
Abstract
Infectious diseases impose serious public health burdens and continue to be a global public health crisis. The treatment of infections caused by multidrug-resistant pathogens is challenging because only a few viable therapeutic options are clinically available. The emergence and risk of drug-resistant superbugs and the dearth of new classes of antibiotics have drawn increasing awareness that we may return to the pre-antibiotic era. To date, lipopeptides have been received considerable attention because of the following properties: They exhibit potent antimicrobial activities against a broad spectrum of pathogens, rapid bactericidal activity and have a different antimicrobial action compared with most of the conventional antibiotics used today and very slow development of drug resistance tendency. In general, lipopeptides can be structurally classified into two parts: a hydrophilic peptide moiety and a hydrophobic fatty acyl chain. To date, a significant amount of design and synthesis of lipopeptides have been done to improve the therapeutic potential of lipopeptides. This review will present the current knowledge and the recent research in design and synthesis of new lipopeptides and their derivatives in the last 5 years.
Similar content being viewed by others
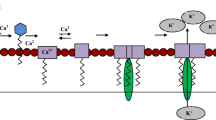
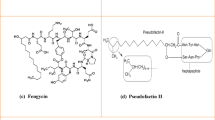
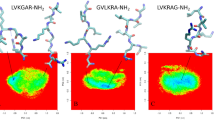
Avoid common mistakes on your manuscript.
Introduction
Resistance of pathogens to conventional antibiotics continues to be a global public health crisis (Cooper and Shlaes 2011). Antibiotic resistance is widely associated with failure of clinical treatment, additional mortality and healthcare costs (Resch et al. 2009; Hanberger et al. 2011). Emergence of drug-resistant superbugs and the dearth of new classes of antibiotics have drawn increasing awareness that we may return to the pre-antibiotic era. Lipopeptides have shown particular efficacy in treating resistant infections (Grossman et al. 2015; Nkongolo et al. 2014; Luo et al. 2015; Huang and Yousef 2014).
Antimicrobial lipopeptides are produced non-ribosomally in bacteria and fungi during cultivation on various carbon sources (Makovitzki et al. 2006). The lipopeptides can be structurally characterized by a hydrophilic peptide (cyclic or linear form) and a hydrophobic fatty acyl chain of an amphiphilic nature. Most of the native lipopeptides have complex cyclic structures. Broadly, there are several major types of lipopeptides: polymyxins, daptomycin, surfactin, iturin and fengycin (Meena and Kanwar 2015; Patel et al. 2015). Figure 1 shows the general structures of surfactin, iturin and fengycin. They can be structurally classified by their combination of fatty acid, peptide moiety and the link between the two parts.

General structures of three main classes of lipopeptides: surfactin, iturin and fengycin
In contrast to cationic antimicrobial peptides, many lipopeptides are not broad spectrum in their activity. For instance, polymyxin B and daptomycin are only active against Gram-negative and Gram-positive bacteria, respectively. In general, the lipopeptides are a very promising class of compounds with potent antimicrobial activities against a panel of pathogens, rapid time-kill and active against drug-resistant pathogen. These properties suggest that they could be useful for clinical antibiotic development. For instance, Polymyxin B and E, daptomycin, caspofungin, micafungin and anidulafungin have all been developed into commercial drugs.
Most lipopeptides kill bacteria via perturbation of the bacterial cell membranes (Jerala 2007). For instance, the initial step of antimicrobial action is governed by the electrostatic interaction between positively charged residues of lipopeptides and negative charged lipoteichoic acid of Gram-positive bacteria, and negatively charged lipopolysaccharide (LPS)-lipid A complex of Gram-negative bacteria, respectively. Then, the large fraction of anionic phospholipids in the cytoplasmic membrane further attracts the lipopeptide traversely into the inner membrane and destabilizes the membrane architecture (Epand 1997). In addition, the lipid moiety of these compounds provides lipophilicity that facilitates bacterial membrane interaction (Malina and Shai 2005). However, a lack of cell selectivity which may be translated as toxicity to mammalian cells is a limitation for intravenous administration. For instance, polymyxin B, is used as the last resort for the treatment as administration of polymyxins is closely associated with adverse renal (nephrotoxicity) and neurological (neurotoxicity) effects in a considerably large number of patients (Falagas and Kasiakou 2006).
A significant amount of design and synthesis of lipopeptides have been devoted in last 5 years (since 2012) to improve the therapeutic potential of the lipopeptides. In addition to the classic approach by simply conjugating the lipid chain to the peptide residues, there are many other unique and interesting designs of synthetic lipopeptides with potent antimicrobial properties. This review will discuss bioactive synthetic lipopeptides that have been presented within the last 5 years. The design and chemistry of synthetic lipopeptides with potential use in microbial infections are the focus. Daptomycin and polymyxin B have been the subject of several good reviews and will only be referred to occasionally in this review to illustrate some points (Cai et al. 2015; Nation et al. 2014). However, their recent mimics as novel synthetic cyclic lipopeptides are relevant to this review. There are many other insightful literature reviews encompassing other areas of lipopeptide research (Patel et al. 2015; Hamley 2015; Mandal et al. 2013; Giuliani and Rinaldi 2011; Cochrane and Vederas 2016; Meena and Kanwar 2015; Mangoni and Shai 2011; Schneider et al. 2014). There is also an useful review on synthetic lipopeptides published in 2007 (Jerala 2007; Azmi et al. 2016b). Recently, we have also published an extensive review on membrane interaction of cationic antimicrobial peptides with the bacterial membrane (Li et al. 2017).
Mimics of natural lipopeptides: semi-synthetic approaches
Natural lipopeptides possess significant values in microbial control for biomedical and agriculture technology. It is noteworthy that myriad examples of cationic antimicrobial peptides (CAMPs, also named host-defense cationic peptides, HDPs), which are ribosomally produced by almost all forms of life are also potential therapeutic agents. Acylation of natural AMPs with fatty acids has been proven to be a useful approach to improve their antimicrobial activity (Mangoni and Shai 2011). In general, mimicking natural peptides could be classified into two main categories: mimicking cyclic peptides and mimicking linear peptides.
Daptomycin analogs
Daptomycin is a clinically important lipopeptide antibiotic that kills Gram-positive bacteria via membrane depolarization. Daptomycin was approved by FDA in 2003 for skin and skin structure infections. Surotomycin (CB-183,315) is an orally dosed, non-absorbed cyclic lipopeptide analog of daptomycin (Fig. 2). As an attempt to further improve the potency against Clostridium difficile (Knight-Connoni et al. 2016) and minimize the potential for resistance selection by C. difficile and other gastrointestinal species, a series of semisynthetic lipopeptides based on daptomycin was designed and synthesized (Yin et al. 2015). The structural modifications were based on the lipid moiety of daptomycin via two main categories: aliphatic tail and aromatic ring containing acyl tails. Among all the analogs synthesized, surotomycin exhibited the most potent overall in vitro antimicrobial profile. A fourfold improvement of C. difficile MIC over daptomycin was measured. In addition, surotomycin was also eightfold more potent against daptomycin-resistant E. faecium and E. faecalis. In the in vivo hamster C. difficile-associated diarrhea (CDAD) model efficacy studies, surotomycin protected animals at an oral dose of 0.5 mg/kg, which even showed comparable or better protection than vancomycin. Surotomycin shows very good activity against anaerobic Gram-positive bacteria, including C. difficile (Citron et al. 2012). Snydman et al. found that surotomycin was effective against C. difficile with MICs for a panel of 55 isolates <0.5 μg/ml. Isolates included in the study were both DR and MDR and resistant to moxifloxacin, clindamycin, vancomycin and metronidazole. Surotomycin was active against enterococcal isolates (n = 60) at MICs of ≤2 µg/mL. Activity against both MSSA (n = 12) and MRSA (n = 12) was noted at concentrations of ≤1 µg/mL. The MIC range against Clostridium sp. (n = 33) was ≤0.125–4 µg/mL (Snydman et al. 2012). Surotomycin displayed concentration-dependent killing of both logarithmic-phase and stationary-phase cultures at a concentration that was ≤16 × MIC. Like daptomycin, at bactericidal concentration, surotomycin dissipated the membrane potential without inducing changes in membrane permeability to small molecules (Mascio et al. 2012). Selection for resistance by serial passage also showed that emergence of resistance to surotomycin against C. difficile, E. faecalis and E. faecium was likely to be rare (Mascio et al. 2014). Evaluation of phase two study of surotomycin has shown that its oral doses at 125 and 250 mg twice a day, is safe and well-tolerated. It is in a phase three clinical development for the CDAD treatment. Surotomycin provides stability, high aqueous solubility and low oral bioavailability, which are important to be applied as desirable pharmaceutical properties for gastrointestinal use. Low oral bioavailability is important to ensure high local concentrations to be achieved at the site of action-gastrointestinal tract (Knight-Connoni et al. 2016).

Structure of daptomycin and surotomycin. Surotomycin is a modified analog of daptomycin with a different lipid moiety
Hart et al. has synthesized analogs of daptomycin created with amino acids at several positions. The ester linkage between the residue-4 and residue-13 was replaced with an amide bond. However, the replacement did not optimize daptomycin as the MICs against S. aureus were 100.8–201.2 µM. However, the replacement of ester linkage to an amide linkage greatly increased the hydrolytic stability (t Hart et al. 2014). Structures of the synthetic daptomycin analogs are shown in Table S1.
Polymyxin analogs
Polymyxin B is a membrane active cyclic lipopeptide that permeabilizes both the outer and inner membranes of Gram-negative bacteria via self-promoting uptake mechanism. The peptide portion of polymyxin B is poly-cationic, which interacts with the anionic head groups of phospholipids found in lipid A. The lipid tail is critical, however, and antimicrobial activity is lost without this fatty acid moiety (Deris et al. 2014). Polymyxin B nonapeptide (PMBN) is a proteolytic product of polymyxin B, which lacks the fatty acid residue and the terminal diaminobutyric acid. PMBN has diminished antimicrobial properties against Gram-negative bacteria, but was active on the outer membrane of Gram-negative bacteria. However, the major therapeutic concerns regarding polymyxin B and its more often used analog colistin are nephrotoxicity and neurotoxicity (Biswas et al. 2012). Thus, Polymyxin B and colistin are generally considered as the last resort treatment for MDR Gram-negative infections. Resistance may be due to a change in the number of phospholipid head groups in lipid A. Polymyxin or colistin resistance is problematic if the bacteria carry other mutations making them resistant to other classes of antibiotics.
Yamamura et al. has designed and synthesized polymycin B mimicking molecule using cyclodextrin (CD). Polyamino CD derivatives possessing amino groups on C-6 were prepared to mimic the polycationic peptidic region of polymyxin B. The number of repeating units (amino groups) used in the study was 6, 7 and 8. To investigate membrane activity of these compounds, K+ efflux from the bacterial cell cytosol was measured. The compound with eight amino groups induced the strongest K+ efflux from both S. aureus FDA209P and E. coli K12 strain W3110. Antimicrobial assays showed that the MICs against E. coli were moderate (MICs = 32–64 µg/mL) while those against S. aureus (MICs = 4–8 µg/mL) were better. The K+ efflux assay also showed that the benzylamino CDs caused higher disruption of the bacterial membrane than polyamino derivatives. Interestingly, the K+ efflux profile of benzylamino CDs was also very similar to gramicidin S. Overall, the addition of hydrophobic benzyl groups to polymyxin B nonapeptide-like amino CDs led to gramicidin S-like membrane activity, instead of polymyxin B (Yamamura et al. 2012).
Cajal’s group has designed and synthesized analogs of polymyxin B (Rabanal et al. 2015). Three analogs shown in Table S1 were studied: sp-34, sp-96 and sp-100. In general, the three compounds share a (D)-tryptophan in position 6, replacing (D)-phenylalanine. The presence of (D) Trp favored membrane interaction and conferred the molecule intrinsic fluorescence properties that allow the determination of membrane binding. Several key Dab residues were mutated to Arg, which has better hydrogen bonding properties than Dab or Lys and favored the insertion in the bacterial lipid. Other modifications include a (D)-cysteine in position 10 (sp-96 and sp-100), and Norleucine (Nleu) instead of Leu7 (sp-100). Nleu increased flexibility in this strategic hydrophobic residue and increased the permeabilizing and fusogenic capacity. Biophysical studies showed that the three polymyxin B analogs bound with high affinity to LPS. Thus, the mutations sp-34, sp-96 and sp-100 exhibited similar membrane targeting action to polymycin B, and were able to extend the antimicrobial spectrum toward Gram-positive bacteria (Grau-Campistany et al. 2016).
Li’s group is the first to use a new polymyxin SAR based mechanistic model to design novel polymyxin-like lipopeptides that specifically target polymyxin-resistant Gram-negative bacteria. They proposed that polymyxin SAR data need to be interpreted based on a mechanistic model of the polymyxin-lipid A target complex (Velkov et al. 2010, 2013). Then, they showed that introduction of hydrophobic modifications at position R6 or R7 of polymyxin B could overcome the polymyxin resistance (R6 and R7 for polymyxin B are D-Phe and Leu, respectively). Incorporation of aliphatic chains greater than 5 carbons at position 6 and 7 significantly increased antibacterial activity. FADDI-002, FADDI-003, FADDI-016, FADDI-019, FADDI-020, FADDI-039 and FADDI-047 showed potent MICs of 2–8 µg/mL against polymyxin-resistant clinical isolates of P. aeruginosa (Table S1). SAR analysis also revealed that the structure of the aliphatic side chain does have a small effect on activity with straight chain aliphatic groups giving the best results. Stereochemistry analysis revealed that the D-analog at position 6 and L-analog at position 7 gave better antimicrobial activities. With regard to safety and tolerability, these new lipopeptides showed that they had similar tolerability as to polymyxin B in rodents. They also exhibited lower nephrotoxicity than polymyxin, probably because of their high plasma protein binding (Velkov et al. 2014).
Magee et al. has also designed and synthesized a series of new polymyxin B analogs by replacement of the Dab-3 residue with a Dap-3 (l-2,3-diaminoproprionic acid) in a combination with a relatively polar 6-oxo-1-pheyl-1-6-dihyropyridine-3-carbonyl side chain as a fatty replacement (Fig. 3). The compound displayed an improved antimicrobial activity against Gram-negative bacteria (MICs = 0.125–4 µg/mL), including polymyxin B-resistant strains, relative to polymyxin B. It also displayed improved in vitro renal cytotoxicity profiles. However, the newly synthesized compound showed inferior in the direct PK/PD comparison with polymyxin B in the murine neutropenic thigh model. Consequently, this compound is not likely to surpass polymyxin B for the overall therapeutic index (Magee et al. 2013).

