in resolved structures; these are often over-expressed in systems quite different from their original organisms.
Because of all the above-mentioned reasons, care must be taken when analyzing and interpreting PTMs data in public repositories. Further advances in this field will help building a better understanding of PTMs implications in biological processes. Finally, we expect that the unprecedented performance of AlphaFold2 (Jumper et al. 2021) in CASP14 and their partnership with EMBL-EBI in releasing the most complete database of predicted protein 3D structures, AlphaFold DB, covering almost the entire human proteome (98.5% of human proteins). (Tunyasuvunakool et al. 2021), will be a great hub of information to assist scientists in studying and modeling PTMs in their three-dimensional context. It is possible to model some of the PTM annotated with care in UniProt on the AlphaFold models as done recently for glycosylations (Bagdonas et al. 2021). However, it is always necessary to carefully check the experimental data behind the annotation carried out (difference between publication and sequence analysis) and the proposed modeling which should be analyzed in a precise manner (some models are erroneous).
Abbreviations
- 3D:
-
Three-dimensional
- ADP:
-
Adenosine diphosphate
- CNN:
-
Convolutional neural network
- DNA:
-
Deoxyribonucleic acid
- GAG:
-
Glycosaminoglycan
- HPP:
-
Human Proteome Project
- IDP:
-
Intrinsically Disordered Protein
- IDR:
-
Intrinsically Disordered Region
- MBP:
-
Myelin basic protein
- MD:
-
Molecular dynamics
- MS:
-
Mass Spectrometry
- nsSNP:
-
Non-synonymous single nucleotide polymorphism
- PCA:
-
Pyrrolidone carboxylic acid
- PPI:
-
Protein–protein interaction
- P-site:
-
Phosphorylation site
- PTM:
-
Post-translational modification
- RF:
-
Random Forest
- RNA:
-
Ribonucleic acid
- SNO:
-
S-nitrosylation
- SVM:
-
Support Vector Machine
- TM:
-
Transmembrane
References
Adhikari S, Nice EC, Deutsch EW, Lane L, Omenn GS, Pennington SR, Paik YK, Overall CM, Corrales FJ, Cristea IM, Van Eyk JE, Uhlen M, Lindskog C, Chan DW, Bairoch A, Waddington JC, Justice JL, LaBaer J, Rodriguez H, He F, Kostrzewa M, Ping P, Gundry RL, Stewart P, Srivastava S, Srivastava S, Nogueira FCS, Domont GB, Vandenbrouck Y, Lam MPY, Wennersten S, Vizcaino JA, Wilkins M, Schwenk JM, Lundberg E, Bandeira N, Marko-Varga G, Weintraub ST, Pineau C, Kusebauch U, Moritz RL, Ahn SB, Palmblad M, Snyder MP, Aebersold R, Baker MS (2020) A high-stringency blueprint of the human proteome. Nat Commun 11(1):5301. https://doi.org/10.1038/s41467-020-19045-9
Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, Costello CE, Cravatt BF, Fenselau C, Garcia BA, Ge Y, Gunawardena J, Hendrickson RC, Hergenrother PJ, Huber CG, Ivanov AR, Jensen ON, Jewett MC, Kelleher NL, Kiessling LL, Krogan NJ, Larsen MR, Loo JA, Ogorzalek Loo RR, Lundberg E, MacCoss MJ, Mallick P, Mootha VK, Mrksich M, Muir TW, Patrie SM, Pesavento JJ, Pitteri SJ, Rodriguez H, Saghatelian A, Sandoval W, Schlüter H, Sechi S, Slavoff SA, Smith LM, Snyder MP, Thomas PM, Uhlén M, Van Eyk JE, Vidal M, Walt DR, White FM, Williams ER, Wohlschlager T, Wysocki VH, Yates NA, Young NL, Zhang B (2018) How many human proteoforms are there? Nat Chem Biol 14(3):206–214. https://doi.org/10.1038/nchembio.2576
Aggarwal S, Banerjee SK, Talukdar NC, Yadav AK (2020) Post-translational modification crosstalk and hotspots in sirtuin interactors implicated in cardiovascular diseases. Front Genet 11:356. https://doi.org/10.3389/fgene.2020.00356
Ajit D, Trzeciakiewicz H, Tseng JH, Wander CM, Chen Y, Ajit A, King DP, Cohen TJ (2019) A unique tau conformation generated by an acetylation-mimic substitution modulates P301S-dependent tau pathology and hyperphosphorylation. J Biol Chem 294(45):16698–16711. https://doi.org/10.1074/jbc.RA119.009674
