
Abstract
The metabolic roles of mitochondria go far beyond serving exclusively as the major producer of ATP in tissues and cells. Evidence has shown that mitochondria may function as a key regulator of skeletal muscle fiber types and overall well-being. Maintaining skeletal muscle mitochondrial content and function is important for sustaining health throughout the lifespan. Of great importance, β-hydroxy-β-methylbutyrate (HMB, a metabolite of l-leucine) has been proposed to enhance the protein deposition and efficiency of mitochondrial biogenesis in skeletal muscle, as well as muscle strength in both exercise and clinical settings. Specifically, dietary supplementation with HMB increases the gene expression of peroxisome proliferator-activated receptor gamma co-activator 1-alpha (PGC-1α), which represents an upstream inducer of genes of mitochondrial metabolism, coordinates the expression of both nuclear- and mitochondrion-encoded genes in mitochondrial biogenesis. Additionally, PGC-1α plays a key role in the transformation of skeletal muscle fiber type, leading to a shift toward type I muscle fibers that are rich in mitochondria and have a high capacity for oxidative metabolism. As a nitrogen-free metabolite, HMB holds great promise to improve skeletal muscle mass and function, as well as whole-body health and well-being of animals and humans.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The mitochondrion has key roles in cell metabolism. The most important function of this organelle is to oxidize energy substrates and produce ATP via the Krebs cycle and the electron transport system (Joseph et al. 2012; Dai et al. 2013). Additionally, the mitochondria play a critical role in maintaining health via their involvement in regulating skeletal muscle fiber size, metabolism, and function (Russell et al. 2014). Particularly, mitochondria may act as a new mediator of skeletal muscle fiber types (Venhoff et al. 2012). Oxidative capacity of fibers is strongly linked with muscle health and overall well-being (Wenz et al. 2009). The protective effects of enhanced mitochondrial oxidative capacity in disease states, such as obesity and diabetes, are largely ascribed to enhanced mitochondrial content and function (Bazer et al. 2015; Scheffler et al. 2014, Tekwe et al. 2013). An increase in mitochondrial mass and/or number is termed “mitochondrial biogenesis.” Multiple genes expressed in mitochondria, such as silent information regulator transcript 1 (SIRT1), AMP-activated protein kinase (AMPK), and peroxisome proliferator-activated receptor gamma co-activator 1-alpha (PGC-1α), may be involved in the regulation of mitochondrial biogenesis and fiber-type transformation (Lin et al. 2002). Therefore, mitochondria in skeletal muscle can improve muscle health and overall well-being through regulating skeletal muscle fiber types and expression of key genes involved in this process.
β-Hydroxy-β-methylbutyrate (HMB), a metabolite of the nutritionally essential branched-chain amino acid (BCAA) leucine (Wu 2013), has been proposed as a nutritional supplement to enhance skeletal muscle mass and strength in both exercise and clinical settings by promoting mitochondrial biogenesis ad fatty acid oxidation (Wilson et al. 2008; Stancliffe 2012). Therefore, this review aims to focus on the relationship between mitochondria and skeletal muscle health, as well as the effects of dietary HMB supplementation on promoting mitochondrial biogenesis and protecting against skeletal muscle loss.
Mitochondria: an overview
Mitochondria are highly organized and dynamic organelles, performing myriad roles in bioenergetics, metabolism, and cell signaling. The primary function of mitochondria is ATP production from the oxidation of energy substrates (fatty acids, glucose, and amino acids) (Dai et al. 2013). The reducing equivalents (NADH and FADH2) generated via the oxidation of acetyl-CoA in the mitochondrial Krebs cycle are oxidized to water via the electron transport system. This biochemical event of oxidative phosphorylation produces most ATP in cells that possess mitochondria. Moreover, mitochondria contain enzymes that are essential for the synthesis of cholesterol and play a key role in amino acid metabolism (Wu 2013). In addition, the controlled generation of reactive oxygen species (ROS) by the mitochondria is crucial for both cell signaling and apoptosis (Hock and Kralli 2009; Stancliffe 2012).