Replacement of the Dab-3 residue of polymyxin B to Dap-3 (L-2,3-diaminoproprionic acid) in combination with a relatively polar 6-oxo-1-phenyl-1-6-dihydropyridine-3-carbonyl side chain improved in vitro antimicrobial activity of polymyxin B against Gram-negative bacteria (Magee et al. 2013)
Rabanal et al. has utilized cysteine to form a disulfide bridge to mimic and simplify polymyxin B structure. In addition, they also substituted the branched natural fatty acid tail with a linear fatty acid. Further addition of two or three arginines conferred moderate activity against S. aureus. However, when arginines were introduced, activity against Gram-negative bacteria reduced (MICs = 8–32 µg/mL). SAR analysis revealed that using d-cysteinamide at the C-terminal and neoleucine at position-7 was able to maintain the antimicrobial activities against Gram-negative bacteria (MICs = 1–4 µg/mL), while the spectrum of activity apparently broadened toward S. aureus (1–4 µg/mL). They also exhibited very potent antimicrobial activities against drug-resistant Gram-negative bacteria (MICs = 0.5–4 µg/mL). In vivo acute toxicity test using subcutaneous administration showed that the lethal dose (LD50) was 283 mg/kg. Mice treated with 200 mg/kg of the modified peptide survived and no signs of pathology in vital organs. By contrast, LD50 reported for subcutaneous administration of polymyxin B was 59.5 mg/kg.
In addition to the direct substitution approach as discussed above, Ferguson et al. has developed a nanomedicine-based delivery system to target the delivery of colistin, polymyxin E, by conjugating the colistin with dextrin. Dextrin is a FDA-approved biodegradable polymer that can be used as a formulation aid. Microbiology assays showed that the dextrin–colistin conjugates exhibited weaker antibacterial activities against a panel of Gram-negative bacteria. Significantly, dextrin conjugation could mask colistin’s cytotoxicity and systemic toxicity. The administration of 0.5 mg/kg via single IV bolus to rat had no sign of systemic toxicity, but administration of the colistin at the equivalent concentration was fatal. Pharmacokinetic studies revealed that dextrin–colistin conjugates could prolong the half-life of the colistin (Ferguson et al. 2014). The dextrin–colistin conjugates also effectively inhibited LPS-induced hemolysis in a concentration-dependent manner, without additive toxicity at higher concentrations. Thus, the conjugates could also represent effective neutralizers of endotoxin with application in the treatment of sepsis (Roberts et al. 2016).
Mimicking other natural lipopeptide analogs
Fusaricidines are a family of cyclic lipodepsipeptide antibiotics isolated from Paenibacillus sp. In contrast to typical CAMPs and daptomycin, fusaricidin and its synthetic derivatives possess a neutral amino acid sequence and a single positive charge located at the termini of their lipidic tails. Bionda et al. has synthesized three cyclic lipopeptides (designated as LI-Fs-1, LI-Fs-2 and LF-Fs-3) derived from fusaricidin (Table S1). These three peptides were different in amino acid at position 1. LI-Fs-2 exhibited similar antibacterial activity against Gram-positive bacteria compared with LI-Fs-1, but with improved stability and decreased human cell cytotoxicity. Interestingly, LI-Fs-3 showed a complete loss of activity. Time-kill assays showed that LI-Fs-1 killed Gram-positive bacteria with more than a 3-log reduction within the first hour. In contrast, LI-Fs-2 was bacteriostatic under the same experimental conditions. Biophysical studies further revealed that LI-Fs-1 depolarized bacterial membranes more efficiently. However, membrane depolarization did not correlate with bacterial cell lethality, suggesting that membrane targeting activity is not the main mode of action for this class of antibacterial peptides. It is also noteworthy that linear analogs of fusaricidin were also synthesized, but no antibacterial activity was detected. Cyclic lipopeptides LI-Fs-1 and LI-Fs-2 improved the survival of G. mellonella larvae infected with MRSA (Bionda et al. 2013). From Li-Fs-2, Bionda et al. developed a positional scanning combinatorial library to screen for enhanced antibacterial activity. Activities were screened against the ESKAPE pathogens (Bionda et al. 2016). The results showed that a new cyclic peptide, designated as LI-Fs-4 in this review, was discovered. LI-Fs-4 exhibited the most broad and potent antibacterial activity in this series of cyclic lipopeptide. Interestingly, LI-Fs-4 was active against bacterial biofilms. It was able to decrease the number of cells within biofilms, and disrupted mature 2-day-old biofilms formed by MRSA and P. aeruginosa at 4 µg/mL. Taken together, an increase in overall hydrophobicity of cyclic lipopeptides and net positive charge resulted in improved antibacterial activity. LI-Fs-4 also showed a low propensity for bacteria to develop resistance compared with vancomycin and ciprofloxacin. Significantly, no non-specific toxicity was observed using Cell TiterGlo Luminescent cell viability assay on BJ cells (ATCC CRL-2522) (Bionda et al. 2016).
De Zoysa et al. has synthesized a series of linear lipopeptides based on a cyclic lipopeptide, battacin. The results showed that the linear analogs had higher potency than cyclic analogs. One linear lipopeptide (sequence = CH3-CH2CH(CH3)(CH2)2CO-Dab-Dab-Dab-Leu-(D)Phe-Dab-Dab-Leu-NH2) exhibited good broad-spectrum antimicrobial activities (MICs = 1–5 µM) against both plant pathogens and human pathogens E. coli, P. aeruginisosa and S. aureus. By contrast, its cyclic counterpart showed higher MICs of 2.5–507 µM. Further SAR analysis using alanine scanning method has identified a potent truncated linear lipopeptide with a sequence of (CH3)2CH(CH2)3CO-(D)Dab–Dab-Leu-(D)Phe-Dab-NH2. The truncated lipopeptide also showed similar potency against P. aeruginosa, S. aureus, E. coli and Psa with MICs of 1–15 µM (De Zoysa et al. 2015).
Arylomycins are a class of non-ribosomally synthesized lipopeptide antimicrobials that inhibit bacterial type I signal peptidase (SPase). Spase is an essential serine–lysine dyad protease that is anchored to the outer leaflet of the cytoplasmic membrane. It removes the N-terminal signal peptides from proteins that are transported out of the cytoplasm. However, the spectrum of the arylomycin is very limited, because of the presence of a specific Pro residue in the target peptidase that disrupts interactions with the lipopeptide tail. To optimize the activity of arylomycins, one of the possible approaches is to obtain an arylomycin analog that could bind to SPase regardless of the resistance-conferring Pro. As an attempt to achieve a broad-spectrum arylomycins analog, Roberts et al. have synthesized a series of arylomycin analogs focusing on derivatives with altered lipopeptide tails. Antimicrobial assays were performed on susceptible and resistant strains of S. epidermidis, S. aureus, E. coli and P. aeruginosa. Lipid tail modifications were performed to investigate the following: minimal tail length, one or more aromatic rings, phenyl substituted tail mimetics, effects of lipopeptide methylation, effects of lipopeptide tail rigidity and flexibility. Overall, both the methylation state and the length of the straight chain fatty acid of the lipopeptide tail (C16) are already optimized for activity (Fig. 4). Nevertheless, the unnatural phenyl analogs are more promising scaffolds with an expanded spectrum against wild type S. aureus strains (Roberts et al. 2011). Liu et al. has further modified arylomycin via glycosylation with a deoxy-α-mannose substituent (Fig. 4). They found that glycosylation did not affect the antimicrobial activity significantly. Structural analysis revealed that the lipoglycopeptides bound to SPase in a manner analogous to the arylomycin. The hydrophobic part of the sugar interacts with active site residues and that glycosylation affected the interactions between the peptidic position of the inhibitors tail and SPase. Glycosylation also improved the water solubility of the arylomycin (Liu et al. 2011).

Chemical structure of arylomycin A-C16 and its glycol-modified derivative, arylomycin C-C16
Amphomycin is a naturally occurring lipopeptide active against Gram-positive bacteria. MX-2401 (Fig. 5) is a novel semisynthetic lipopeptide antibiotic that comprises a chemically modified amphomycin core to which an aromatic linker and C12 lipid side chain are added. In general, MX-2401 showed potent antimicrobial activity against a variety of Gram-positive bacteria associated with serious infections, including MRSA and VRE, with MIC90 ranges of 2–4 µg/mL. Compared to daptomycin, MX-2401 had a longer half-life and was not as strongly dependent on the concentration of Ca2+ (Dugourd et al. 2011). Although MX-2401 displayed poor oral availability, it displayed favorable PK profile in IV administration (Pasetka et al. 2010).