Ayyappan V, Wat R, Barber C, Vivelo CA, Gauch K, Visanpattanasin P, Cook G, Sazeides C, Leung AKL (2021) ADPriboDB 2.0: an updated database of ADP-ribosylated proteins. Nucleic Acids Res 49(D1):D261–D265. https://doi.org/10.1093/nar/gkaa941
Bagdonas H, Ungar D, Agirre J (2020) Leveraging glycomics data in glycoprotein 3D structure validation with Privateer. Beilstein J Org Chem 16:2523–2533. https://doi.org/10.3762/bjoc.16.204
Bagdonas H, Fogarty CA, Fadda E, Agirre J (2021) The case for post-predictional modifications in the AlphaFold Protein Structure Database. Nat Struct Mol Biol 28(11):869–870. https://doi.org/10.1038/s41594-021-00680-9
Bah A, Forman-Kay JD (2016) Modulation of intrinsically disordered protein function by post-translational modifications. J Biol Chem 291(13):6696–705. https://doi.org/10.1074/jbc.R115.695056
Beattie JF, Breault GA, Ellston RP, Green S, Jewsbury PJ, Midgley CJ, Naven RT, Minshull CA, Pauptit RA, Tucker JA, Pease JE (2003) Cyclin-dependent kinase 4 inhibitors as a treatment for cancer. Part 1: identification and optimisation of substituted 4,6-bis anilino pyrimidines. Bioorg Med Chem Lett 13(18):2955–2960. https://doi.org/10.1016/s0960-894x(03)00202-6
Berezovsky IN, Guarnera E, Zheng Z, Eisenhaber B, Eisenhaber F (2017) Protein function machinery: from basic structural units to modulation of activity. Curr Opin Struct Biol 42:67–74. https://doi.org/10.1016/j.sbi.2016.10.021
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242. https://doi.org/10.1093/nar/28.1.235
Bignon E, Allega MF, Lucchetta M, Tiberti M, Papaleo E (2018) Computational structural biology of S-nitrosylation of cancer targets. Front Oncol 8:272. https://doi.org/10.3389/fonc.2018.00272
Bode AM, Dong Z (2004) Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 4(10):793–805. https://doi.org/10.1038/nrc1455
Bohm M, Bohne-Lang A, Frank M, Loss A, Rojas-Macias MA, Lutteke T (2019) Glycosciences.DB: an annotated data collection linking glycomics and proteomics data (2018 update). Nucleic Acids Res 47(D1):D1195–D1201. https://doi.org/10.1093/nar/gky994
Chen YJ, Lu CT, Su MG, Huang KY, Ching WC, Yang HH, Liao YC, Chen YJ, Lee TY (2015) dbSNO 2.0: a resource for exploring structural environment, functional and disease association and regulatory network of protein S-nitrosylation. Nucleic Acids Res 43(Database issue):503–511. https://doi.org/10.1093/nar/gku1176
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325(5942):834–840. https://doi.org/10.1126/science.1175371
Craveur P, Rebehmed J, de Brevern AG (2014) PTM-SD: a database of structurally resolved and annotated posttranslational modifications in proteins. Database. https://doi.org/10.1093/database/bau041
Craveur P, Narwani TJ, Rebehmed J, de Brevern AG (2019) Investigation of the impact of PTMs on the protein backbone conformation. Amino Acids 51(7):1065–1079. https://doi.org/10.1007/s00726-019-02747-w
Dai C, Gu W (2010) p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med 16(11):528–536. https://doi.org/10.1016/j.molmed.2010.09.002
Deribe YL, Pawson T, Dikic I (2010) Post-translational modifications in signal integration. Nat Struct Mol Biol 17(6):666–672. https://doi.org/10.1038/nsmb.1842
Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng Z, Green RK, Flippen-Anderson JL, Westbrook J, Berman HM, Bourne PE (2005) The RCSB Protein Data Bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res 33(Databse issue):D233-237. https://doi.org/10.1093/nar/gki057
Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, Moritz RL, Carver JJ, Wang M, Ishihama Y, Bandeira N, Hermjakob H, Vizcaino JA (2017) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 45(D1):D1100–D1106. https://doi.org/10.1093/nar/gkw936