Due to these essential roles of mitochondria, there is a tight regulation of mitochondrial mass and function. In response to physical activity, availability of nutrients, and anabolic hormones, this well-coordinated regulation of mitochondrial dynamics permits mitochondria to fulfill their physiological functions through mitochondrial biogenesis. It is an essential biological process required for growth and development, while meeting energy requirements of the cell (Stefano et al. 2012). Of note, the tight control of mitochondrial biogenesis is mainly through transcriptional regulation, particularly, through the synchronized transcription of mitochondrial genes in both the nucleus and the mitochondria (Poyton and McEwen 1996; Mootha et al. 2003). Given these essential roles of the mitochondria in the cell, it is no surprise that damage to mitochondria ultimately leads to disease (Duchen 2004). Abnormal mitochondrial metabolism or activity is termed “mitochondrial dysfunction.” An increasing body of literature identifies mitochondrial dysfunction as a contributing factor to numerous diseases, including liver disease, cardiovascular disease, obesity, diabetes, cancer, and dementia (Zhang et al. 2003; Duchen 2004; Joseph et al. 2012; Stancliffe 2012). Moreover, the accumulation of mitochondrial damage and the resultant oxidative stress has been regarded as a potential mechanism responsible for aging (Weber and Reichert 2010). Previous studies have demonstrated that mitochondrial biogenesis can ameliorate metabolic disorders and promote longevity by decreasing risk of age-related diseases (Filhiol 2012). Accordingly, maintenance of abundant, functional mitochondria is fundamental to life. Some key genes involved in the regulation of mitochondrial biogenesis play important roles in maintaining abundant and functional mitochondria. Therefore, we will discuss key genes involved in the regulation of mitochondrial biogenesis in more detail below.
Key genes involved in the regulation of mitochondrial biogenesis
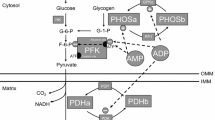
In response to a stimulus such as excise or skeletal muscle contractile activity, intracellular Ca2+ and AMP levels increase resulting in the activation of signaling molecules. These signaling pathways converge to promote mitochondrial biogenesis (Fig. 1; Joseph et al. 2012).

A proposed model of mitochondrial biogenesis. In response to a stimulus such as excise or skeletal muscle contractile activity, intracellular Ca2+ and AMP levels increase, resulting in the activation of signaling molecules. These signaling pathways converge on peroxisome proliferator-activated receptor-γ co-activator-1α (PGC-1α) to promote mitochondrial biogenesis. PGC-1α activates the expression of oxidative phosphorylation (OXPHOS) genes and the nuclear respiratory factor-1 and 2 (NRF1–2). NRF-1 and NRF-2 bind and upregulate the expression of mitochondrial transcription factor A (TFAM). Additionally, PGC-1α activates silent information regulator transcript 3 (SIRT3) and SIRT5 in peroxisome proliferator-activated receptor α (PPARα)- and estrogen-related receptor α (ERRα)-dependent manners. Consequently, mitochondrial biogenesis, oxidative capacity, and ATP production within the muscle cell are enhanced by PGC-1α
PGC-1α
PGC-1α is often regarded as the master regulator of mitochondrial biogenesis because of its ability to stimulate the expression of mitochondrial genes (Hock and Kralli 2009). PGC-1α represents an upstream inducer of genes of mitochondrial metabolism by positively influencing some hormone nuclear receptors and nuclear transcription factors, such as peroxisome proliferator-activated receptor α (PPARα), estrogen-related receptor α (ERRα), and nuclear respiratory factor 1–2 (NRF1–2) (Wu et al. 1999; Huss et al. 2004; Narkar et al. 2008; Scarpulla 2011). There is evidence that expression of PGC-1α in cells is enhanced by dietary supplementation with l-arginine (Fu et al. 2005). NFR-1 induces the expression of a segment of the mitochondrial genome, specifically the genes that encode oxidative phosphorylation components and mitochondrial ribosomal proteins (Virbasius et al. 1993; Scarpulla 2008). Co-activation of NRF1 and NRF2 by PGC-1α also stimulates mitochondrial transcription factor A (TFAM) expression (Wu et al. 1999). TFAM is a nucleus-encoded gene that is partially responsible for the coordinated transcription of the nuclear and mitochondrial genomes such as mitochondrial DNA during mitochondrial biogenesis (Pagliarini et al. 2008; Scarpulla 2008). Moreover, PGC-1α induces oxidative phosphorylation (OXPHOS) genes that involved in the final step of electron transport chain and ATP synthesis, thereby allowing for the complete oxidation of fatty acids to water and CO2 in the mitochondria (Wu et al. 1999; Koves et al. 2005). Thus, PGC-1α coordinates the expression of both nuclear- and mitochondrial-encoded genes in mitochondrial biogenesis (Yan et al. 2011).