Structure of MX-2401. The figure was extracted and modified from a published report (Pasetka et al. 2010)
Nisin is a potent cyclic peptide with potent antimicrobial activity produced by various strains of Lactococcus lactis. The presence of five lanthionine rings (A–E) imparts a predefined conformation for nisin’s mode of action. Nisin’s antibacterial action involves its ability to bind to lipid II with the N-terminal A/B ring system. Then, C–E rings from the C-terminal region will insert into the bacterial inner membrane, leading to pore formation and bacterial cell death (Breukink et al. 1999). The main limitations of nisin are its susceptibility to proteolytic degradation in vivo, toxicity and poor pharmacokinetics. The presence of the protease-sensitive C-terminal region of nisin is required for full antimicrobial activity and is proposed to interact with bacterial membranes upon lipid II binding by the A/B ring moiety. As an attempt to maintain antibacterial properties and improve protease stability of nisin, Koopmans et al. substituted the C-terminus of nisin with a simple membrane active lipid. Antimicrobial assays showed that analogs containing the C10, C14 and farnesyl lipids showed the most potent antimicrobial activities (MICs = 4–64 µg/mL) against Gram-positive bacteria. A triazole linked analog by incorporation of a terphenyl lipid, exhibited the most potent antimicrobial activities (MICs = 4–8 µg/mL). These new analogs induced a clear loss of structural integrity in bacterial cells, with an indication of cell death via lysis. Significantly, the stabilities of the new analogs in human serum were greatly improved. For instance, while only 33% of nisin remained intact after 24 h, the new analogs were 94% intact over the same time period (Koopmans et al. 2015).
BPC194 (sequence = c(KKLKKFKKLQ) is a synthetic cyclic peptide with a high antimicrobial activity, low hemolysis and high phytotoxicity. It is derived from a de novo designed cyclic peptide and then optimized via a combinatorial chemistry approach (Monroc et al. 2006a, b). Vila et al. has designed and synthesized a series of peptides to explore the selective activity against microbial, plant and animal cells. In one study, they investigated: the length of the fatty acid chain and the lysine residue that was derivatized, incorporation of d-amino acid and the replacement of Phe residue with His residue. In general, the results show that cyclolipopeptides with differential biological activity profile were developed. For instance, cyclolipopeptides with a d-amino acid or a His residue displayed low hemolysis at concentration up to 250 µM. By contrast, the parent cyclopeptide, BPC194 is highly hemolytic. However, all the newly developed cyclolipopeptides showed poorer antimicrobial activities against plant bacteria (Erwinia amylovora PMV6076 and P. syringae pv. syringae EPS94) and fungus (Fusarium oxysporum f. sp. lycopersici FOL race 2 ATCC201829) tested than the parent cyclic peptide. Structures of selected cyclolipopeptides (BPC500, BPC676, BPC686, BPC714 and BPC728 are shown in Table S1 (Vila et al. 2016).
Mimicking linear natural peptides analogs
In addition to cyclic lipopeptides, there are also many successful examples of synthetic lipopeptides mimicking natural linear AMPs with potential therapeutic values. Lactoferricin is CAMP derived by pepsin digestion of lactoferrin. Lactoferricin displays antimicrobial activity against a wide range of microorganisms (Gifford et al. 2005). The incorporation of an acyl group into the peptide backbone only resulted in a compound with improved MIC (LF-215; sequence = FWRIRIRR). Acylation of LF11-215, an 11-mer peptide derived from human lactoferricin, with octanoic acid (O-LF11-215) and 2,2-dimethylbutanoic acid (DI-MB-LF11-215) slightly improved the MICs from 64 µg/mL to 32 and 16 µg/mL, respectively. The possible reason is that N-acylation enhances the binding to LPS of the outer membrane of P. aeruginosa, which can counteract the membrane permeabilization by reducing the effective peptide concentration at the inner membrane. The acylated analogs killed significantly faster than their parental non-acylated counterparts when tested at their respective MBCs (Sanchez-Gomez et al. 2015). The N-acylated peptides were found to significantly affect the distribution of the cardiolipin domains. Before E. coli was treated with the N-acylated peptides, cardiolipin was mainly localized to poles and septum area. O-LF11-215 significantly affected the distribution of cardiolipin domains as the cardiolipin was distributed along the cell membrane. For the LF11-215-treated cells, cardiolipin was still mainly distributed at septum and poles. N-acylated peptides induced numerous small membrane domains, creating more defects at domain borders and caused a more severe membrane perturbation. In addition, N-acylated peptides were also found to interact strongly with both anionic and neutral lipids, which further hinder the formation of CL domains at septum and poles. Overall, N-acylated LF-11 had stronger bactericidal activity and induced stronger defects in membrane perturbation (Zweytick et al. 2011, 2014).
Defensins are classified into α- and β-defensins, based on the spatial distribution of the three cysteine intramolecular bonds. Human α-defensins, such as HD5 are key components and key contributors to innate immunity. HD5 exhibits broad-spectrum antibacterial activity. A full-length HD5 has 32 amino acids with three disulphide bridges. Interestingly, several reports propose that full-length HD5 may not be a stringent requirement for antibacterial activity (Varkey and Nagaraj 2005; Lundy et al. 2008). However, the linear analog of HD5, designated D5R (sequence = ATYRTGRATRESLSGVEISGRLYRLR; MIC = 20–100 µM) was less active than HD5 (MIC = 2.5–20 µM, except for P. aeruginosa). Similarly, its D-residues, D5r (sequence = ATYrTGrATrESLSGVEISGrLYrLR; r = d-arginine) had similar activity to D5R. As an attempt to improve the antimicrobial activity of the linear analogs, D5R and D5r were conjugated with myristic acid and lauric acid. Fatty acylation with both myristic and lauric acids results in dramatic increase in antibacterial activity against both Gram-negative, Gram-positive bacteria and fungi. The peptides conjugated with both myristic acid and lauric acid are also active against P. aeruginosa (MIC = 7.5–10 µM) unlike full-length HD5 (inactive). Confocal microscopy revealed that HD5 did not cause extensive damage to bacterial membrane. This was further proven by Sytox Green assay, which showed that HD5 did not permeabilize the bacterial inner membrane. Similarly, non-acylated peptides, D5R and D5r also did not induce inner membrane perturbation. By contrast, acylated peptides caused extensive membrane disruption. Interestingly, HD5 had ability to bind with plasmic DNA. Thus, HD5 bound to DNA and further resulted in shut down of bacterial metabolism leading to cell death. Both linear analogs and acylated analogs exhibited reduced drastic reduction of affinity to DNA. Incorporation of d-arginines resulted in further reduction in binding. In summary, acylation of HD5 linear analogs shifted the antibacterial action from DNA binding to membrane disruption (Mathew and Nagaraj 2015).
Tridecaptins are a group of linear cationic lipopeptides analogs of the polymyxins. Tridecaptin A1 is a lipopeptide isolated from Paenibacillus terrae NRRL B-30644, which displays antimicrobial activity against the common foodborne pathogen Campylobacter jenuni (Lohans et al. 2012, 2014). Tridecaptin A1 has a structure of CH3CH2CH(CH3)(CH2)2CH(OH)CH2CO-vD-Dab-GswS-Dab-D-Dab-FEV-D-alle-A. As an attempt to make structurally simple analogs and improve antimicrobial activities, Cochrane et al. has synthesized a library of lipid tail modified Tridecaptin A1. Tridecaptin A1 is more potent against Gram-negative pathogens (MICs = 1.56–12.5 µg/mL). However, it is much less active against Gram-positive bacteria (MICs = 0–>100 µg/mL). In that study, eleven analogs with different lipid tails were synthesized. Significantly, the unacylated peptide was much less active than the natural lipopeptide. Analogs in which the lipid tail was shorter than six-carbon units or longer than twelve-carbon units were also significantly less active than the natural lipopeptide. The optimal length for the lipid tail was 8- to 12-carbon units, displaying the strongest antimicrobial activities against Gram-negative bacteria. However, none of the analogs exhibited potent antimicrobial activity against Gram-positive bacteria. Interestingly, when the lipid tail was conjugated with triethylene glycol, so the carbon unit was in the range of 8–12 carbon units, the compound was inactive against all bacteria at the highest tested concentration (MIC > 100 µg/mL). The results suggested that the presence of heteroatoms in the lipid tail possibly prevents effective interaction with the cell membrane or alters the conformation of the peptide. Overall, the lipid tail of the tridecaptin A1 proved to be an ideal site for modification. Octanoic acid found to be an optimal fatty acid yielded an analog that retained strong activity against food-borne pathogen and Gram-negative pathogens. The synthesis of tridecaptin A1 is also simplified by replacing the chiral lipid tail of the natural tridecaptin A1 (Cochrane et al. 2014).
Bacteriocins are antimicrobial peptides secreted by many lactic acid bacteria. These peptides are usually cationic and membrane permeabilizing. Plantaricin is a bacteriocin produced by Lactobacillus plantarum NRIC 149 isolated from pineapple. Siano et al. has synthesized a series of peptide analogs based on region 6–22 of Plantaricin 149 (Pln149a, sequence = YSLQMGATAIKQVKKLFKKKGG). Peptide derived from region 6–22 of Pln149a was designated as CT (sequence = GATAIKQVKKLFKKKGG). CT peptide was acylated with octanoic acid (C8-CT) and dodecanoic acid (C12-CT). CT(W17) corresponds to a CT-substituted analog where Phe17 was replaced by Trp17 (sequence = GATAIKQVKKLWKKKGG). Antimicrobial activity showed that acylated peptides exhibited much improved antimicrobial activity against S. aureus. In general, acylation could improve antimicrobial activity of CT peptide against both strains of bacteria tested. The substitution of Phe by Trp had an unfavorable effect on the antimicrobial property. Membrane interaction studies with dipalmitoylphosphatidylcholine-polydiacetylene vesicles showed that lipopeptides interacted to a greater extent with both biological and biomimetic membranes (Siano et al. 2011).
The studies discussed above show that amino acid replacement could also impact the antimicrobial properties of a peptide. Zotti et al. has explored the role of the Aib (α-aminoisobutyric acid) by substituting the Aib residues with Leu in trichogin GA IV, an antimicrobial lipopeptaibiotic. Trichogin GA IV belongs to a sub-class of peptaibiotics termed lipopeptaibols, carrying a fatty acyl moiety at the N-terminus. Trichogin GA IV has a primary peptide sequence of: n-Oct-Aib-Gly-LeuAib-Gly-Gly-Leu-Aib-Gly-Ile-Lol, where Lol is leucinol. Trichogin GA IV is an antimicrobial active against Gram-positive bacteria. Aib is known to be responsible for the adoption of particularly stable helical structures. In that study, four new trichogin Ga IV analogs were synthesized: C8-LGL-Aib-GGL-Aib-GI-Lol (L1), C8-Aib-GL-LGGL-Aib-GI-Lol (L4), C8-Aib-GL-Aib-GGLLGI-Lol (L8) and C8-LGL-Aib-GGLLGI-Lol (L1,8). All L1, L4 and L8 analogs removed the activity of trichogin GA IV against E. coli (MIC = 32 µg/mL), as all Leu-substituted analogs were inactive against E. coli at the highest concentration tested (>64 µg/mL). The results suggested that Aib residues present in the native sequence were important against E. coli. By contrast, L1 and L8 analogs showed an enhanced activity against S. aureus, with MICs of 2 and 4 µg/mL, respectively. Native Trichogin Ga IV had MICs of 8–16 µg/mL against S. aureus. The L4 analog was inactive against S. aureus (MIC > 64 µg/mL). 2D-NMR and X-ray diffraction studies showed that the absence of the helix-stabilizing Aib residue at 4th position, in the core of the primary structure motif, is probably mainly responsible for the conformation behavior of this analog. Conversely, substitution of Aib to Leu at 1st and 8th positions only affected the helical structure to a minor extent. Both L1 and L8 could interact with lipid bilayer more efficiently too. Lastly, the Aib to Leu replacements do not affect the resistance to proteolysis (De Zotti et al. 2012).
Anoplin (sequence = GLLKAIKTLL) is a purified CAMP from the venom of the solitary wasp Anoplius samariensis (Konno et al. 2001). From a SAR analysis, Ifrah et al. found that the overall charge of +4 is essential for selectivity, while hydrophobicity is required for activity (Ifrah et al. 2005). To further improve the antimicrobial activity of anoplin, Slootweg et al. has incorporated (S)-2-aminoundecanoic acid (lipoamino acid, LAA C9) on positions 2, 6 or 10. Antimicrobial assay showed that LAA C9 substituted analogs exhibited a four- to eightfold improved antimicrobial activities against S. aureus (MICs = 2.5 µg/mL) compared to anoplin (MIC = 20.6 µg/mL). In addition, they also showed improved MICs against E. coli (MICs = 5.2–20.6 µg/mL). MIC of anoplin against E. coli was 41 µg/mL. Hemolytic activity of LAA C9-substituted analogs was higher than anoplin (39–108 vs >500 µg/mL). N-decanoylated anoplin (C9-GLLKAIKTLL) was used as a control for non-selective lipopeptides. Although it showed very potent antimicrobial activities against both E. coli and S. aureus (MIC = 2.5 µg/mL), the membrane selectivity was poor. Biophysical assays using carboxyfluorescein leakage assay showed that LAA C9 substituted analogs increased membrane permeabilization to anionic lipid containing membranes. Overall, lipoamino acid could be used to increase hydrophobicity of a peptide and antimicrobial activity of the peptides. Compared to N-acetylation with saturated fatty acid, lipoamino acid showed better selectivity and as membrane selectivity was not significantly disrupted (Slootweg et al. 2013).
Tryptophan- and arginine-rich antimicrobial peptides possess high potencies against both Gram-positive and Gram-negative bacteria. Several reports have shown that peptides comprised of only tryptophan, arginine and phenylalanine have potent antimicrobial activities (He et al. 2007; Haug et al. 2008). Fang et al. hypothesized that effective AMPs could be prepared by combining the advantages of lipopeptides with those of RW rich peptides. K and I were used to tune the peptide hydrophobicity and charge. A series of short lipopeptides containing RWKI residues were synthesized. Antimicrobial assays showed that most of these peptides were more active against S. aureus than E. coli. Interestingly, no significant differences between all sequence variations ranging from simple RW or KW containing sequences to the peptides mixed with R, K, W and I residues. The results clearly showed that antimicrobial activity is related to their physiochemical properties especially in charge and hydrophobicity. With regard to fatty acid length, both C10− and C14 conjugated peptides were active against S. aureus (MICs = 3.9–31.2 µg/mL). However, when the fatty acid length increased to C18, the antimicrobial activities were greatly weakened (MICs = 15.6–>31.2 µg/mL). SAR analysis of hemolytic activity showed that the number and the position of the W residue were important. More W residues resulted in increased lytic effects. However, greater lytic effects were also associated with a longer lipid tail. By contrast, increase in charge would favor its discrimination ability between more neutral mammalian cell membranes and more negatively charged bacterial membrane. Overall, SAR analysis showed that hemolytic activity on human erythrocyte was tuneable by varying their physicochemical parameters especially charge, hydrophobicity and residual position. These lipopeptides were also membrane targeting. It is noteworthly that several peptides such as AMP-C10-3 (sequence = C10-RKWWK) and AMP-C10-4 (sequence = C10-RIKWWK) were able to retard the migration of DNA on agarose gel in the DNA binding assay. This signified that multiple modes of action were involved. However, for those peptides with fast killing property, bacterial membrane is still the primary target (Fang et al. 2014).
L-to-D substitution of a α-helix peptides modulates the α-helical structure, resulting in a change in interface localization of the peptides, which further affects the membrane targeting properties. To investigate the effects of L-to-D on their antimicrobial activities, Albada et al. has synthesized two groups of 32 peptides each of diastereomeric lipidated RW-peptides. One group was lipidated with octanoic acid while the other was lipidated with decanoic acid. The peptide backbone used in this study was based on WRWRW, and subsequently a L-analog was substituted with a d-analog In this section, capital letter represents L-analog, whereas small letter represents d-analog). Antibacterial assays showed that all peptides modified with d-analogs were comparable in activity against Gram-positive bacteria. For instance, almost no difference was observed between the diastereomeric C8- and C10-lipidated peptides against B. subtilis and MRSA. For diastereomeric C8-and C10 lipidated peptides, most derivatives were similarly or less active than the all-L peptides. Encouragingly, the L-to-D transformation could be observed for their hemolytic activity. All L-analogs (C8-WRWRW) displayed 15.6 ± 3.2% hemolysis at 142 µM, whereas most of the stereoisomers of this peptide were significantly less hemolytic. For example, C8-WRwRw, C8-WrwRW, C8-WrWrw, C8-wRwrW, C8-Wrwrw, C8-wRwrw, and C8-wrwRw were almost not hemolytic and still possessed strong activity against Gram-positive bacteria. Taken together, L-to-D transformation could increase the potential therapeutic window of these compounds (Albada et al. 2013). Albada et al. also investigated the effects of C- or N-terminal lipidation on RW-peptides. Both N- and C-lipidated peptides showed similar activities against Gram-positive bacteria. Interestingly, C-lipidated peptides, especially C-C8(RW)3, C-C10(RW)3 and C-C12(RW)3, exhibited improved antimicrobial activities against Gram-negative bacteria, with MIC ranged of 2–5 µM and 5–9 µM against E. coli and A. baumannii, respectively. Their N-lipidated analog showed MIC ranged of 5–18 µM and 9–35 µM against E. coli and A. baumannii, respectively. Hemolytic assay also showed that C-lipidated peptides had higher HC50 value (Albada et al. 2012).
A summary of selected examples of the synthetic peptides described above are shown in Table S2.
Design of new lipopeptides with different scaffold and structural properties
Natural lipopeptides have several limitations such as poor stability and high cost of production (Giuliani and Rinaldi 2011). Particularly, although the use of peptides composed of d-amino acids is sometimes contemplated owing to their high resistance to proteolysis, this approach often significantly increases the cost of manufacturing with only limited pharmacokinetic advantages. To overcome these limitations, focus has shifted from the production of these naturally occurring AMPs or their sequence analogs to developing mimics endowed with better properties such as ultrashort peptides, multimeric peptides or peptidomimetics.
Ultrashort lipopeptides
CAMPs are not ideal drugs as they are prone to peptidase degradation (Hancock and Sahl 2006; Straus and Hancock 2006). Ultra-short lipopeptides are engineered antimicrobial peptides with 2–4 amino acids linked to a fatty acid. The net charge is positive, ranging from 1 to 4. Interestingly, substituting only one out of the two to four amino acids is sufficient to create molecules with different biological functions. They represent the shortest lipopeptides reported so far. Even an ultra-short peptide with KK conjugated to palmitic acid is able to demonstrate both bactericidal and fungicidal activities (Baranska-Rybak et al. 2013). Fatty acid is a very important component in ultra-short lipopeptides. For instance, Avrahami and Shai produced a series of active ultra-short lipopeptides by acylating the inactive short peptides (Avrahami and Shai 2003, 2004). C16-KGGk is one of the most potent ultra-short lipopeptides (Makovitzki et al. 2006). Recently, Lin and Grossfield have investigated the antimicrobial action of C16-KGGk using umbrella sampling. The results showed that C16 hydrophobic tail is the main contributor to its affinity to lipid membrane, whereas the peptide part is important for its selectivity (bacterial mimicked membrane over the mammalian mimicked membrane). Electrostatic interaction contributed about 40% to the overall ΔG of binding, suggesting that electrostatics play a significant role in selectivity. Moreover, their studies also revealed that the ultrashort lipopeptides can bind to lipid membrane via different paths: hydrophobic interactions with the acyl chains or the electrostatic interaction between lysine side chains and the lipid phosphates (Lin and Grossfield 2014). After bound to the membrane, these lipopeptides altered the local lipid organization by recruiting negatively charged POPG lipids to the site of binding. This drastic reorganization of the membrane bilayer has major impacts on bilayer dynamics and cellular process that depend on specific bilayer compositions (Horn et al. 2012).
KYR is one of the shortest AMPs reported. KYR is the amino acid sequence of the bovine hemoglobin alpha chain strain, which was part of the longer amino acid sequence that was obtained from hydrolysing the hemoglobin alpha chain. As an attempt to further improve antimicrobial activities of this ultra-short peptide, C-terminus was amidated and the N-terminus was acylated with fatty acid of chain length C10, C12, C14 and C16. Antimicrobial assay indicated that C14-KYR-NH2 (10–27 µg/mL) was the most effective ultra-short peptides in this series. (Catiau et al. 2011). Interestingly, haemolytic assay showed that all the ultra-short peptides were non-hemolytic at the highest tested concentration of 256 µM. Cytotoxicity assay (MTT assay) using Vero cells indicated that no cytotoxicity was detected at 40 µM. However, strong cytotoxicity could be observed at 80 µM. Mechanism studies suggested that C12- and C14-KYR-NH2 first bound to the outer membrane of the bacterial cell and further permeabilized into the outer membrane rapidly. Then, they reached inner membrane and induce membrane permeabilization and depolarization, resulted in rapid time-kill in 10 min. Taken together, high selectivity and rapid killing of bacteria make them promising candidates as drug lead compounds (Nasompag et al. 2015).
Sikorska et al. has investigated the activity, self-organization and interactions of three ultrashort peptides: C16-KK-NH2, C16-KGK-NH2 and C16-KKKK-NH2. These ultra-short lipopeptides exhibited broad-spectrum antimicrobial activities, with MICs ranged from 4 to 16 µg/mL against Gram-positive and Gram-negative bacteria. Their bactericidal concentrations ranged from 4 to 32 µg/mL. As the MBC/MIC ratios are less than or equal 2, this further asserting the bactericidal power of the tested compounds. C16-KK displayed the most potent hemolytic activity. HC50 of C16-KK was 25 mg/mL, whereas a five times higher concentrations needed to induce 50% hemolysis for both C16-KGK-NH2 and C16-KKKK-NH2. Overall, they show a good selectivity index (HC50/MIC) of >3000. Similar to other ultra-short lipopeptides, initial electrostatic and subsequent hydrophobic interactions enabled the lipopeptides to interact with the lipid bilayer. Both types of interactions were important to provide a sufficient energy to anchor the peptide into the membrane (Sikorska et al. 2014).
Findlay et al. have investigated the window of activity of the lipopeptides C16-KGK and C16-KKK. In addition to normal carbon–hydrogen hydrophobic domain, they also investigated effects of carbon–fluorine (CF) bonds on the ultra-short lipopeptides. Molecules with CF bonds are both hydrophobic and lipophobic. Amphiphiles heavy with CF bonds might, therefore, prefer to self-associate, reducing the effect of their hydrophobic character whilst in solution and potentially creating areas of high peptide concentration within the membrane. In this study, hydrophobic tails of the ultra-short peptides were constructed by saturated hydrocarbons and fluorocarbons (Table S3). C16-KGK was the most potent compound against MSSE and MRSE strains with MICs of ≤0.25 µg/mL. Antimicrobial activity of the longest fluorinated compounds, such as F11-KGK(sequence = CF3(CF2)9CO-KKK-NH2), was moderate against Gram-positive bacteria (32 µg/mL). Other compounds with shorter fluorinated tails were less active as the lipids became shorter. For instance, F7-KGK and F7-KKK were broadly inactive against all the strains tested (Findlay et al. 2012).
Lohan et al. has reported antimicrobial activities of a series of non-natural ultrashort lipopeptides composed of non-genetically coded amino acid ornithine (1–5 ornithines), covalently attached to fatty acids of different chain lengths (C8–C18). All of the synthesized lipopeptides were tested against a range of microbes including Gram-positive, Gram-negative bacteria and fungi. Lipopeptides with single Orn residue and short aliphatic tail were inactive with MICs of >100 µg/mL. Improved MICs could be observed when number of the Orn residues increased to from two to five units. The most potent ultrashort lipopeptide in this series was LP16, with a structure of C14–Orn–Orn–Orn–Orn. LP16 exhibited potent and broad-spectrum antimicrobial activities against E. coli, P. aeruginosa and S. aureus with MICs of 1.5 and 6.25 µg/mL against B. subtilis. It also displayed potent activities against drug-resistant pathogens such as methicillin-resistant S. epidermidis (MRSE; MIC = 6.25 µg/mL) and MRSA (12.5 µg/mL). SAR analysis also revealed that three ornithine residues and C14 and C16 were the optimal ornithine number and chain length, respectively. Any higher number of ornithine residue or chain length longer than that did not provide further enhancement in activity. LP16 was also active against a panel of fungal strains including C. albican, A. fumigates, A. niger and C. neoformans (MICs = 3.1–12.5 µg/mL). Lipopeptides with comparatively bulky aliphatic tail were found to be more active toward fungal strains. For instance, MICs of LP24 (C18-Orn–Orn–Orn–Orn) and LP29 (C16-Orn–Orn–Orn–Orn-Orn) showed MICs in the range of 1.5–3.1 µg/mL against all the tested strains. The most potent compound, LP16 showed significant selectivity ratio (HC50/MIC) ratio of 500. Compounds with longer aliphatic chain, such as LP24 and LP30 suffered from a poor selectivity ratio of 20. LP16 an LP23 (C16-Orn–Orn–Orn–Orn) were further assayed for their time-kill kinetics. Both LP16 and LP23 were able to completely eradicate 7-log reduction of S. aureus at 2 × MIC in 3 h, whereas complete killing was achieved in 2 h against E. coli. Significantly, both LP16 and LP23 did not induce drug resistance in S. aureus even after 15 rounds of passaging. LP16 was also proven to be stable against trypsin for 24 h (Lohan et al. 2013).
Ghosh et al. has designed and synthesized a series of ultra-short peptides that only involve the use of one single lysine and two lipid tails. In contrast to most of the lipopeptides with only one lipid tail, Ghosh reported that two short lipid tails instead of a single long one significantly improve the membrane selectivity against bacteria. For instance, C8-K-C8 showed similar antibacterial activities to C16-K, but with a better of HC50 value (108 vs 73 µM). In this study, C8-K-C10, a non-symmetrical analog was identified as the most potent compound with MICs of 5–10 µM against Gram-positive and Gram-negative bacteria. This compound did not induce significant toxicity toward human embryonic kidney cells at the minimum bactericidal concentration at 2 × MIC. Similar to other ultrashort lipopeptides, this compound was also able to depolarize and permeabilize bacterial cell membranes. Interestingly, C8-K-C10 reduced a moderate bacterial burden in a mouse burn-infection model. However, colistin still showed superior activity against C8-K-C10, so, further improvements against Gram-negative bacteria were required (Ghosh et al. 2016).
Domalaon et al. has investigated the effects of ring constraint in the amino acid side chain on the ultra-short lipopeptides. Most of the ultra-short lipopeptides possess a flexible peptide backbone. Lysine, l-diaminobutyric acid (Dab) and l-homoserine (HSe) were selected for amino acid with flexible side-chain. To constraint the peptide backbone, l-proline with a ring-constrained side chain, was used. In this study, l-4R-aminoproline (Pcat) and l-4R-hydroxyproline (PHyp) were used as conformationally constrained analogs of Dab and Hse, respectively. Their side chains were constrained in the form of a five-membered pyrrolidine ring. To further explore the effect of increased hydrophobicity, l-4R-hexyloxy-based proline analog (PHex) was also synthesized. All the structures of unnatural amino acids used in that study are shown in Fig. 6. Nine ultra-short lipopeptides were synthesized (Table S3). USCL-K1 (C16-KKKK-NH2) was used as a reference peptide. To study how the nature of the lipid tail affects the properties of the lipopeptides, 16-hydroxypalmitic acid (C16OH) was conjugated onto ultrashort peptides with peptide backbone of KKKK (USCL-K2), DabDabDabDab (USCL-Dab2) and Pcat Pcat Pcat Pcat.(USCL-Pcat2). All other ultrashort peptides were conjugated with palmitic acid. Hse and Phyp were used to study the hydrophobic effects in the peptide sequence. Antimicrobial assays using a panel of reference and clinical Gram-positive and Gram-negative pathogens showed that flexible amino acids are beneficial to the activity in comparison to a ring-constrained amino acid. For instance, USCL-Pcat1 exhibited fourfold higher MIC than its flexible analog, USCL-Dab1. This observation was also observed for USCL-Dab3 (C16-Dab-HSE-HSE-Dab-NH2) against USCL Pcat3. C16OH-analogs also showed a decrease of antibacterial activity, because of the disturbance within lipopeptide hydrophobic domain. Overall, these synthetic ultra-short lipopeptides were more active against Gram-positive bacteria. Only USCL-K1 and USCL-Dab1 exhibited relatively potent MICs against E. coli (MICs = 8–16 µg/mL). For example, more hydrophobic analogs, such as USCL-Pcat4 (C16-PcatPHexPHexPCat-NH2), only showed antimicrobial activities against Gram-positive bacteria. A possible reason was that the hydrophobicity of the peptide was above the threshold to penetrate or interact with the outer membrane of Gram-negative bacteria. Cytotoxicity studies using LDH assay against human monocytic THP-1 cells showed that no cytotoxicity was observed up to 20–40 µM. These lipopeptides also did not induce the production of the pro-inflammatory cytokines TNF-α and IL-1β, thus indicating that these compounds will not mediate an inflammatory response in blood-derived mononuclear cells. No significant production of chemokines IL-8 and Gro-α were observed too (Domalaon et al. 2014).