Diella F, Cameron S, Gemund C, Linding R, Via A, Kuster B, Sicheritz-Ponten T, Blom N, Gibson TJ (2004) Phospho.ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinform 5:79. https://doi.org/10.1186/1471-2105-5-79
Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F (2011) Phospho.ELM: a database of phosphorylation sites—update 2011. Nucleic Acids Res 39(Database issue):D261-267. https://doi.org/10.1093/nar/gkq1104
Donnelly C, Williams A (2020) Investigating the potential impact of post translational modification of auto-antigens by tissue transglutaminase on humoral islet autoimmunity in type 1 diabetes. Metabol Open 8:100062. https://doi.org/10.1016/j.metop.2020.100062
Egorova KS, Kondakova AN, Toukach PV (2015) Carbohydrate Structure Database: tools for statistical analysis of bacterial, plant and fungal glycomes. Database. https://doi.org/10.1093/database/bav073
Eisenhaber B, Eisenhaber F (2010) Prediction of posttranslational modification of proteins from their amino acid sequence. Methods Mol Biol (clifton, NJ) 609:365–384. https://doi.org/10.1007/978-1-60327-241-4_21
Etchebest C, Benros C, Hazout S, de Brevern AG (2005) A structural alphabet for local protein structures: improved prediction methods. Proteins 59(4):810–827. https://doi.org/10.1002/prot.20458
Farriol-Mathis N, Garavelli JS, Boeckmann B, Duvaud S, Gasteiger E, Gateau A, Veuthey AL, Bairoch A (2004) Annotation of post-translational modifications in the Swiss-Prot knowledge base. Proteomics 4(6):1537–1550. https://doi.org/10.1002/pmic.200300764
Gao J, Xu D (2012) Correlation between posttranslational modification and intrinsic disorder in protein. In: Pacific Symposium on Biocomputing, pp 94–103
Gao J, Shao K, Chen X, Li Z, Liu Z, Yu Z, Aung LHH, Wang Y, Li P (2020) The involvement of post-translational modifications in cardiovascular pathologies: focus on SUMOylation, neddylation, succinylation, and prenylation. J Mol Cell Cardiol 138:49–58. https://doi.org/10.1016/j.yjmcc.2019.11.146
Gibson AE, Arris CE, Bentley J, Boyle FT, Curtin NJ, Davies TG, Endicott JA, Golding BT, Grant S, Griffin RJ, Jewsbury P, Johnson LN, Mesguiche V, Newell DR, Noble ME, Tucker JA, Whitfield HJ (2002) Probing the ATP ribose-binding domain of cyclin-dependent kinases 1 and 2 with O(6)-substituted guanine derivatives. J Med Chem 45(16):3381–3393. https://doi.org/10.1021/jm020056z
Glaser F, Rosenberg Y, Kessel A, Pupko T, Ben-Tal N (2005) The ConSurf-HSSP database: the mapping of evolutionary conservation among homologs onto PDB structures. Proteins 58(3):610–617. https://doi.org/10.1002/prot.20305
Gong CX, Liu F, Grundke-Iqbal I, Iqbal K (2005) Post-translational modifications of tau protein in Alzheimer’s disease. J Neural Transm (vienna) 112(6):813–838. https://doi.org/10.1007/s00702-004-0221-0
Gu Y, Rosenblatt J, Morgan DO (1992) Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J 11(11):3995–4005
Gupta R, Birch H, Rapacki K, Brunak S, Hansen JE (1999) O-GLYCBASE version 4.0: a revised database of O-glycosylated proteins. Nucleic Acids Res 27(1):370–372. https://doi.org/10.1093/nar/27.1.370
Gutmanas A, Alhroub Y, Battle GM, Berrisford JM, Bochet E, Conroy MJ, Dana JM, Fernandez Montecelo MA, van Ginkel G, Gore SP, Haslam P, Hatherley R, Hendrickx PM, Hirshberg M, Lagerstedt I, Mir S, Mukhopadhyay A, Oldfield TJ, Patwardhan A, Rinaldi L, Sahni G, Sanz-Garcia E, Sen S, Slowley RA, Velankar S, Wainwright ME, Kleywegt GJ (2014) PDBe: protein data bank in Europe. Nucleic Acids Res 42(Database issue):D285-291. https://doi.org/10.1093/nar/gkt1180