Due to its vital role in mitochondrial biogenesis, PGC-1α overexpression in skeletal muscle leads to increased mitochondrial abundance and gene expression, and improved exercise performance (Lin et al. 2002; Calvo et al. 2008). Conversely, PGC-1α null mice show a decrease in mitochondrial gene expression and impaired mitochondrial function (Leone et al. 2005; Arany et al. 2006). Moreover, the PGC-1α activity is regulated by SIRT1 and AMPK, which are activated by an increase in cellular energy needs (Gerhart-Hines et al. 2007; Jager et al. 2007).
SIRTs
The SIRTs (SIRTs; SIRT1–7) are a family of NAD+-dependent enzymes that dynamically regulate mitochondrial function. Mammalian SIRTs belongs to class III histone deacetylates with specific subcellular localization and protein substrates (Rodgers et al. 2008). The enzymatic activity of the SIRTs is dependent on the NAD+/NADH ratio and is associated with cellular energy charge (Feldman et al. 2012). Function of SIRT1 is required for the stimulation of mitochondrial biogenesis in skeletal muscle (Price et al. 2012). Moreover, SIRT1 promotes mitochondrial biogenesis via deacetylation and, therefore, activation of PGC-1α (Rodgers et al. 2005; Gerhart-Hines et al. 2007).
Apart from SIRT1, SIRT3 and SIRT5 are also implicated in regulating mitochondrial function. Previous studies have shown that SIRT3 controls the global acetylation status of mitochondrial proteins, increases cellular ATP levels, and induces mitochondrial biogenesis. Moreover, PGC-1α has been reported to induce expression of SIRT3 in an ERRα-dependent manner (Lombard et al. 2007; Ahn et al. 2008; Hirschey et al. 2010; Kong et al. 2010). Recently, SIRT5 has also been identified as a novel factor that controls mitochondrial function (Buler et al. 2014). Overexpression of PGC-1α in mouse primary hepatocytes increases SIRT5 mRNA expression (fourfold) and its protein abundance in a PPARα- and ERRα-dependent manner. Additionally, SIRT5 is also under the modulation of AMPK, but in the opposite direction of PGC-1α. Overexpression of SIRT5 promotes ATP synthesis and oxygen consumption in HepG2 cells, but does not influence mitochondrial mass. Thus, SIRT5 may have a positive effect on oxidative phosphorylation (Buler et al. 2014). Overall, during mitochondrial biogenesis, SIRT1 induces the activation of PGC-1α that is pivotal to orchestrate the activation of a broad set of transcription factors and nuclear hormone receptors, leading to enhanced expression of the nucleus-encoded mitochondrial genes (Aquilano et al. 2010).