Unnatural amino acids used in the study of Domalaon et al. to investigate the effect of amino acid ring constraint on antibacterial activity. Dab l-diaminobutyric acid; HSe l-homoserine; P cat l-4R-aminoproline, P Hyp l-4R-hydroxyproline; P Hex l-4R-hexyloxy-based proline analog
α,β-Unsaturated γ-amino acids (E-vinylogous amino acids) are also geometrically constrained amino acids because of the double bond. E-vinylogous amino acids are important components of many biological active peptides (Linington et al. 2009). They are potential candidates for the design of inhibitors of serine and cysteine proteases. E-vinylogous amino acids in hybrid peptides tend to adopt an extended β-sheet type of structures because of the geometrical constrains of the double bonds. Shankar et al. has designed and synthesized two series of short hybrid E-vinylogous lipopeptides containing C12 and C8 fatty acids (Fig. 7). Antimicrobial assays showed that, except for S7 (C12-dgPhe-Lys-dgPhe-Lys, dgPhe = α,β-dehydro-γ-amino acid), all peptides displayed useful antimicrobial activities against Gram-positive, Gram-negative bacteria and fungal strains (MICs = 3.125–12.5 µg/mL). For the hybrid peptide with C8 lipid tail, S8 (C8-dhPhe-Lys-dgPhe-Lys) was the most potent compound (1.56–3.12 µg/mL). Control peptides with an N-acetyl group and the α-peptide with a C8-fatty acid, showed no activity even at higher concentrations. These results suggest that E-vinylogous hybrid lipopeptides can be considered as potential antimicrobial candidates. Atomic force microscopy revealed that the different nanostructures and assemblies of these lipopeptides may be influencing the disparity in their biological activities. The hybrid peptides with moderate hydrophobicity and high electrical potential of self-assemblies showed better antimicrobial and low hemolytic activities. Peptides that adopted nanofiber structures displayed less hemolytic activity, while peptides that adopted nanoneedle structures displayed the highest hemolytic activity. The robust growth of nanoneedles from the hybrid peptides containing aromatic amino acids may be responsible for the disruption of bacterial membranes. Taken together, E-vinylogous amino acids shifted the self-assembly properties of the lipopeptides, which further affected the antimicrobial and hemolytic properties of the hybrid peptides. This provides a unique opportunity to further design potent antimicrobial candidates to combat bacterial resistant (Shankar et al. 2013).

General structure of a short hybrid E-vinylogous lipopeptides. X is the fatty acid conjugated to the peptide backbone. AA1 and AA3 are dg-amino acids (α,β-dehydro-γ-amino acid). AA2 and AA4 are L-amino acids
Greber et al. has synthesized five new lipopeptides with double fatty acid chains. The acylation was done at α and ɛ positions of a peptide backbone of KKKK with saturated fatty acids (Fig. 8). Antimicrobial assay showed that the double-chain lipopeptides with fatty acid chains of 10 and 12 exhibited potent antimicrobial activities against Gram-positive bacteria (MICs = 2–16 µg/mL). C8-, C14- and C16-conjugated peptides showed much weaker activities (MICs = 32–512 µg/mL). All of the lipopeptides synthesized showed much weaker activities against Gram-negative bacteria and fungus (MICs = 32–512 µg/mL). Their surface active properties were also investigated. As expected, lipopeptides with longer hydrophobic chains are more susceptible to aggregation in bulk and showed lower CMC values. Overall, double-chain lipidated peptides displayed good antimicrobial activities against Gram-positive bacteria. However, their significant haemolytic activity prevented them from further consideration (Greber et al. 2014).

Molecular structure of lipopeptide with double fatty acid chains, N-α-acyl-N-ɛ-acyl lysine analogs. n = 6, 8, 10, 12 and 14. Peptide backbone is KKKK
Previously, we have discussed lipoamino acid-modified anoplin, which enhanced the antimicrobial activity of native anoplin while retaining promising membrane selectivity. Lipoamino acids are an unnatural amino acid with structural properties of lipids and amino acids, allowing them to be easily incorporated into a peptide sequence. In this section, utilization of lipoamino acid in ultra-short peptide is reviewed. Azmi et al. has utilized lipoamino acids with 12 carbons (LAAs C12) to modulate the lipophilicity of the molecules. Lysine was chosen to represent the cationic residue as it is positively charged at physiological pH. In this study, four unnatural ultrashort lipopeptides were synthesized (Fig. 9). Dynamic light scattering showed that all lipopeptides formed nanoparticles within the range of 40–80 nm, except Lys-LAAs C12, which had a highly polydispersed particle size distribution. Antimicrobial activities showed that a single copy of LAAs C12 was insufficient to generate any significant antibacterial activity. Compounds with two copies of LAAs C12 showed potent antimicrobial activities against Gram-positive bacteria (MICs = 0.1–7.5 µM). All of the peptides were inactive against Gram-negative bacteria (MICs ≥ 30 µM). Double copies of LAAs C12 could be further classified structurally: Linear, branch and cyclic. The cyclic compound appeared to be the most effective in comparison to its linear or branched counterparts, especially for self-assembling lipopeptides with a potent activity against multidrug-resistant Gram-positive bacteria (Azmi et al. 2015). The antibacterial activities of linear and branched lipopeptides did not differ significantly across most of the tested bacterial strains. Toxicity assay also showed that these lipopeptides were non-toxic up to 15 µM. They were also stable against trypsin for a prolonged period of time even at 12 h. The results are consistent with other reports that the use of unnatural amino acids in peptide scaffold often increases their enzymatic stability. Transmission electron microscopy also revealed they could induce membrane disruption. Taken together, a branched lipopeptide with two lipoamino acids and four cationic moieties demonstrated the most potent antimicrobial activity against multidrug-resistant bacteria (MIC 0.1–0.2 µM) (Azmi et al. 2016a).