Hanan EJ, Eigenbrot C, Bryan MC, Burdick DJ, Chan BK, Chen Y, Dotson J, Heald RA, Jackson PS, La H, Lainchbury MD, Malek S, Purkey HE, Schaefer G, Schmidt S, Seward EM, Sideris S, Tam C, Wang S, Yeap SK, Yen I, Yin J, Yu C, Zilberleyb I, Heffron TP (2014) Discovery of selective and noncovalent diaminopyrimidine-based inhibitors of epidermal growth factor receptor containing the T790M resistance mutation. J Med Chem 57(23):10176–10191. https://doi.org/10.1021/jm501578n
Hatos A, Hajdu-Soltesz B, Monzon AM, Palopoli N, Alvarez L, Aykac-Fas B, Bassot C, Benitez GI, Bevilacqua M, Chasapi A, Chemes L, Davey NE, Davidovic R, Dunker AK, Elofsson A, Gobeill J, Foutel NSG, Sudha G, Guharoy M, Horvath T, Iglesias V, Kajava AV, Kovacs OP, Lamb J, Lambrughi M, Lazar T, Leclercq JY, Leonardi E, Macedo-Ribeiro S, Macossay-Castillo M, Maiani E, Manso JA, Marino-Buslje C, Martinez-Perez E, Meszaros B, Micetic I, Minervini G, Murvai N, Necci M, Ouzounis CA, Pajkos M, Paladin L, Pancsa R, Papaleo E, Parisi G, Pasche E, Barbosa Pereira PJ, Promponas VJ, Pujols J, Quaglia F, Ruch P, Salvatore M, Schad E, Szabo B, Szaniszlo T, Tamana S, Tantos A, Veljkovic N, Ventura S, Vranken W, Dosztanyi Z, Tompa P, Tosatto SCE, Piovesan D (2020) DisProt: intrinsic protein disorder annotation in 2020. Nucleic Acids Res 48(D1):D269–D276. https://doi.org/10.1093/nar/gkz975
Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B (2004) PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 4(6):1551–1561. https://doi.org/10.1002/pmic.200300772
Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43(Database issue):D512-520. https://doi.org/10.1093/nar/gku1267
Huang Q, Chang J, Cheung MK, Nong W, Li L, Lee MT, Kwan HS (2014) Human proteins with target sites of multiple post-translational modification types are more prone to be involved in disease. J Proteome Res 13(6):2735–2748. https://doi.org/10.1021/pr401019d
Huang KY, Su MG, Kao HJ, Hsieh YC, Jhong JH, Cheng KH, Huang HD, Lee TY (2016) dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins. Nucleic Acids Res 44(D1):D435-446. https://doi.org/10.1093/nar/gkv1240
Huang KY, Lee TY, Kao HJ, Ma CT, Lee CC, Lin TH, Chang WC, Huang HD (2019) dbPTM in 2019: exploring disease association and cross-talk of post-translational modifications. Nucleic Acids Res 47(D1):D298-d308. https://doi.org/10.1093/nar/gky1074
Hubbard T, Andrews D, Caccamo M, Cameron G, Chen Y, Clamp M, Clarke L, Coates G, Cox T, Cunningham F, Curwen V, Cutts T, Down T, Durbin R, Fernandez-Suarez XM, Gilbert J, Hammond M, Herrero J, Hotz H, Howe K, Iyer V, Jekosch K, Kahari A, Kasprzyk A, Keefe D, Keenan S, Kokocinsci F, London D, Longden I, McVicker G, Melsopp C, Meidl P, Potter S, Proctor G, Rae M, Rios D, Schuster M, Searle S, Severin J, Slater G, Smedley D, Smith J, Spooner W, Stabenau A, Stalker J, Storey R, Trevanion S, Ureta-Vidal A, Vogel J, White S, Woodwark C, Birney E (2005) Ensembl 2005. Nucleic Acids Res 33(Database issue):D447-453. https://doi.org/10.1093/nar/gki138
Jimenez JL, Hegemann B, Hutchins JR, Peters JM, Durbin R (2007) A systematic comparative and structural analysis of protein phosphorylation sites based on the mtcPTM database. Genome Biol 8(5):R90. https://doi.org/10.1186/gb-2007-8-5-r90
Jo S, Kim T, Iyer VG, Im W (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29(11):1859–1865. https://doi.org/10.1002/jcc.20945
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D (2021) Highly accurate protein structure prediction with AlphaFold. Nature. https://doi.org/10.1038/s41586-021-03819-2
Jungblut PR, Holzhutter HG, Apweiler R, Schluter H (2008) The speciation of the proteome. Chem Cent J 2:16. https://doi.org/10.1186/1752-153X-2-16
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22(12):2577–2637. https://doi.org/10.1002/bip.360221211