AMPK
AMPK is another key factor that mediates mitochondrial biogenesis and is a key element in maintaining energy homeostasis. AMPK responds to AMP, and hence it is sensitive to even discrete changes in cellular charge (Lage et al. 2008; Hardie et al. 2012). Apart from AMP, AMPK (Thr-172) can be phosphorylated and activated by a rise in intracellular Ca2+ via the Ca2+-activated kinase calmodulin-dependent kinase kinase-β (CaMKKβ). Compound C, an inhibitor of AMPK, markedly suppresses leucine’s effects on mitochondrial biogenesis, suggesting that AMPK is required for elevated mitochondrial biogenesis and fatty acid oxidation by leucine in C2C12 myotubes (Liang et al. 2014). There is ample evidence indicating that AMPK can promote mitochondrial biogenesis and oxidative capacity, and prevent the mitochondrial dysfunction in skeletal muscle (Canto et al. 2009, 2010). AMPK might serve as a downstream target of SIRT1. SIRT1 activation is essential for AMPK phosphorylation and improvement of mitochondrial function via deacetylation and activation of liver kinase B1 (LKB1). LKB1 directly phosphorylates Thr-172 of AMPK and activates its kinase activity (Shaw et al. 2004). Thus, it raises the possibility that AMPK might also serve as a downstream target of SIRT1 (Price et al. 2012; Liang et al. 2014). Conversely, AMPK may activate SIRT1 through indirectly increasing cellular NAD+ levels (Canto et al. 2009). Collectively, activated AMPK and SIRT1 further activate PGC-1α through phosphorylation and deacetylation mechanisms, promoting mitochondrial biogenesis and oxidative capacity and preventing mitochondrial dysfunction in skeletal muscle (Jager et al. 2007; Canto et al. 2010).
Taken together, PGC-1α, SIRT1, and AMPK are all involved in the regulation of mitochondrial biogenesis. Their function and regulation are closely intertwined. SIRT1 activates AMPK via deacetylation and activation of LKB1, whereas AMPK activates SIRT1 through indirectly increasing cellular NAD+ levels. Activated SIRT1 and AMPK further activate PGC-1α through phosphorylation and deacetylation, leading to stimulate the expression of mitochondrial genes. In addition, PGC-1α induces the expression of SIRT3 and SIRT5 in a PPARα- and ERRα-dependent manner. Consequently, mitochondrial biogenesis and oxidative capacity within the cell are enhanced.
Mitochondrial function and skeletal muscle health
Skeletal muscle and its fiber type
Lean body mass (LBM) accounts for almost 75 % of normal body weight, and includes all tissues except adipose tissue (Demling 2009). Skeletal muscle comprises the majority of LBM, the maintenance of which is important for supporting whole-body protein metabolism, physical strength, wound healing, immune function, and organ function (Wolfe 2006; Demling 2009). Naturally, a progressive loss of skeletal muscle mass (also known as sarcopenia) occurs with aging. However, injury or/and illness (such as infection, AIDS, cancer, non-healing wounds, and congestive heart failure) can lead to or accelerate this loss of LBM (Hou et al. 2015a; Lin et al. 2002; Demling 2009; Kim et al. 2010; Yi et al. 2015). Besides a loss of skeletal muscle, the sarcopenic phenotype is often linked with a shift in muscle fiber type, a reduced ability to perform activities of daily living, and an increased morbidity and mortality regardless of age (Johnston et al. 2008; Kim et al. 2010). Previous studies have demonstrated that sarcopenia leads to an approximately 50 % reduction in the number of total muscle fibers between the ages of 20–80, accompanied by a disproportionate loss of fast-twitch muscle fibers (Verdijk et al. 2010; Drey 2011).