Ultra-short lipopeptides involved an unnatural amino acid lipoamino acid (LAAs C12). The structures were extracted and modified from published reports (Azmi et al. 2016a)
Ultra-short lipopeptide could also be conjugated with other molecules to enhance antimicrobial properties. For example, cationic polyamines spermine, spermidine and putrescine are ubiquitous components of eukaryotic and prokaryotic cells with multiple roles in modulating functions of DNA, RNA and protein inside cells. These molecules carry positive charges. Joshi et al. has anticipated that incorporation of spermidine as a positively charged moiety to hydrophobic dipeptides may lead to small peptidomimetics with better antibacterial activity. Three different types of fatty acids were used as hydrophobic moieties: 3-(4-hydroxy phenyl)-propionic acid (HPPA), linoleic acid (LIN) and stearic acid (STER). For the dipeptide portion, three different combinations of tryptophan (W) and ornithine (O) amino acids were explored: WW, WO and OO. Taken together, a series of compounds (n = 19) with a template of hydrophobic-dipeptide-spermidine were synthesized and evaluated. Compounds #14–#19 (Table S3) showed very promising antimicrobial activities against both Gram-positive and Gram-negative bacteria. Their MIC ranges were 0.88–28.4 µg/mL. Compounds conjugated with HPPA were much less active with MICs of ≥56.8 µg/mL. Other compounds, without either spermidine conjugates or fatty acid moieties, most of them were inactive at highest tested concentration of 227.2 µg/mL. SAR analysis revealed that a hydrophobicity window of 50–70% with a minimum +2 charges to be crucial for activity and cell selectivity. These compounds showed different extents of membrane perturbation (Joshi et al. 2012).
There are also some synthetic ultra-short peptides that possess anti-biofilm activity. C14-KK and C14-RRR were sufficient enough to make these compounds antimicrobials. They also exhibited anti-biofilm properties against clinically relevant MRSA strains, such as S. aureus USA300 and USA400, which were comparable to daptomycin and vancomycin. C14-RRR was also found to synergize effectively with vancomycin to prevent biofilm formation (Mishra et al. 2015). In addition, C12-OOWW-NH2 was found to be very effective against methicillin-resistant S. epidermidis (MRSE). At 15.63 µg/mL, a complete eradication of 24-h established biofilm of MRSE ATCC35984 was achieved within 4 h. A dose-dependent activity was observed as a complete eradication was observed within 2 h if the concentration increased to 200 µg/mL. C12-OOWW-NH2 was also active to eradicate the biofilms formed by MSSA and MRSA. It is also noteworthy that the licensed antimicrobials clindamycin, daptomycin, linezolid, tigecycline and vancomycin were unable to completely kill biofilms of MRSA (Laverty et al. 2015).
Peptidomimetics
A peptidomimetic is a small peptide-like chain designed to mimic a natural peptide. Oligoacyllysines (OAKs) are linear peptidomimetics consisting of alternating acyl chains and cationic amino acids, a design that prevents the formation of stable secondary structure (Radzishevsky et al. 2007). The tandem repeats of acyllysines are designed to mimic the primary structure and function of natural antimicrobial peptides. The basic structure of OAKs could be classified into two categories: α-OAKs and β-OAKs (Fig. 10). In this review, only the recent sensitization studies involved OAKs will be discussed, as other studies of OAKs have been extensively reviewed (Giuliani and Rinaldi 2011). OAK C12(ω7)K-β12 rapidly achieved a transient membrane depolarization, which deprived bacteria of the proton-motive force required for active efflux at sub-MIC level. Targeting membrane potential could represent an efficient means to achieve selective control of cell proliferation, and simultaneously alter a wide range of crucial processes that drive their energy from the proton-motive force, rendering emergence of resistance a difficult task to accomplish. For instance, resistance–nodulation–division (RND) drug efflux pumps depend on the proton-motive force as energy source for extruding out from the periplasmic space a wide range of substrates, including antibiotics. OAKs were able to sensitive erythromycin and rifampicin against Gram-negative bacteria both in vitro and in vivo. Although the exact depolarization mechanism of OAKs is still unknown, it is generally thought that OAK increased the access of antibiotic into the cytoplasm and inhibited growth. Significantly, PK data also supports that required concentrations of OAKs to exert depolarization effect could be achieved via subcutaneous administration. The results suggested that sensitization property of OAK could be a useful approach to expand the currently dwindling antibiotic armamentarium for therapeutic treatment of Gram-negative bacteria (Goldberg et al. 2013; Jammal et al. 2015).

Primary structure of a α-OAK and b β-OAK. The figure was modified from a published report (Goldberg et al. 2013)
AApeptides are peptides that contain N-acylated-N-aminoethylamino acid units derived from chiral PNA backbones. There are two types of AApeptides: α-AApeptides and γ-AApeptides, depending on the position of the side chain (Fig. 11). AApeptides have identical number of functional groups or side groups as conventional peptides of the same length. AApeptides are good candidate to develop novel antimicrobial agents because of mimicking the structure, function and mechanism of AMPs, stability and unlimited derivatizing potential. AApeptides are also more flexible in their backbone as they have more dihedral angles compared to canonical peptides. Biological activities of AApeptides could be tuned readily with the introduction of a variety of additional hydrophobic building blocks, and the nature of the hydrophobic and cationic groups (Niu et al. 2013). In general, AApeptides could be classified into three categories: α-AApeptides, γ-AApeptides and cyclic AApeptides. The design of α-AApeptides is based on the following principles: global distribution of cationic and hydrophobic side chains is key to antimicrobial activity, a defined secondary structure is not necessary and the side groups have to segregate into hydrophobic and cationic clusters. Thus, α-AApeptides building blocks were designed to be either amphiphilic or cationic, which were then assembled and lipidated with fatty acid to produce lipidated α-AApeptides. Antimicrobial assays showed that short lipidated α-AApeptides with 2-mer oligomer only were active against Gram-positive bacteria. When more building blocks were added (3-mer oligomer), the α-AApeptides showed improved antimicrobial activity against Gram-negative bacteria. Further addition of more α-AApeptides building blocks improved the broad spectrum and antimicrobial activities against Gram-negative bacteria. SAR analysis also revealed that both certain degree of hydrophobicity has to be reached for the α-AApeptides to be active. Lipidation was important to increase the antimicrobial activity of lipo-α-AApeptides. For instance, the lipidated 3-mer α-AApeptide already displayed an improved activity than the linear 7-mer α1 made from the same building block, signified that lipidation significantly enhanced the antimicrobial properties. In addition, both lipidation and addition of more α-AApeptides building blocks increase the hydrophobicity of sequences enhance the broad-spectrum activity against Gram-negative bacteria. Fluorescence microscopy showed that lipo-α-AApeptides were membrane targeting (Hu et al. 2012). Lipo-γ-AApeptides also displayed similar activity to lipo-α-AApeptides: broad spectrum and membrane targeting. A series of lipo-γ-AApeptides were synthesized. Almost all the cationic lipo-γ-AApeptides were found to be broad spectrum and active against Gram-negative bacteria. Lipo-γ-AApeptides with negatively charged were inactive, which further suggested that electrostatic repulsion with negatively charged bacteria membrane would reduce antimicrobial properties. Compared to lipo-α-AApeptides, 1-mer oligomer displayed potent antimicrobial activity against Gram-negative bacteria (2.5–35 µg/mL), provided the alkyl length was >12 carbon units. Conjugation with the unsaturated tail to γ-AApeptides would have less propensity for aggregation and further improved antimicrobial properties and the lipo-γ-AApeptides were much less hemolytic. Multi-passage resistance selection study also showed that no drug resistance in S. aureus was induced even after 17 rounds of testing (Niu et al. 2012). Electron paramagnetic resonance spectroscopy showed that γ-AApeptides selectively permeate and structurally modify negatively charged bacterial-mimic membranes. By contrast, cholesterol-containing neutral membranes mimicking mammalian cells were minimally affected by the γ-AApeptides. γ-AApeptides may disrupt bacterial membrane via a carpet-like mechanism (Kaur et al. 2016). However, most of the lead α- and γ-AApeptides are long sequences and require a number of steps to synthesize. To generate short antimicrobial peptides with simplified synthetic scheme, Li et al. has utilized a hybrid backbone of lipo-α/γ hybrid peptides. Antimicrobial assay showed that the short peptidomimetic, containing just one γ-AApeptide building block and one lysine residue, showed patented antimicrobial activities against a panel of pathogens including MRSA, MRSE, E. faecalis, P. aeruginosa and E. coli (MICs = 2–5 µg/mL). SAR analysis showed that increases in hydrophobicity would enhance the antimicrobial activity. In addition, hybrid peptides containing two C16 alkyl chains were not active against any bacteria at the highest concentrations tested (MIC > 50 µg/mL). The hybrid peptides displayed rapid time-kill kinetics against both Gram-positive and Gram-negative bacteria, mimicking AMP behavior. Similar to α-and γ-AApeptides, the hybrid AApeptides also killed bacteria via membrane targeting pathway (Li et al. 2014b). As an attempt to further improve antimicrobial activities, cyclic antimicrobial γ-AApeptides were also designed and synthesized. Cyclic peptides are expected to have improved antimicrobial activity compared with their linear counterparts, as their structures are more rigid, which results in more stable amphipathic structures. Cyclic lipo-γ-AApeptides exhibited good broad-spectrum activity against a range of multidrug-resistant Gram-negative and Gram-positive bacteria (MICs = 1–10 µg/mL). However, in general, cyclic lipo-γ-AApeptides were more hemolytic compared to the linear analogs. Interestingly, these peptides also imitated AMPs in their immunomodulatory capabilities. These γ-AApeptides have been shown to antagonize the LPS activated NF-κB signaling response and suppress harmful pro-inflammatory cytokines (Li et al. 2014a). Overall, lipo-AApeptides with potent broad-spectrum antimicrobial activities, enhanced stability and limitless derivation potential make them a very potential candidate to be further developed as therapeutic agent to combat with multidrug-resistant pathogens.

Lipo-AApeptides could be structurally classified into α-AApeptide and γ-AApeptide. α/γ-AApeptide is a hybrid formed of AApeptide. AApeptide could be modified to cyclic peptide too
Reduced amide-containing peptides have been used in peptide sequences to mimic AMPs to improve resistance against proteolytic degradation (Kim et al. 2009). To minimize the molecular weight of the reduced –amide-containing peptide, Teng et al. designed and synthesized a series of acylated reduced amide pseudopeptide. The general chemical structures of the pseudopeptides used in that study are shown in Fig. 12. SAR analysis revealed that R2, R3 and R4 have to be hydrophobic moieties to exert antimicrobial activities against Gram-positive and Gram-negative bacteria. In general, compounds with a hydrophilic moiety at R2 and R3 positions would reduce or diminish antibacterial activity. R1 is a hydrophilic moiety, so the overall structures of the active compounds are amphiphilic. In this study, two compounds (Fig. 12) were identified as the most potent compound against MRSA. Further studies revealed that they are membrane targeting antimicrobials, could kill MRSA rapidly and did not induce resistance in bacteria. Significantly, they also showed anti-inflammatory potential in the MRSA-induced pneumonia-bearing rat model. Overall, facile synthesis, high cost-effectiveness and the structures are readily tuneable make them appealing classes of membrane targeting antimicrobials (Teng et al. 2016).

General structure of acylated reduced amide. R1 is a hydrophilic moiety while R2, R3 and R4 are hydrophobic moieties. Moieties used to identify two potent compounds are also shown
Multivalent lipopeptides
Defensins show useful antibacterial properties, such as rapid time-kill, broad spectrum of activity and low rate in selecting resistant mutants in vitro. Our group has identified and modified the 10-amino acid long C-terminal analogs of human-β-defensin 3 (hBD-3) as an active sequence unit or scaffold for multivalent modification via lysine [B2088, sequence = (RGRKVVRR)2KK] for its antimicrobial activity (Liu et al. 2008; Bai et al. 2009, 2012; Zhou et al. 2011). B2088 exhibits interesting antimicrobial properties including rapid killing of bacteria, promising selectivity to discriminate the bacterial membrane and low propensity for bacteria to develop resistance. To further improve the antimicrobial activity against Gram-negative bacteria, two fatty acid tails were conjugated at N-terminal of the B2088 to improve the interaction with LPS. In this study, the peptide dimers were modified by conjugating fatty acids with various numbers of carbons ranging from 2 to 16 (Fig. 13). Antimicrobial assay showed that peptide dimers conjugated with C8 and C10 exhibited the most potent activities (C8-B2088 and C10-B2088; MICs = 3–6 µM) against Gram-negative bacteria. N-lipidated monomers (C8-B1088 and C10-B1088), exhibited weaker MICs with 6–20 µM against P. aeruginosa and E. coli. The results also revealed that N-lipidation with ≥14 carbon units generally resulted in less activity (MICs = 1.5–>40 µM). Hemolytic activities also demonstrated no hemolysis was observed up to peptide dimers conjugated to carbon unit of 10. C8- and C10-B2088 were selected for further screening against a panel of 10 strains of P. aeruginosa. The MICs measured were 1.5–3 µM, which an overall improvement of 80% compared to their non-acylated analog (B2088). Encouragingly, C8-B2088 and C10-B2088 were also active against a panel of carbapenem-resistant Enterobacteriaceae (CRE), with MICs of 3–10 µM. In addition, C8-B2088 also exhibited significantly improved bactericidal activity compared to that of the nonlipidated form. In addition, C8-B2088 exhibited significantly improved bactericidal activity compared to that of the non-lipidated form. 3-log reduction could be achieved in 30 min at 2 × MIC. No 3-log reduction was observed for non-acylated analog. Biophysical studies revealed that N-lipidated peptide dimers showed stronger LPS permeabilization, which further induced self-promoted uptake to disrupt inner membrane. C8-B2088 also showed better synergistic action. At longer lipid length, self-aggregation dominated and therefore poorer antimicrobial activities were observed. It is also noteworthy that replacement of valine to glycine [C8-B2099, sequence = (C8-RGRKGGRR)2KK] has no significant effects on antimicrobial activities. Taken together, C8-B2088 displayed promising antimicrobial activity with maximum selectivity. It displayed rapid killing, excellent biocompatibility, and synergism with conventional antibiotics (Koh et al. 2015).