Kawahara R, Recuero S, Srougi M, Leite KRM, Thaysen-Andersen M, Palmisano G (2021) The complexity and dynamics of the tissue glycoproteome associated with prostate cancer progression. Mol Cell ProteomMCP 20:100026. https://doi.org/10.1074/mcp.RA120.002320
Kern F, Fehlmann T, Keller A (2020) On the lifetime of bioinformatics web services. Nucleic Acids Res 48(22):12523–12533. https://doi.org/10.1093/nar/gkaa1125
Khater S, Mohanty D (2015) novPTMenzy: a database for enzymes involved in novel post-translational modifications. Database J Biol Databases Curation. https://doi.org/10.1093/database/bav039
Kozma D, Simon I, Tusnady GE (2013) PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res 41(Database issue):D524-529. https://doi.org/10.1093/nar/gks1169
Lee TY, Huang HD, Hung JH, Huang HY, Yang YS, Wang TH (2006) dbPTM: an information repository of protein post-translational modification. Nucleic Acids Res 34(Database issue):D622-627. https://doi.org/10.1093/nar/gkj083
Lee TY, Hsu JB, Chang WC, Wang TY, Hsu PC, Huang HD (2009) A comprehensive resource for integrating and displaying protein post-translational modifications. BMC Res Notes 2:111. https://doi.org/10.1186/1756-0500-2-111
Lee TY, Chen YJ, Lu CT, Ching WC, Teng YC, Huang HD, Chen YJ (2012) dbSNO: a database of cysteine S-nitrosylation. Bioinformatics (oxford, England) 28(17):2293–2295. https://doi.org/10.1093/bioinformatics/bts436
Legrain P, Aebersold R, Archakov A, Bairoch A, Bala K, Beretta L, Bergeron J, Borchers CH, Corthals GL, Costello CE, Deutsch EW, Domon B, Hancock W, He F, Hochstrasser D, Marko-Varga G, Salekdeh GH, Sechi S, Snyder M, Srivastava S, Uhlen M, Wu CH, Yamamoto T, Paik YK, Omenn GS (2011) The human proteome project: current state and future direction. Mol Cell Proteom MCP 10:M111 009993. https://doi.org/10.1074/mcp.M111.009993
Lernmark A (2013) Is there evidence for post-translational modification of beta cell autoantigens in the aetiology and pathogenesis of type 1 diabetes? Diabetologia. https://doi.org/10.1007/s00125-013-3041-7
Li F, Fan C, Marquez-Lago TT, Leier A, Revote J, Jia C, Zhu Y, Smith AI, Webb GI, Liu Q, Wei L, Li J, Song J (2020) PRISMOID: a comprehensive 3D structure database for post-translational modifications and mutations with functional impact. Brief Bioinform 21(3):1069–1079. https://doi.org/10.1093/bib/bbz050
Lo A, Cheng CW, Chiu YY, Sung TY, Hsu WL (2011) TMPad: an integrated structural database for helix-packing folds in transmembrane proteins. Nucleic Acids Res 39(Database issue):D347-355. https://doi.org/10.1093/nar/gkq1255
Lodish HF (2013) Molecular cell biology, 7th edn. W.H. Freeman and Co., New York
Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40(Database issue):D370-376. https://doi.org/10.1093/nar/gkr703
Lu CT, Huang KY, Su MG, Lee TY, Bretaña NA, Chang WC, Chen YJ, Chen YJ, Huang HD (2013) DbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res 41(Database issue):D395–D305. https://doi.org/10.1093/nar/gks1229
Mann M, Jensen ON (2003) Proteomic analysis of post-translational modifications. Nat Biotechnol 21(3):255–261. https://doi.org/10.1038/nbt0303-255
Margreitter C, Petrov D, Zagrovic B (2013) Vienna-PTM web server: a toolkit for MD simulations of protein post-translational modifications. Nucleic Acids Res 41(Web Server issue):W422-426. https://doi.org/10.1093/nar/gkt416
Minguez P, Letunic I, Parca L, Bork P (2013) PTMcode: a database of known and predicted functional associations between post-translational modifications in proteins. Nucleic Acids Res 41(Database issue):D306-311. https://doi.org/10.1093/nar/gks1230
Minguez P, Letunic I, Parca L, Garcia-Alonso L, Dopazo J, Huerta-Cepas J, Bork P (2015) PTMcode v2: a resource for functional associations of post-translational modifications within and between proteins. Nucleic Acids Res 43(Database issue):D494-502. https://doi.org/10.1093/nar/gku1081
Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Biswas M, Bradley P, Bork P, Bucher P, Copley R, Courcelle E, Durbin R, Falquet L, Fleischmann W, Gouzy J, Griffith-Jones S, Haft D, Hermjakob H, Hulo N, Kahn D, Kanapin A, Krestyaninova M, Lopez R, Letunic I, Orchard S, Pagni M, Peyruc D, Ponting CP, Servant F, Sigrist CJ, InterPro C (2002) InterPro: an integrated documentation resource for protein families, domains and functional sites. Brief Bioinform 3(3):225–235. https://doi.org/10.1093/bib/3.3.225
Muller MM (2018) Post-translational modifications of protein backbones: unique functions, mechanisms, and challenges. Biochemistry 57(2):177–185. https://doi.org/10.1021/acs.biochem.7b00861
Nekooki-Machida Y, Hagiwara H (2020) Role of tubulin acetylation in cellular functions and diseases. Med Mol Morphol 53(4):191–197. https://doi.org/10.1007/s00795-020-00260-8
Pérez S, Sarkar A, Rivet A, Breton C, Imberty A (2015) Glyco3D: a portal for structural glycosciences. Methods Mol Biol (clifton, NJ) 1273:241–258. https://doi.org/10.1007/978-1-4939-2343-4_18
Perez S, Bonnardel F, Lisacek F, Imberty A, Ricard Blum S, Makshakova O (2020) GAG-DB, the new interface of the three-dimensional landscape of glycosaminoglycans. Biomolecules. https://doi.org/10.3390/biom10121660
Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Perez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz S, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaino JA (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47(D1):D442–D450. https://doi.org/10.1093/nar/gky1106
Piovesan D, Hatos A, Minervini G, Quaglia F, Monzon AM, Tosatto SCE (2020) Assessing predictors for new post translational modification sites: a case study on hydroxylation. PLoS Comput Biol 16(6):e1007967. https://doi.org/10.1371/journal.pcbi.1007967
Piovesan D, Necci M, Escobedo N, Monzon AM, Hatos A, Micetic I, Quaglia F, Paladin L, Ramasamy P, Dosztanyi Z, Vranken WF, Davey NE, Parisi G, Fuxreiter M, Tosatto SCE (2021) MobiDB: intrinsically disordered proteins in 2021. Nucleic Acids Res 49(D1):D361–D367. https://doi.org/10.1093/nar/gkaa1058
Radivojac P, Baenziger PH, Kann MG, Mort ME, Hahn MW, Mooney SD (2008) Gain and loss of phosphorylation sites in human cancer. Bioinformatics (oxford, England) 24(16):i241-247. https://doi.org/10.1093/bioinformatics/btn267
Ramasamy P, Turan D, Tichshenko N, Hulstaert N, Vandermarliere E, Vranken W, Martens L (2020) Scop3P: a comprehensive resource of human phosphosites within their full context. J Proteome Res 19(8):3478–3486. https://doi.org/10.1021/acs.jproteome.0c00306
Rao RM, Wong H, Dauchez M, Baud S (2021) Effects of changes in glycan composition on glycoprotein dynamics: example of N-glycans on insulin receptor. Glycobiology. https://doi.org/10.1093/glycob/cwab049
Rigden DJ, Fernandez XM (2021) The 2021 Nucleic Acids Research database issue and the online molecular biology database collection. Nucleic Acids Res 49(D1):D1–D9. https://doi.org/10.1093/nar/gkaa1216
Rose AS, Hildebrand PW (2015) NGL Viewer: a web application for molecular visualization. Nucleic Acids Res 43(W1):W576-579. https://doi.org/10.1093/nar/gkv402
Scherbinina SI, Toukach PV (2020) Three-dimensional structures of carbohydrates and where to find them. Int J Mol Sci. https://doi.org/10.3390/ijms21207702
Schwartz D (2012) Prediction of lysine post-translational modifications using bioinformatic tools. Essays Biochem 52:165–177. https://doi.org/10.1042/bse0520165
Sidney J, Vela JL, Friedrich D, Kolla R, von Herrath M, Wesley JD, Sette A (2018) Low HLA binding of diabetes-associated CD8+ T-cell epitopes is increased by post translational modifications. BMC Immunol 19(1):12. https://doi.org/10.1186/s12865-018-0250-3