In mammals, skeletal muscle is a mosaic of different types of muscle fibers with diverse structural properties and functional capabilities. Functional and phenotypic diversity of skeletal muscle is attributed to the heterogeneous composition of fibers (Scheffler et al. 2014). Traditionally, muscle fibers have been classified by immunofluorescence analyses of myosin heavy chain (MHC) isoforms. Based on the expression of the predominant MHC isoforms, humans have three fiber types (I, IIa, and IId/x), while rodents have four fiber types (I, IIa, IId/x, and IIb) (Smerdu et al. 1994; Ennion et al. 1995; Horton et al. 2001; Yan et al. 2011). Type I slow-twitch, oxidative fibers are slow in force generation and have a high capacity for oxidation of energy substrates. These muscle fibers are rich in oxidative enzymes, mitochondria, and capillary (Prince et al. 1981). Type IIa fast-twitch, oxidative fibers are fast in force generation but have similar oxidative profiles to type I fibers (Eddinger and Moss 1987). Type IId/x fibers, which are fast-twitch muscle fibers with a glycolytic metabolic profile, are rich in glycolytic enzymes but poor in mitochondria and capillary blood supply (Prince et al. 1981). Type IIb fibers have an even more fast-twitch, glycolytic phenotype than type IId/x fibers (Termin et al. 1989; Rivero et al. 1998). Muscle fibers are now commonly distinguished as slow-twitch (red) and fast-twitch (white) (Zierath and Hawley 2004). The slow-twitch myofibers contain mainly the type I MHC isoform, which are characterized by the high content of mitochondria and high rates of oxidative metabolism. The fast-twitch myofibers contain type IIa, IId/x, and IIb MHC isoforms, which are mainly glycolytic and perform quick contractions. These muscle fibers are required for movements involving strength and speed, but are easily fatigued (Berchtold et al. 2000; Olson and Williams 2000; Pette and Staron 2001). Each skeletal muscle fiber contains several hundred to several thousand myofibrils which comprise large polymerized protein molecules (actin and myosin) responsible for contraction (Karagounis and Hawley 2010). A shift in fiber distribution from slow-twitch to fast-twitch results from altered activities of key oxidative and glycolytic enzymes (Pette and Hofer 1979; Lin et al. 2002).
Mitochondrial function as a new mediator of skeletal muscle fiber type
Skeletal muscle fiber-type phenotype is mediated by several independent signaling pathways. These include pathways involved with PGC-1α (Lin et al. 2002), calcium/calmodulin-dependent protein kinase (CaMK) (Wu et al. 2002), calcineurin (Naya et al. 2000), and the ras/mitogen-activated protein kinase (Murgia et al. 2000). This review will mainly focus on PGC-1α. In response to mitochondrial dysfunction, skeletal muscle changes fiber-type composition via decreasing the proportion of slow fibers and increasing fast fibers. This transformation in the absence of fiber-type regeneration and observed adjustments from oxidative to glycolytic metabolism provides evidence for mitochondrial function as a new mediator of skeletal muscle fiber type (Venhoff et al. 2012). These authors demonstrated, for the first time, an important role for mitochondrial function in muscle fiber-type transformation. As stated above, PGC-1α plays a particularly robust role in mitochondrial biogenesis and function. It is interesting to speculate that PGC-1α might promote mitochondrial biogenesis and function, leading to increased oxidative capacity of fibers and up-regulated proportion of slow muscle fibers.
Much evidence shows that PGC-1α is a key regulator of fiber-type determination (Gouspillou et al. 2014). It is noteworthy that PGC-1α is expressed preferentially in skeletal muscles rich in type I fibers. Elevation of PGC-1α expression in skeletal muscles rich in type II fibers results in marked changes in tissue morphology, gene expression, and function (Lin et al. 2002). In skeletal muscles, PGC-1α has been reported to cause a shift toward type I fibers that are rich in mitochondria and highly oxidative. Of note, type I fibers contain a high mitochondria content and use oxidative metabolism as a primary source of energy (Lin et al. 2002; Rasbach et al. 2010). Further evidence shows that the beneficial effects of resveratrol on skeletal muscle are likely due to PGC-1α-mediated increases in mitochondrial biogenesis and a shift toward more oxidative muscle fibers (Price et al. 2012). Moreover, muscle-specific PGC-1α knock-out mice show a shift from oxidative type I and IIa toward type IIx and IIb muscle fibers, increasing muscle damage (Handschin et al. 2007). Intriguingly, SIRT1 transgenic muscle exhibits a fiber shift from fast-to-slow-twitch, increases expression of PGC-1α and decreases expression of genes associated with muscle atrophy (Chalkiadaki et al. 2014). The mechanisms whereby PGC-1α promotes a shift toward type I fibers are as follows. PGC-1α serves as a target downstream for calcium/calcineurin/CaMK signaling. CaMK signaling induces PGC-1 expression at a transcriptional level (Lin et al. 2002; Wu et al. 2002). Nfat and myocyte enhancer factor 2 (Mef2) transcription factor families are the two known effectors of the calcineurin signaling on gene expression in skeletal muscle. Moreover, a slow fiber-specific element of the slow troponin l gene contains a canonical Nfat site that is essential for maximal expression in slow-twitch muscle fibers and for maximal responsiveness to calcineurin (Calvo et al. 1999; Wu et al. 2000, 2001). PGC-1α directly interacts with Mef2, which physically binds the Nfat site in a slow fiber-specific element of the slow troponin l gene, increasing expression of type I fibrillar proteins. Consequently, the proportion of slow-twitch fibers is enhanced (Wu et al. 2001; Crabtree and Olson 2002; Lin et al. 2002; Gouspillou et al. 2014).