Structures of the peptide monomers (B1088 and B1099) and their corresponding peptide dimers, B2088 (X = V), B2099 (X = G) and N-lipidated B2088 and B2099
Multivalent β-Boomerangs are peptides that adopt a boomerang-like structure in complex with LPS. β-Boomerang was designed de novo as an attempt to disrupt the LPS outer membrane as a mode of action (Bhattacharjya et al. 2007; Bhunia et al. 2009). To further improve the antimicrobial properties and LPS neutralization of β-boomerang peptide, Mohanram and Bhattacharjya have further modified the most potent second generation β-boomerang peptide, YI12WF (sequence = YVLWKRKRFIFI-amide) as a starting template. Butanoic acid and octanoic acid were acylated at the N-termini. In addition, residue I11 was replaced by residue cystein to prepare a multivalent β-boomerangs linked by a disulphide bridge. To improve water solubility, a lysine residue was introduced into the cationic loop of the YI12WF, and the new peptide was designated as YI13C (sequence = YVLWKRKRKFCFI-amide). There are three new lipopeptides were used in the study: C4YI13C (sequence = C4-YVLWKRKRKFCFI), C8YI13C (sequence = C8-YVLWKRKRKFCFI) and C8YI13CAA (sequence = C8-YVLAKRKRKACFI). C8YI13CAA was designed to investigate the role of aromatic amino acid in antimicrobial activity, as alanine residues were used to replace tryptophan and phenylalanine residues. Antimicrobial assay showed that both C4YI13C and C8YI13C improved antibacterial activity of the non-acylated analog against Gram-negative bacteria, whereas only C8YI13C could improve the antibacterial activity against Gram-positive bacteria. Poor antimicrobial activity was observed for C8YI13CAA against both Gram-positive and Gram-negative bacteria, signified that aromatic residues were important in determining activity of the β-boomerang peptides. Biophysical studies such as LPS neutralization assay, NPN outer membrane permeabilization assay and FITC-LPS dissociation assay showed that acylated analogs were more active to interact and neutralize LPS (Mohanram and Bhattacharjya 2014).
Kamysz et al. has designed three multivalent peptides based on dimerization of peptides via intermolecular disulphide bond. In this study, two monomers were used: synthetic N-terminal peptide of human LF (sequence: ACWQVSRRRRG, designated as peptide I) and a short lipopeptide (COO-C12, where O = ornithine, designated as peptide III). Then, homo- and heterodimers were created by intermolecular disulphide bonds: peptide II and peptide IV were homo-dimers of peptide I and peptide III, respectively, while heterodimer V was formed by intermolecular disulphide bond of monomers peptide I and III. Antimicrobial assays were performed against Gram-positive bacteria, Gram-negative bacteria and fungi. All the tested micro-organisms were human pathogens. The results show that peptide III was the most effective compound (MICs = 2–8 µg/mL), but also the most haemolytic (16 µg/mL). Both homodimeric forms of peptides I and III exerted the same antimicrobial and haemolytic activity as their monomeric forms. Interestingly, heterodimer peptide V exhibited similar antimicrobial activities to the peptide III, but it was much less haemolytic (128 µg/mL). All compounds were relatively inactive against Candida albicans. Taken together, heterodimerization of antimicrobial lipopeptides via intermolecular disulphide bond might be a powerful modification in design of antimicrobial peptides for improved properties (Kamysz et al. 2015).
Dendrimer formation is also another approach to obtain a multivalent form of peptide. Janiszewska et al. has synthesized a series of dendrimers using the Boc chemistry in solution. The structure of the dendrimers used in the study is shown in Fig. 14. In general, the dendrimers could be classified into two categories: Group A with more branched and group B with less branched (Fig. 14). Compounds from group A have more branched structure of the amphiphilic heads, while compounds from group B have long lipophilic arms, terminated with 2-chlorocarbobenzoxy groups (2-Cl-Z). Two lipid moieties were used in this study: octyl (C8) and dodecyl (C12) chain length. Antimicrobial assay showed that a significant enhancement of antimicrobial potency along with increased selectivity against Candida strains was detected for derivatives with the C12 residues (MIC 7–8 µM). These compounds exhibited similar antimicrobial activities against Gram-positive and Gram-negative bacteria as their C8-analogs. Biophysical analysis showed that these compounds induced potassium leakage from fungal cells and induced cell death. This was further proven by SEM microscopy, which shows that the morphological of fungal cells were disrupted. The authors also found that the C12-dendrimers inhibited activity of candida β(1,3)-glucan synthase in a concentration-dependant manner (Janiszewska et al. 2012).

Structure of the dendrimers designed and synthesized by Janiszewska et al. The dendrimers could be divided into two main groups: General structure A with more branched and general structure B with less branched. General structure A was represented by a compound with R1 = 2-Cl-Z, R2 = NH2, R3 = 2-Cl-Z and R4 = NH2; General structure B was represented by a compound with R1 = NH2, R2 = 2-Cl-Z, R3 = NH2 and R4 = 2-Cl-Z. The figure was modified from (Janiszewska et al. 2012)
The summary of structure of the multivalent lipopeptides described above is listed on Table S4.
Stapled lipopeptide
Hydrocarbon stapling involves a ring-closing metathesis reaction to form a cyclic peptide in between two α-substituted non-natural amino acids containing unbranched side chains with terminal olefinic groups at i and i + 4 positions. Hydrocarbon stapling could stabilize a peptide’s secondary structure, enhance conformational rigidity and enhance peptide’s biological function (Schafmeister et al. 2000). Jenner et al. has synthesized two stapled lipopeptides analogs (labeled as C5-WWVXARAXRR* and C6-WWVXARAXRR*), based on lipopeptide 1 (C5-WWVAARAARR). In this study, their non-stapled analogs with the sequence of C5-WWVXARAXRR and C6-WWVXARAXRR were also studied. Antimicrobial assays showed that both stapled and non-stapled analogs exhibited potent antimicrobial activity against Gram-positive bacteria. The stapled peptide had a slightly enhanced antimicrobial activity (1.25–5 µM) than their non-stapled analogs (MICs = 5–10 µM). By contrast, the parent peptide, LP1 was inactive against all the bacteria studied up to 20 µM. However, none of these peptides were active against Gram-negative bacteria up to 20 µM. Both stapled and stapled analogs showed low and similar HC50 values (3.59–14.5 µM). CD revealed that LP1 adopts a random-coil confirmation while only slightly higher β-strand characteristics were observed for the stapled peptides in 30% TFE. Only little α-helix content was observed. In addition, the stapled peptides were also membrane targeting as they could disrupt phospholipid membrane. The synthesis and the peptides used in this study are shown in Fig. 15 (Jenner et al. 2017).