Su MG, Huang KY, Lu CT, Kao HJ, Chang YH, Lee TY (2014) topPTM: a new module of dbPTM for identifying functional post-translational modifications in transmembrane proteins. Nucleic Acids Res 42(Database issue):D537-545. https://doi.org/10.1093/nar/gkt1221
Timofeev O, Cizmecioglu O, Settele F, Kempf T, Hoffmann I (2010) Cdc25 phosphatases are required for timely assembly of CDK1-cyclin B at the G2/M transition. J Biol Chem 285(22):16978–16990. https://doi.org/10.1074/jbc.M109.096552
Tompa P, Davey NE, Gibson TJ, Babu MM (2014) A million peptide motifs for the molecular biologist. Mol Cell. 17;55(2):161–169: https://doi.org/10.1016/j.molcel.2014.05.032
Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Zidek A, Bridgland A, Cowie A, Meyer C, Laydon A, Velankar S, Kleywegt GJ, Bateman A, Evans R, Pritzel A, Figurnov M, Ronneberger O, Bates R, Kohl SAA, Potapenko A, Ballard AJ, Romera-Paredes B, Nikolov S, Jain R, Clancy E, Reiman D, Petersen S, Senior AW, Kavukcuoglu K, Birney E, Kohli P, Jumper J, Hassabis D (2021) Highly accurate protein structure prediction for the human proteome. Nature 596(7873):590–596. https://doi.org/10.1038/s41586-021-03828-1
Tusnady GE, Kalmar L, Simon I (2008) TOPDB: topology data bank of transmembrane proteins. NucleicAcids Res 36(Database issue):D234-239. https://doi.org/10.1093/nar/gkm751
UniProt C (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47(D1):D506–D515. https://doi.org/10.1093/nar/gky1049
Van Eyk JE (2011) Overview: the maturing of proteomics in cardiovascular research. Circ Res 108(4):490–498. https://doi.org/10.1161/CIRCRESAHA.110.226894
Velankar S, Best C, Beuth B, Boutselakis CH, Cobley N, Sousa Da Silva AW, Dimitropoulos D, Golovin A, Hirshberg M, John M, Krissinel EB, Newman R, Oldfield T, Pajon A, Penkett CJ, Pineda-Castillo J, Sahni G, Sen S, Slowley R, Suarez-Uruena A, Swaminathan J, van Ginkel G, Vranken WF, Henrick K, Kleywegt GJ (2010) PDBe: Protein Data Bank in Europe. Nucleic Acids Res 38(Database issue):D308-317. https://doi.org/10.1093/nar/gkp916
Vidal CJ (2011) Post-translational modifications in health and disease. Springer, New York
Vivelo CA, Wat R, Agrawal C, Tee HY, Leung AK (2017) ADPriboDB: the database of ADP-ribosylated proteins. Nucleic Acids Res 45(D1):D204–D209. https://doi.org/10.1093/nar/gkw706
Walsh CT, Garneau-Tsodikova S, Gatto GJ Jr (2005) Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl 44(45):7342–7372. https://doi.org/10.1002/anie.200501023
Walsh I, Fishman D, Garcia-Gasulla D, Titma T, Pollastri G, Harrow J, Psomopoulos FE, Tosatto SCE, Group EMLF (2021) DOME: recommendations for supervised machine learning validation in biology. Nat Methods 18(10):1122–1127. https://doi.org/10.1038/s41592-021-01205-4
Wang D, Liu D, Yuchi J, He F, Jiang Y, Cai S, Li J, Xu D (2020a) MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res 48(W1):W140-w146. https://doi.org/10.1093/nar/gkaa275
Wang H, Wang Z, Li Z, Lee TY (2020b) Incorporating deep learning with word embedding to identify plant ubiquitylation sites. Front Cell Dev Biol 8:572195. https://doi.org/10.3389/fcell.2020.572195
Wang R, Wang Z, Wang H, Pang Y, Lee TY (2020c) Characterization and identification of lysine crotonylation sites based on machine learning method on both plant and mammalian. Sci Rep 10(1):20447. https://doi.org/10.1038/s41598-020-77173-0
Welburn JP, Tucker JA, Johnson T, Lindert L, Morgan M, Willis A, Noble ME, Endicott JA (2007) How tyrosine 15 phosphorylation inhibits the activity of cyclin-dependent kinase 2-cyclin A. J Biol Chem 282(5):3173–3181. https://doi.org/10.1074/jbc.M609151200