Taken together, mitochondria regulate muscle fiber-type transformation via PGC-1α (Fig. 2). On one hand, PGC-1α mediates mitochondrial biogenesis and activates oxidative metabolism, driving fast-to-slow fiber switch. On the other hand, PGC-1α directly interacts with Mef2, which physically binds the Nfat site in a slow-twitch fiber-specific element of the slow troponin l gene, increasing the expression of type I fibrillar proteins. Accordingly, the proportion of slow-twitch fiber types is up-regulated, and muscular atrophy is prevented, when PGC-1α is activated.

Mitochondria regulate fiber-type transformation via PGC-1α. CaMK signaling can induce PGC-1α gene expression at the transcriptional level. PGC-1α promotes mitochondrial biogenesis and enhances oxidative capacity of skeletal muscle fibers, leading to a shift toward slow-twitch fibers. In addition, PGC-1α directly interacts with Mef2. Mef2 physically binds the Nfat site in a slow fiber-specific element of the slow troponin l gene, increasing the expression of type I fibrillar proteins. Adapted from Lin et al. (2002) and Zierath and Hawley (2004)
Mitochondrial biogenesis improves skeletal muscle health
Skeletal muscle plays a major role in oxidizing fatty acids and glucose into water and CO2 (Jobgen et al. 2006). Oxidative capacity of fibers is strongly linked with skeletal muscle health and overall well-being. Enhanced oxidative capacity attenuates muscle loss during aging (Wenz et al. 2009), and affords protection against insulin resistance and metabolic dysregulation (Wang et al. 2004; Scheffler et al. 2014). The protective effects of enhanced oxidative capacity in disease states are largely ascribed to enhanced mitochondrial content and increased mitochondrial function. To accomplish this, mitochondria possess an increased ability to oxidize fatty acids and glucose, augmenting ATP generation and protecting against cellular stress (Scheffler et al. 2014). Therefore, augmenting the proportion of slow-twitch oxidative muscle fibers can improve health via increasing the mitochondrial function and ameliorating metabolic syndrome (Zierath and Hawley 2004). Indeed, the function of skeletal muscle highly depends on the ATP production by mitochondria (Verdijk et al. 2010). As noted previously, mitochondria play a critical role in influencing skeletal muscle fiber size and number to modulate substrate metabolism and the concentrations of lipids and glucose in plasma (Russell et al. 2014). Furthermore, sarcopenic muscle is associated with mitochondrial dysfunction (Johnston et al. 2008; Verdijk et al. 2010; Drey 2011). Accordingly, maintaining mitochondrial content and function of skeletal muscle is of great importance for sustained health throughout the lifespan.