Hydrocarbon-stapling to form cyclic lipopeptide in between two amino acids containing free olefin group at position i and i + 4
Conclusion
Lipopeptides are very useful molecules as both of their fatty acid and peptide moieties could be modified for different applications. Particularly, they are potential candidates to be used in pharmaceutical, agriculture, plants and food industries, where antimicrobial properties are applied. This review has described and discussed different designs and chemical approaches to obtain potent synthetic lipopeptides against a panel of pathogens, such as bacteria, fungi, biofilm since last 5 years (since 2012). In general, modifying a peptide via acylation and lipidation with an appropriate lipid length (carbon number 4–14) lead to better antimicrobial profile, including improved antimicrobial activity, improved antimicrobial spectrum, improved time-kill kinetics, improved anti-biofilm activity, enhanced interaction and peptide penetration with bacterial membrane. In addition, fluorinated tails did not improve the antimicrobial activities significantly. The minimal charge required is +2 with at least two cationic amino acids in the peptide moiety. The exception is the ultrashort peptide designed and synthesized by Ghosh et al., where two lipid moieties are conjugated to a single lysine. Hemolytic activity also increases with number of hydrophobic residues in the peptide chain, reduced in charged and longer lipid moiety. For a more detailed description on the effects of each modification described in this review, please refer to Supporting information, Table S1–S4.
Although the number of promising candidates is very high and continues to increase, however, several main concerns for the lipopeptides such as systemic toxicity and poor pharmacokinetic profiles are still the major limitation for them to be applied clinically. Moreover, there are also limitations on the bioavailability of lipopeptides as they can be degraded easily by the body’s peptidase. Consequently, lipopeptides are mainly still limited to topical use (Mandal et al. 2013). Moving forward, more studies on development of new generation of lipopeptides with improved systemic toxicity and bioavailability and PK profiles are extremely important and necessary to further broaden the therapeutic applications of lipopeptides in the treatment of infectious diseases.
References
Albada HB, Prochnow P, Bobersky S, Langklotz S, Schriek P, Bandow JE, Metzler-Nolte N (2012) Tuning the activity of a short arg-trp antimicrobial peptide by lipidation of a C- or N-terminal lysine side-chain. ACS Med Chem Lett 3(12):980–984. doi:10.1021/ml300148v
Albada HB, Prochnow P, Bobersky S, Langklotz S, Bandow JE, Metzler-Nolte N (2013) Short antibacterial peptides with significantly reduced hemolytic activity can be identified by a systematic L-to-D exchange scan of their amino acid residues. ACS Comb Sci 15(11):585–592. doi:10.1021/co400072q
Avrahami D, Shai Y (2003) Bestowing antifungal and antibacterial activities by lipophilic acid conjugation to D, L-amino acid-containing antimicrobial peptides: a plausible mode of action. Biochemistry 42(50):14946–14956. doi:10.1021/bi035142v
Avrahami D, Shai Y (2004) A new group of antifungal and antibacterial lipopeptides derived from non-membrane active peptides conjugated to palmitic acid. J Biol Chem 279(13):12277–12285. doi:10.1074/jbc.M312260200
Azmi F, Elliott AG, Khalil ZG, Hussein WM, Kavanagh A, Huang JX, Quezada M, Blaskovich MA, Capon RJ, Cooper MA, Skwarczynski M, Toth I (2015) Self-assembling lipopeptides with a potent activity against Gram-positive bacteria, including multidrug resistant strains. Nanomedicine (Lond) 10(22):3359–3371
Azmi F, Elliott AG, Marasini N, Ramu S, Ziora Z, Kavanagh AM, Blaskovich MA, Cooper MA, Skwarczynski M, Toth I (2016a) Short cationic lipopeptides as effective antibacterial agents: design, physicochemical properties and biological evaluation. Bioorg Med Chem 24(10):2235–2241. doi:10.1016/j.bmc.2016.03.053
Azmi F, Skwarczynski M, Toth I (2016b) Towards the development of synthetic antibiotics: designs inspired by natural antimicrobial peptides. Curr Med Chem 23(41):4610–4624
Bai Y, Liu S, Jiang P, Zhou L, Li J, Tang C, Verma C, Mu Y, Beuerman RW, Pervushin K (2009) Structure-dependent charge density as a determinant of antimicrobial activity of peptide analogues of defensin. Biochemistry 48(30):7229–7239. doi:10.1021/bi900670d
Bai Y, Liu S, Li J, Lakshminarayanan R, Sarawathi P, Tang C, Ho D, Verma C, Beuerman RW, Pervushin K (2012) Progressive structuring of a branched antimicrobial peptide on the path to the inner membrane target. J Biol Chem 287(32):26606–26617. doi:10.1074/jbc.M112.363259
Baranska-Rybak W, Pikula M, Dawgul M, Kamysz W, Trzonkowski P, Roszkiewicz J (2013) Safety profile of antimicrobial peptides: camel, citropin, protegrin, temporin a and lipopeptide on HaCaT keratinocytes. Acta Pol Pharm 70(5):795–801
Bhattacharjya S, Domadia PN, Bhunia A, Malladi S, David SA (2007) High-resolution solution structure of a designed peptide bound to lipopolysaccharide: transferred Nuclear Overhauser effects, micelle selectivity, and anti-endotoxic activity. Biochemistry 46(20):5864–5874. doi:10.1021/bi6025159
Bhunia A, Mohanram H, Domadia PN, Torres J, Bhattacharjya S (2009) Designed beta-boomerang antiendotoxic and antimicrobial peptides: structures and activities in lipopolysaccharide. J Biol Chem 284(33):21991–22004. doi:10.1074/jbc.M109.013573
Bionda N, Fleeman RM, Shaw LN, Cudic P (2013) Effect of ester to amide or N-methylamide substitution on bacterial membrane depolarization and antibacterial activity of novel cyclic lipopeptides. ChemMedChem 8(8):1394–1402. doi:10.1002/cmdc.201300173
Bionda N, Fleeman RM, de la Fuente-Nunez C, Rodriguez MC, Reffuveille F, Shaw LN, Pastar I, Davis SC, Hancock RE, Cudic P (2016) Identification of novel cyclic lipopeptides from a positional scanning combinatorial library with enhanced antibacterial and antibiofilm activities. Eur J Med Chem 108:354–363. doi:10.1016/j.ejmech.2015.11.032
Biswas S, Brunel JM, Dubus JC, Reynaud-Gaubert M, Rolain JM (2012) Colistin: an update on the antibiotic of the 21st century. Expert Rev Anti Infect Ther 10(8):917–934. doi:10.1586/eri.12.78
Breukink E, Wiedemann I, van Kraaij C, Kuipers OP, Sahl HG, de Kruijff B (1999) Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 286(5448):2361–2364
Cai Y, Lee W, Kwa AL (2015) Polymyxin B versus colistin: an update. Expert Rev Anti Infect Ther 13(12):1481–1497. doi:10.1586/14787210.2015.1093933
Catiau L, Traisnel J, Delval-Dubois V, Chihib NE, Guillochon D, Nedjar-Arroume N (2011) Minimal antimicrobial peptidic sequence from hemoglobin alpha-chain: KYR. Peptides 32(4):633–638. doi:10.1016/j.peptides.2010.12.016
Citron DM, Tyrrell KL, Merriam CV, Goldstein EJ (2012) In vitro activities of CB-183,315, vancomycin, and metronidazole against 556 strains of Clostridium difficile, 445 other intestinal anaerobes, and 56 Enterobacteriaceae species. Antimicrob Agents Chemother 56(3):1613–1615. doi:10.1128/AAC.05655-11
Cochrane SA, Vederas JC (2016) Lipopeptides from Bacillus and Paenibacillus spp.: a gold mine of antibiotic candidates. Med Res Rev 36(1):4–31. doi:10.1002/med.21321
Cochrane SA, Lohans CT, Brandelli JR, Mulvey G, Armstrong GD, Vederas JC (2014) Synthesis and structure-activity relationship studies of N-terminal analogues of the antimicrobial peptide tridecaptin A(1). J Med Chem 57(3):1127–1131. doi:10.1021/jm401779d
Cooper MA, Shlaes D (2011) Fix the antibiotics pipeline. Nature 472(7341):32. doi:10.1038/472032a
De Zotti M, Biondi B, Park Y, Hahm KS, Crisma M, Toniolo C, Formaggio F (2012) Antimicrobial lipopeptaibol trichogin GA IV: role of the three Aib residues on conformation and bioactivity. Amino Acids 43(4):1761–1777. doi:10.1007/s00726-012-1261-7
De Zoysa GH, Cameron AJ, Hegde VV, Raghothama S, Sarojini V (2015) Antimicrobial peptides with potential for biofilm eradication: synthesis and structure activity relationship studies of battacin peptides. J Med Chem 58(2):625–639. doi:10.1021/jm501084q
Deris ZZ, Swarbrick JD, Roberts KD, Azad MA, Akter J, Horne AS, Nation RL, Rogers KL, Thompson PE, Velkov T, Li J (2014) Probing the penetration of antimicrobial polymyxin lipopeptides into gram-negative bacteria. Bioconjug Chem 25(4):750–760. doi:10.1021/bc500094d
Domalaon R, Yang X, O’Neil J, Zhanel GG, Mookherjee N, Schweizer F (2014) Structure-activity relationships in ultrashort cationic lipopeptides: the effects of amino acid ring constraint on antibacterial activity. Amino Acids 46(11):2517–2530. doi:10.1007/s00726-014-1806-z
Dugourd D, Yang H, Elliott M, Siu R, Clement JJ, Straus SK, Hancock RE, Rubinchik E (2011) Antimicrobial properties of MX-2401, an expanded-spectrum lipopeptide active in the presence of lung surfactant. Antimicrob Agents Chemother 55(8):3720–3728. doi:10.1128/AAC.00322-11
Epand RM (1997) Biophysical studies of lipopeptide-membrane interactions. Biopolymers 43(1):15–24. doi:10.1002/(SICI)1097-0282(1997)43:1<15:AID-BIP3>3.0.CO;2-3
Falagas ME, Kasiakou SK (2006) Toxicity of polymyxins: a systematic review of the evidence from old and recent studies. Crit Care 10(1):R27
Fang Y, Zhong W, Wang Y, Xun T, Lin D, Liu W, Wang J, Lv L, Liu S, He J (2014) Tuning the antimicrobial pharmacophore to enable discovery of short lipopeptides with multiple modes of action. Eur J Med Chem 83:36–44. doi:10.1016/j.ejmech.2014.06.003
Ferguson EL, Azzopardi E, Roberts JL, Walsh TR, Thomas DW (2014) Dextrin–colistin conjugates as a model bioresponsive treatment for multidrug resistant bacterial infections. Mol Pharm 11(12):4437–4447. doi:10.1021/mp500584u
Findlay B, Zhanel GG, Schweizer F (2012) Investigating the antimicrobial peptide ‘window of activity’ using cationic lipopeptides with hydrocarbon and fluorinated tails. Int J Antimicrob Agents 40(1):36–42. doi:10.1016/j.ijantimicag.2012.03.013
Ghosh C, Konai MM, Sarkar P, Samaddar S, Haldar J (2016) Designing simple lipidated lysines: bifurcation imparts selective antibacterial activity. ChemMedChem. doi:10.1002/cmdc.201600400
Gifford JL, Hunter HN, Vogel HJ (2005) Lactoferricin: a lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell Mol Life Sci 62(22):2588–2598. doi:10.1007/s00018-005-5373-z
Giuliani A, Rinaldi AC (2011) Beyond natural antimicrobial peptides: multimeric peptides and other peptidomimetic approaches. Cell Mol Life Sci 68(13):2255–2266. doi:10.1007/s00018-011-0717-3
Goldberg K, Sarig H, Zaknoon F, Epand RF, Epand RM, Mor A (2013) Sensitization of gram-negative bacteria by targeting the membrane potential. FASEB J 27(9):3818–3826. doi:10.1096/fj.13-227942
Grau-Campistany A, Manresa A, Pujol M, Rabanal F, Cajal Y (2016) Tryptophan-containing lipopeptide antibiotics derived from polymyxin B with activity against Gram positive and Gram negative bacteria. Biochim Biophys Acta 1858(2):333–343. doi:10.1016/j.bbamem.2015.11.011
Greber KE, Dawgul M, Kamysz W, Sawicki W, Lukasiak J (2014) Biological and surface-active properties of double-chain cationic amino acid-based surfactants. Amino Acids 46(8):1893–1898. doi:10.1007/s00726-014-1744-9
Grossman TH, O’Brien W, Kerstein KO, Sutcliffe JA (2015) Eravacycline (TP-434) is active in vitro against biofilms formed by uropathogenic Escherichia coli. Antimicrob Agents Chemother 59(4):2446–2449. doi:10.1128/AAC.04967-14
Hamley IW (2015) Lipopeptides: from self-assembly to bioactivity. Chem Commun (Camb) 51(41):8574–8583. doi:10.1039/c5cc01535a
Hanberger H, Walther S, Leone M, Barie PS, Rello J, Lipman J, Marshall JC, Anzueto A, Sakr Y, Pickkers P, Felleiter P, Engoren M, Vincent JL (2011) Increased mortality associated with methicillin-resistant Staphylococcus aureus (MRSA) infection in the intensive care unit: results from the EPIC II study. Int J Antimicrob Agents 38(4):331–335. doi:10.1016/j.ijantimicag.2011.05.013
Hancock RE, Sahl HG (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 24(12):1551–1557. doi:10.1038/nbt1267
Haug BE, Stensen W, Kalaaji M, Rekdal O, Svendsen JS (2008) Synthetic antimicrobial peptidomimetics with therapeutic potential. J Med Chem 51(14):4306–4314. doi:10.1021/jm701600a
He J, Eckert R, Pharm T, Simanian MD, Hu C, Yarbrough DK, Qi F, Anderson MH, Shi W (2007) Novel synthetic antimicrobial peptides against Streptococcus mutans. Antimicrob Agents Chemother 51(4):1351–1358. doi:10.1128/AAC.01270-06
Horn JN, Sengillo JD, Lin D, Romo TD, Grossfield A (2012) Characterization of a potent antimicrobial lipopeptide via coarse-grained molecular dynamics. Biochim Biophys Acta 1818(2):212–218. doi:10.1016/j.bbamem.2011.07.025
Hu Y, Amin MN, Padhee S, Wang RE, Qiao Q, Bai G, Li Y, Mathew A, Cao C, Cai J (2012) Lipidated peptidomimetics with improved antimicrobial activity. ACS Med Chem Lett 3(8):683–686. doi:10.1021/ml3001215
Huang E, Yousef AE (2014) Paenibacterin, a novel broad-spectrum lipopeptide antibiotic, neutralises endotoxins and promotes survival in a murine model of Pseudomonas aeruginosa-induced sepsis. Int J Antimicrob Agents 44(1):74–77. doi:10.1016/j.ijantimicag.2014.02.018
Ifrah D, Doisy X, Ryge TS, Hansen PR (2005) Structure-activity relationship study of anoplin. J Pept Sci 11(2):113–121. doi:10.1002/psc.598
Jammal J, Zaknoon F, Kaneti G, Goldberg K, Mor A (2015) Sensitization of Gram-negative bacteria to rifampin and OAK combinations. Sci Rep 5:9216. doi:10.1038/srep09216
Janiszewska J, Sowinska M, Rajnisz A, Solecka J, Lacka I, Milewski S, Urbanczyk-Lipkowska Z (2012) Novel dendrimeric lipopeptides with antifungal activity. Bioorg Med Chem Lett 22(3):1388–1393. doi:10.1016/j.bmcl.2011.12.051
Jenner ZB, Crittenden CM, Gonzalez M, Brodbelt JS, Bruns KA (2017) Hydrocarbon-stapled lipopeptides exhibit selective antimicrobial activity. Biopolymers. doi:10.1002/bip.23006
Jerala R (2007) Synthetic lipopeptides: a novel class of anti-infectives. Expert Opin Investig Drugs 16(8):1159–1169. doi:10.1517/13543784.16.8.1159
Joshi S, Dewangan RP, Yadav S, Rawat DS, Pasha S (2012) Synthesis, antibacterial activity and mode of action of novel linoleic acid-dipeptide-spermidine conjugates. Org Biomol Chem 10(41):8326–8335. doi:10.1039/c2ob26393a
Kamysz E, Sikorska E, Dawgul M, Tyszkowski R, Kamysz W (2015) Influence of dimerization of lipopeptide Laur-Orn-Orn-Cys-NH2 and an N-terminal peptide of human lactoferricin on biological activity. Int J Pept Res Ther 21:39–46. doi:10.1007/s10989-014-9423-y
Kaur P, Li Y, Cai J, Song L (2016) Selective membrane disruption mechanism of an antibacterial gamma-AApeptide defined by EPR spectroscopy. Biophys J 110(8):1789–1799. doi:10.1016/j.bpj.2016.02.038
Kim SM, Kim JM, Joshi BP, Cho H, Lee KH (2009) Indolicidin-derived antimicrobial peptide analogs with greater bacterial selectivity and requirements for antibacterial and hemolytic activities. Biochim Biophys Acta 1794(2):185–192. doi:10.1016/j.bbapap.2008.10.009
Knight-Connoni V, Mascio C, Chesnel L, Silverman J (2016) Discovery and development of surotomycin for the treatment of Clostridium difficile. J Ind Microbiol Biotechnol 43(2–3):195–204. doi:10.1007/s10295-015-1714-6
Koh JJ, Lin H, Caroline V, Chew YS, Pang LM, Aung TT, Li J, Lakshminarayanan R, Tan DT, Verma C, Tan AL, Beuerman RW, Liu S (2015) N-lipidated peptide dimers: effective antibacterial agents against gram-negative pathogens through lipopolysaccharide permeabilization. J Med Chem 58(16):6533–6548. doi:10.1021/acs.jmedchem.5b00628