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999a) Protein identification and analysis tools in the ExPASy server. Methods Mol Biol (clifton, NJ) 112:531–552. https://doi.org/10.1385/1-59259-584-7:531
Wilkins MR, Gasteiger E, Gooley AA, Herbert BR, Molloy MP, Binz PA, Ou K, Sanchez JC, Bairoch A, Williams KL, Hochstrasser DF (1999b) High-throughput mass spectrometric discovery of protein post-translational modifications. J Mol Biol 289(3):645–657. https://doi.org/10.1006/jmbi.1999.2794
Wu CH, Yeh LS, Huang H, Arminski L, Castro-Alvear J, Chen Y, Hu Z, Kourtesis P, Ledley RS, Suzek BE, Vinayaka CR, Zhang J, Barker WC (2003) The protein information resource. Nucleic Acids Res 31(1):345–347. https://doi.org/10.1093/nar/gkg040
Xin F, Radivojac P (2012) Post-translational modifications induce significant yet not extreme changes to protein structure. Bioinformatics (oxford, England) 28(22):2905–2913. https://doi.org/10.1093/bioinformatics/bts541
Yalinca H, Gehin CJC, Oleinikovas V, Lashuel HA, Gervasio FL, Pastore A (2019) The role of post-translational modifications on the energy landscape of Huntingtin N-Terminus. Front Mol Biosci 6:95. https://doi.org/10.3389/fmolb.2019.00095
Zahn-Zabal M, Michel PA, Gateau A, Nikitin F, Schaeffer M, Audot E, Gaudet P, Duek PD, Teixeira D, Rech de Laval V, Samarasinghe K, Bairoch A, Lane L (2020) The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucleic Acids Res 48(D1):D328–D334. https://doi.org/10.1093/nar/gkz995
Zanzoni A, Ausiello G, Via A, Gherardini PF, Helmer-Citterich M (2007) Phospho3D: a database of three-dimensional structures of protein phosphorylation sites. Nucleic Acids Res 35(Database issue):D229-231. https://doi.org/10.1093/nar/gkl922
Zanzoni A, Carbajo D, Diella F, Gherardini PF, Tramontano A, Helmer-Citterich M, Via A (2011) Phospho3D 2.0: an enhanced database of three-dimensional structures of phosphorylation sites. Nucleic Acids Res 39(Database issue):D268-271. https://doi.org/10.1093/nar/gkq936
Zhang C, Walker AK, Zand R, Moscarello MA, Yan JM, Andrews PC (2012) Myelin basic protein undergoes a broader range of modifications in mammals than in lower vertebrates. J Proteome Res 11(10):4791–4802. https://doi.org/10.1021/pr201196e
Zhang L, Liu M, Qin X, Liu G (2020) Succinylation site prediction based on protein sequences using the IFS-LightGBM (BO) model. Comput Math Methods Med 2020:8858489. https://doi.org/10.1155/2020/8858489
Zhou F, Xue Y, Yao X, Xu Y (2006) A general user interface for prediction servers of proteins’ post-translational modification sites. Nat Protoc 1(3):1318–1321. https://doi.org/10.1038/nprot.2006.209
Acknowledgements
The authors would like to thank Dr Tsikas Dimitros, the editor of this special issue for allowing us to present this review and for his kindness in granting us multiple extensions to finalize the manuscript. The authors also wish to thank all the authors of the various databases and tools whose contributions made it possible to tackle this difficult and arduous research area. AdB acknowledges the French National Research Agency with grant ANR-19-CE17-0021 (BASIN) and the Indo-French Center for the Promotion of Advanced Research/CEFIPRA for collaborative grants (numbers 5302-2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The authors declare that this is a review paper where human ethical approval and informed consent are not applicable. All authors have read and agreed to the publication of the manuscript.
Additional information
Handling editor: D. Tsikas.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
de Brevern, A.G., Rebehmed, J. Current status of PTMs structural databases: applications, limitations and prospects. Amino Acids 54, 575–590 (2022). https://doi.org/10.1007/s00726-021-03119-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-021-03119-z