Mitochondrial activity and function in skeletal muscle is a highly controlled process (Joseph et al. 2012). Mitochondrial dysfunction can result in a lowered mitochondrial mass and oxidative capacity, resulting in an increase in free radical production and consequently oxidative stress (Johnston et al. 2008; Verdijk et al. 2010). Impaired mitochondrial function is characterized by a rapid onset of symptoms commonly seen in the elderly, including muscle loss and insulin resistance (Wallace 2010; Sahin et al. 2011). Numerous studies have shown that a decline in mitochondrial function in normal individuals underlie many common age-related diseases and that treatments aimed at stimulating mitochondrial function can delay the progression of some of these diseases (Figueiredo et al. 2009; Wenz et al. 2009; de Moura et al. 2010; Fillmore et al. 2010; Horvath et al. 2011). Therefore, changes in mitochondrial content and function can directly or indirectly impact skeletal muscle function and consequently whole-body health and well-being. Thus, identifying compounds that can attenuate both the mitochondrial dysfunction and the loss of LBM associated with sarcopenia is a key focus of current medical research.
HMB promotes mitochondrial biogenesis and improves skeletal muscle health
The role of HMB in mitochondrial biogenesis
Branched-chain amino acids have been suggested as potential candidates in promoting survival via regulating mitochondrial function (Valerio et al. 2011). For example, leucine can stimulate protein synthesis in skeletal muscle (Davis et al. 2010) and muscle bass (Columbus et al. 2015; Sun et al. 2015). Of note, long-term dietary supplementation with a specific BCAA-enriched mixture (BCAAem) improves age-related disorders in animals and humans and promotes mice survival (D’Antona et al. 2010). The anti-aging role of BCAAs may be mediated by mitochondrial biogenesis in mammals. Specifically, supplementation of BCAAem increases mitochondrial biogenesis and SIRT1 expression in skeletal muscle, and this is accompanied by enhanced expression of ROS-removing genes and reduced ROS production in middle-aged mice (D’Antona et al. 2010). All of the BCAAem-mediated effects are strongly attenuated in endothelial nitric oxide synthase null mutant mice (Pansarasa et al. 2008; Solerte et al. 2008; D’Antona et al. 2010), as physiological levels of nitric oxide are an activator of mitochondrial biogenesis (McKnight et al. 2010).
Of all the BCAAs, leucine is the most effective in the regulation of many cellular processes such as protein synthesis and energy metabolism, and has received much attention (Yin et al. 2010; Li et al. 2011; Duan et al. 2015a, b, c). Recently, increasing evidence has shown that leucine also plays a critical role in mitochondrial biogenesis (Li et al. 2012). In C2C12 cell models, leucine (0.5 mM) increases mitochondrial mass by 30 %, and stimulates expression of mitochondrial biogenesis genes (SIRT1, PGC-1α, and NRF-1) as well as mitochondrial component genes (UCP3, COX, and NADH) by three- to fivefold (Li et al. 2012). Of great importance, SIRT1 has been shown to be implicated in leucine-induced mitochondrial biogenesis in muscle cells, because transfection of C2C12 myocytes with SIRT1 siRNA leads to parallel inhibition of SIRT1 expression and leucine-stimulated activation of PGC-1α and NRF-1 (Sun and Zemel 2009). Likewise, Vaughan et al. (2013) reported that leucine (0.1–0.5 mM) dose-dependently enhanced PGC-1α expression, mitochondrial density, and oxidative capacity in skeletal muscle cells. Further evidence suggests that leucine (0.5 mM) markedly increases mitochondrial content, expression of mitochondrial biogenesis-related genes, fatty acid oxidation, SIRT1 activity and expression, and AMPK phosphorylation in C2C12 myotubes (Sun and Zemel 2009). Of note, activation of SIRT1 precedes that of AMPK, suggesting that leucine activation of SIRT1, rather than AMPK, is the primary event (Liang et al. 2014). Additionally, using an animal model (high-fat die-fed male C57BL/6J mice), leucine supplementation correlates with increased expression of SIRT1 and decreased acetylation (activation) of PGC-1α, contributing to up-regulation of genes controlling mitochondrial biogenesis and fatty acid oxidation (Li et al. 2012). Of great importance, AMPK is required for SIRT1’s ability to promote mitochondrial biogenesis and fatty acid oxidation by leucine. Thus, addition of leucine within physiological ranges improves mitochondrial function. However, chronic exposure to elevated concentrations of leucine (≥1.0 mM) reduces the synthesis of nitric oxide from l-arginine by endothelial cells (Yang et al. 2015) and should be avoided. This illustrates the need to consider amino acid balance in diets when nutritional strategies are used to improve muscle mass and function (Hou et al. 2015b; Wu et al. 2014).