Konno K, Hisada M, Fontana R, Lorenzi CC, Naoki H, Itagaki Y, Miwa A, Kawai N, Nakata Y, Yasuhara T, Neto JR, de Azevedo WF Jr, Palma MS, Nakajima T (2001) Anoplin, a novel antimicrobial peptide from the venom of the solitary wasp Anoplius samariensis. Biochim Biophys Acta 1550(1):70–80
Koopmans T, Wood TM, t Hart P, Kleijn LH, Hendrickx AP, Willems RJ, Breukink E, Martin NI (2015) Semisynthetic lipopeptides derived from nisin display antibacterial activity and lipid II binding on par with that of the parent compound. J Am Chem Soc 137(29):9382–9389. doi:10.1021/jacs.5b04501
Laverty G, Gorman SP, Gilmore BF (2015) Biofilm eradication kinetics of the ultrashort lipopeptide C12-OOWW-NH2 utilizing a modified MBEC Assay(). Chem Biol Drug Des 85(5):645–652. doi:10.1111/cbdd.12441
Li Y, Smith C, Wu H, Padhee S, Manoj N, Cardiello J, Qiao Q, Cao C, Yin H, Cai J (2014a) Lipidated cyclic gamma-AApeptides display both antimicrobial and anti-inflammatory activity. ACS Chem Biol 9(1):211–217. doi:10.1021/cb4006613
Li Y, Smith C, Wu H, Teng P, Shi Y, Padhee S, Jones T, Nguyen AM, Cao C, Yin H, Cai J (2014b) Short antimicrobial lipo-alpha/gamma-AA hybrid peptides. ChemBioChem 15(15):2275–2280. doi:10.1002/cbic.201402264
Li J, Koh JJ, Liu S, Lakshminarayanan R, Verma CS, Beuerman RW (2017) Membrane active antimicrobial peptides: translating mechanistic insights to design. Front Neurosci 11:73. doi:10.3389/fnins.2017.00073
Lin D, Grossfield A (2014) Thermodynamics of antimicrobial lipopeptide binding to membranes: origins of affinity and selectivity. Biophys J 107(8):1862–1872. doi:10.1016/j.bpj.2014.08.026
Linington RG, Clark BR, Trimble EE, Almanza A, Urena LD, Kyle DE, Gerwick WH (2009) Antimalarial peptides from marine cyanobacteria: isolation and structural elucidation of gallinamide A. J Nat Prod 72(1):14–17. doi:10.1021/np8003529
Liu S, Zhou L, Li J, Suresh A, Verma C, Foo YH, Yap EP, Tan DT, Beuerman RW (2008) Linear analogues of human beta-defensin 3: concepts for design of antimicrobial peptides with reduced cytotoxicity to mammalian cells. ChemBioChem 9(6):964–973. doi:10.1002/cbic.200700560
Liu J, Luo C, Smith PA, Chin JK, Page MG, Paetzel M, Romesberg FE (2011) Synthesis and characterization of the arylomycin lipoglycopeptide antibiotics and the crystallographic analysis of their complex with signal peptidase. J Am Chem Soc 133(44):17869–17877. doi:10.1021/ja207318n
Lohan S, Cameotra SS, Bisht GS (2013) Systematic study of non-natural short cationic lipopeptides as novel broad-spectrum antimicrobial agents. Chem Biol Drug Des 82(5):557–566. doi:10.1111/cbdd.12182
Lohans CT, Huang Z, van Belkum MJ, Giroud M, Sit CS, Steels EM, Zheng J, Whittal RM, McMullen LM, Vederas JC (2012) Structural characterization of the highly cyclized lantibiotic paenicidin A via a partial desulfurization/reduction strategy. J Am Chem Soc 134(48):19540–19543. doi:10.1021/ja3089229
Lohans CT, van Belkum MJ, Cochrane SA, Huang Z, Sit CS, McMullen LM, Vederas JC (2014) Biochemical, structural, and genetic characterization of tridecaptin A(1), an antagonist of Campylobacter jejuni. ChemBioChem 15(2):243–249. doi:10.1002/cbic.201300595
Lundy FT, Nelson J, Lockhart D, Greer B, Harriott P, Marley JJ (2008) Antimicrobial activity of truncated alpha-defensin (human neutrophil peptide (HNP)-1) analogues without disulphide bridges. Mol Immunol 45(1):190–193
Luo S, Kang HS, Krunic A, Chen WL, Yang J, Woodard JL, Fuchs JR, Hyun Cho S, Franzblau SG, Swanson SM, Orjala J (2015) Trichormamides C and D, antiproliferative cyclic lipopeptides from the cultured freshwater cyanobacterium cf. Oscillatoria sp. UIC 10045. Bioorg Med Chem 23(13):3153–3162. doi:10.1016/j.bmc.2015.04.073
Magee TV, Brown MF, Starr JT, Ackley DC, Abramite JA, Aubrecht J, Butler A, Crandon JL, Dib-Hajj F, Flanagan ME, Granskog K, Hardink JR, Huband MD, Irvine R, Kuhn M, Leach KL, Li B, Lin J, Luke DR, MacVane SH, Miller AA, McCurdy S, McKim JM Jr, Nicolau DP, Nguyen TT, Noe MC, O’Donnell JP, Seibel SB, Shen Y, Stepan AF, Tomaras AP, Wilga PC, Zhang L, Xu J, Chen JM (2013) Discovery of Dap-3 polymyxin analogues for the treatment of multidrug-resistant Gram-negative nosocomial infections. J Med Chem 56(12):5079–5093. doi:10.1021/jm400416u
Makovitzki A, Avrahami D, Shai Y (2006) Ultrashort antibacterial and antifungal lipopeptides. Proc Natl Acad Sci USA 103(43):15997–16002
Malina A, Shai Y (2005) Conjugation of fatty acids with different lengths modulates the antibacterial and antifungal activity of a cationic biologically inactive peptide. Biochem J 390(Pt 3):695–702
Mandal SM, Barbosa AE, Franco OL (2013) Lipopeptides in microbial infection control: scope and reality for industry. Biotechnol Adv 31(2):338–345. doi:10.1016/j.biotechadv.2013.01.004
Mangoni ML, Shai Y (2011) Short native antimicrobial peptides and engineered ultrashort lipopeptides: similarities and differences in cell specificities and modes of action. Cell Mol Life Sci 68(13):2267–2280. doi:10.1007/s00018-011-0718-2
Mascio CT, Mortin LI, Howland KT, Van Praagh AD, Zhang S, Arya A, Chuong CL, Kang C, Li T, Silverman JA (2012) In vitro and in vivo characterization of CB-183,315, a novel lipopeptide antibiotic for treatment of Clostridium difficile. Antimicrob Agents Chemother 56(10):5023–5030. doi:10.1128/AAC.00057-12
Mascio CT, Chesnel L, Thorne G, Silverman JA (2014) Surotomycin demonstrates low in vitro frequency of resistance and rapid bactericidal activity in Clostridium difficile, Enterococcus faecalis, and Enterococcus faecium. Antimicrob Agents Chemother 58(7):3976–3982. doi:10.1128/AAC.00124-14
Mathew B, Nagaraj R (2015) Antimicrobial activity of human alpha-defensin 5 and its linear analogs: N-terminal fatty acylation results in enhanced antimicrobial activity of the linear analogs. Peptides 71:128–140. doi:10.1016/j.peptides.2015.07.009
Meena KR, Kanwar SS (2015) Lipopeptides as the antifungal and antibacterial agents: applications in food safety and therapeutics. Biomed Res Int 2015:473050. doi:10.1155/2015/473050
Mishra B, Lushnikova T, Wang G (2015) Small lipopeptides possess anti-biofilm capability comparable to daptomycin and vancomycin. RSC Adv 5(73):59758–59769. doi:10.1039/C5RA07896B
Mohanram H, Bhattacharjya S (2014) beta-Boomerang antimicrobial and antiendotoxic peptides: lipidation and disulfide bond effects on activity and structure. Pharmaceuticals (Basel) 7(4):482–501. doi:10.3390/ph7040482
Monroc S, Badosa E, Besalu E, Planas M, Bardaji E, Montesinos E, Feliu L (2006a) Improvement of cyclic decapeptides against plant pathogenic bacteria using a combinatorial chemistry approach. Peptides 27(11):2575–2584
Monroc S, Badosa E, Feliu L, Planas M, Montesinos E, Bardaji E (2006b) De novo designed cyclic cationic peptides as inhibitors of plant pathogenic bacteria. Peptides 27(11):2567–2574
Nasompag S, Dechsiri P, Hongsing N, Phonimdaeng P, Daduang S, Klaynongsruang S, Camesano TA, Patramanon R (2015) Effect of acyl chain length on therapeutic activity and mode of action of the CX-KYR-NH2 antimicrobial lipopeptide. Biochim Biophys Acta 1848(10 Pt A):2351–2364. doi:10.1016/j.bbamem.2015.07.004
Nation RL, Velkov T, Li J (2014) Colistin and polymyxin B: Peas in a pod, or chalk and cheese? Clin Infect Dis 59(1):88–94. doi:10.1093/cid/ciu213
Niu Y, Padhee S, Wu H, Bai G, Qiao Q, Hu Y, Harrington L, Burda WN, Shaw LN, Cao C, Cai J (2012) Lipo-gamma-AApeptides as a new class of potent and broad-spectrum antimicrobial agents. J Med Chem 55(8):4003–4009. doi:10.1021/jm300274p
Niu Y, Wu H, Li Y, Hu Y, Padhee S, Li Q, Cao C, Cai J (2013) AApeptides as a new class of antimicrobial agents. Org Biomol Chem 11(26):4283–4290. doi:10.1039/c3ob40444g
Nkongolo S, Ni Y, Lempp FA, Kaufman C, Lindner T, Esser-Nobis K, Lohmann V, Mier W, Mehrle S, Urban S (2014) Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J Hepatol 60(4):723–731. doi:10.1016/j.jhep.2013.11.022
Pasetka CJ, Erfle DJ, Cameron DR, Clement JJ, Rubinchik E (2010) Novel antimicrobial lipopeptides with long in vivo half-lives. Int J Antimicrob Agents 35(2):182–185. doi:10.1016/j.ijantimicag.2009.10.013
Patel S, Ahmed S, Eswari JS (2015) Therapeutic cyclic lipopeptides mining from microbes: latest strides and hurdles. World J Microbiol Biotechnol 31(8):1177–1193. doi:10.1007/s11274-015-1880-8
Rabanal F, Grau-Campistany A, Vila-Farres X, Gonzalez-Linares J, Borras M, Vila J, Manresa A, Cajal Y (2015) A bioinspired peptide scaffold with high antibiotic activity and low in vivo toxicity. Sci Rep 5:10558. doi:10.1038/srep10558
Radzishevsky IS, Rotem S, Bourdetsky D, Navon-Venezia S, Carmeli Y, Mor A (2007) Improved antimicrobial peptides based on acyl-lysine oligomers. Nat Biotechnol 25(6):657–659
Resch A, Wilke M, Fink C (2009) The cost of resistance: incremental cost of methicillin-resistant Staphylococcus aureus (MRSA) in German hospitals. Eur J Health Econ 10(3):287–297. doi:10.1007/s10198-008-0132-3
Roberts TC, Schallenberger MA, Liu J, Smith PA, Romesberg FE (2011) Initial efforts toward the optimization of arylomycins for antibiotic activity. J Med Chem 54(14):4954–4963. doi:10.1021/jm1016126
Roberts JL, Cattoz B, Schweins R, Beck K, Thomas DW, Griffiths PC, Ferguson EL (2016) In vitro evaluation of the interaction of dextrin–colistin conjugates with bacterial lipopolysaccharide. J Med Chem 59(2):647–654. doi:10.1021/acs.jmedchem.5b01521
Sanchez-Gomez S, Ferrer-Espada R, Stewart PS, Pitts B, Lohner K, Martinez de Tejada G (2015) Antimicrobial activity of synthetic cationic peptides and lipopeptides derived from human lactoferricin against Pseudomonas aeruginosa planktonic cultures and biofilms. BMC Microbiol 15:137. doi:10.1186/s12866-015-0473-x
Schafmeister CE, Po J, Verdine GL (2000) An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc 122(24):5891–5892. doi:10.1021/ja000563a
Schneider T, Muller A, Miess H, Gross H (2014) Cyclic lipopeptides as antibacterial agents—potent antibiotic activity mediated by intriguing mode of actions. Int J Med Microbiol 304(1):37–43. doi:10.1016/j.ijmm.2013.08.009
Shankar SS, Benke SN, Nagendra N, Srivastava PL, Thulasiram HV, Gopi HN (2013) Self-assembly to function: design, synthesis, and broad spectrum antimicrobial properties of short hybrid E-vinylogous lipopeptides. J Med Chem 56(21):8468–8474. doi:10.1021/jm400884w
Siano A, Humpola MV, Rey MC, Simonetta A, Tonarelli GG (2011) Interaction of acylated and substituted antimicrobial peptide analogs with phospholipid-polydiacetylene vesicles. Correlation with their biological properties. Chem Biol Drug Des 78(1):85–93. doi:10.1111/j.1747-0285.2011.01099.x
Sikorska E, Dawgul M, Greber K, Ilowska E, Pogorzelska A, Kamysz W (2014) Self-assembly and interactions of short antimicrobial cationic lipopeptides with membrane lipids: ITC, FTIR and molecular dynamics studies. Biochim Biophys Acta 1838(10):2625–2634. doi:10.1016/j.bbamem.2014.06.016
Slootweg JC, van Schaik TB, Quarles van Ufford HL, Breukink E, Liskamp RM, Rijkers DT (2013) Improving the biological activity of the antimicrobial peptide anoplin by membrane anchoring through a lipophilic amino acid derivative. Bioorg Med Chem Lett 23(13):3749–3752. doi:10.1016/j.bmcl.2013.05.002
Snydman DR, Jacobus NV, McDermott LA (2012) Activity of a novel cyclic lipopeptide, CB-183,315, against resistant Clostridium difficile and other gram-positive aerobic and anaerobic intestinal pathogens. Antimicrob Agents Chemother 56(6):3448–3452. doi:10.1128/AAC.06257-11
Straus SK, Hancock RE (2006) Mode of action of the new antibiotic for gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim Biophys Acta 1758(9):1215–1223
t Hart P, Kleijn LH, de Bruin G, Oppedijk SF, Kemmink J, Martin NI (2014) A combined solid- and solution-phase approach provides convenient access to analogues of the calcium-dependent lipopeptide antibiotics. Org Biomol Chem 12(6):913–918. doi:10.1039/c3ob42238k
Teng P, Huo D, Nimmagadda A, Wu J, She F, Su M, Lin X, Yan J, Cao A, Xi C, Hu Y, Cai J (2016) Small antimicrobial agents based on acylated reduced amide scaffold. J Med Chem 59(17):7877–7887. doi:10.1021/acs.jmedchem.6b00640
Varkey J, Nagaraj R (2005) Antibacterial activity of human neutrophil defensin HNP-1 analogs without cysteines. Antimicrob Agents Chemother 49(11):4561–4566
Velkov T, Thompson PE, Nation RL, Li J (2010) Structure–activity relationships of polymyxin antibiotics. J Med Chem 53(5):1898–1916. doi:10.1021/jm900999h
Velkov T, Roberts KD, Nation RL, Thompson PE, Li J (2013) Pharmacology of polymyxins: new insights into an ‘old’ class of antibiotics. Future Microbiol 8(6):711–724. doi:10.2217/fmb.13.39
Velkov T, Roberts KD, Nation RL, Wang J, Thompson PE, Li J (2014) Teaching ‘old’ polymyxins new tricks: new-generation lipopeptides targeting gram-negative ‘superbugs’. ACS Chem Biol 9(5):1172–1177. doi:10.1021/cb500080r
Vila S, Badosa E, Montesinos E, Planas M, Feliu L (2016) Synthetic cyclolipopeptides selective against microbial, plant and animal cell targets by incorporation of d-amino acids or histidine. PLoS ONE 11(3):e0151639. doi:10.1371/journal.pone.0151639
Yamamura H, Suzuki K, Uchibori K, Miyagawa A, Kawai M, Ohmizo C, Katsu T (2012) Mimicking an antimicrobial peptide polymyxin B by use of cyclodextrin. Chem Commun (Camb) 48(6):892–894. doi:10.1039/c1cc16369h
Yin N, Li J, He Y, Herradura P, Pearson A, Mesleh MF, Mascio CT, Howland K, Steenbergen J, Thorne GM, Citron D, Van Praagh AD, Mortin LI, Keith D, Silverman J, Metcalf C (2015) Structure–activity relationship studies of a series of semisynthetic lipopeptides leading to the discovery of Surotomycin, a novel cyclic lipopeptide being developed for the treatment of Clostridium difficile-associated diarrhea. J Med Chem 58(12):5137–5142. doi:10.1021/acs.jmedchem.5b00366
Zhou L, Liu SP, Chen LY, Li J, Ong LB, Guo L, Wohland T, Tang CC, Lakshminarayanan R, Mavinahalli J, Verma C, Beuerman RW (2011) The structural parameters for antimicrobial activity, human epithelial cell cytotoxicity and killing mechanism of synthetic monomer and dimer analogues derived from hBD3 C-terminal region. Amino Acids 40(1):123–133. doi:10.1007/s00726-010-0565-8
Zweytick D, Deutsch G, Andra J, Blondelle SE, Vollmer E, Jerala R, Lohner K (2011) Studies on lactoferricin-derived Escherichia coli membrane-active peptides reveal differences in the mechanism of N-acylated versus nonacylated peptides. J Biol Chem 286(24):21266–21276. doi:10.1074/jbc.M110.195412
Zweytick D, Japelj B, Mileykovskaya E, Zorko M, Dowhan W, Blondelle SE, Riedl S, Jerala R, Lohner K (2014) N-acylated peptides derived from human lactoferricin perturb organization of cardiolipin and phosphatidylethanolamine in cell membranes and induce defects in Escherichia coli cell division. PLoS ONE 9(3):e90228. doi:10.1371/journal.pone.0090228
Acknowledgements
This work was supported by the Singhealth foundation research grant (SHF/FG691S/2016), National Medical Research Council (NMRC/CBRG/0080/2015 and NMRC/TCR/R1018) and the Singhealth Foundation (SHF/FG538P/2013).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: J. D. Wade.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Koh, JJ., Lin, S., Beuerman, R.W. et al. Recent advances in synthetic lipopeptides as anti-microbial agents: designs and synthetic approaches. Amino Acids 49, 1653–1677 (2017). https://doi.org/10.1007/s00726-017-2476-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-017-2476-4