The effects of BCAA, particularly leucine, on muscle mitochondrial biogenesis and fatty acid oxidation are actually regulated by the metabolite of HMB (Stancliffe 2012). HMB, a metabolite of leucine, is produced in tissues of animals and humans (Nissen and Abumrad 1997). HMB, which is commercially available, has been claimed to build skeletal muscle and strength in both exercise and clinical settings, while increasing fatty acid oxidation, a marker of mitochondrial function (Cheng et al. 1997, 1998; Slater and Jenkins 2000; Wilson et al. 2008). HMB (0.5 μM) combined with metformin and resveratrol markedly increases fat oxidation, SIRT1 activity, and AMPK in muscle cells (Bruckbauer and Zemel 2013). Additionally, HMB also stimulates the expression of mitochondrial regulatory (PGC-1α and NRF-1) and component (UCP3) genes in murine myotubes, and increases mitochondrial biogenesis by 50 % in C2C12 myotubes (Stancliffe 2012). Moreover, HMB may play a role in the production of coenzyme Q, a downstream metabolite of 3-hydroxy-3-methylglutarylcoenzyme A, which plays a crucial role in myocyte proliferation and mitochondrial function (Evans and Rees 2002). As such, HMB may be able to improve muscle health by promoting mitochondrial biogenesis (Fig. 3). However, in vivo effects of HMB on skeletal muscle mitochondrial biogenesis in animals and humans are not known.

A potential mechanism whereby β-hydroxy-β-methylbutyrate (HMB) prevents muscle wasting. HMB stimulates the expression of PGC-1α and increases mitochondrial biogenesis. Additionally, under the control of PGC-1α, muscle slow-twitch fiber types are increased and muscle wasting is reduced in the body
Clinical effects of HMB supplementation on skeletal muscle health
Exercise induces changes in skeletal muscle by transforming the myofibers from glycolytic to oxidative forms, rendering them more resistant to fatigue and atrophy. Conversely, aging is associated with skeletal muscle atrophy, which is characterized by a progressive loss of oxidative fibers (Aspnes et al. 1997; Chalkiadaki et al. 2014). In recent years, HMB has been an interesting target for studies because of its efficacy as a potent therapeutical supplement for the treatment of muscle disorders (Smith et al. 2004, 2005; Eley et al. 2007, 2008a, b).
The first studies investigating the effect of oral supplementation with different doses of HMB on the mediation of muscle mass in humans were performed in a resistance training study in 1996 (Nissen et al. 1996). The authors reported that dietary supplementation with 0, 1.5, and 3.0 g/day of HMB to humans undergoing resistance training for 3 weeks resulted in a decrease in exercise-associated muscle proteolysis during the first 2 weeks and a reduction in muscle damage during the third week. When subjects were supplemented with 3.0 g/day of HMB, while doing resistance training for 7 weeks, they exhibited a marked increase in fat-free mass and strength (Jowko et al. 2001). Likewise, when 39 men and 36 women between 20- and 40-year old were randomized to either HMB supplementation (3.0 g/days) or placebo in two gender cohorts, HMB increased upper body strength and minimized muscle damage when combined with a 4-week exercise program (Panton et al. 2000). Furthermore, HMB coupled with exercise has been widely used by athletes in an effort to enhance their strength and muscle mass (Nissen and Sharp 2003). In line with these observations, dietary supplementation of HMB was effective in decreasing muscle proteolysis observed in mice implanted with the MAC16 tumor, which is reflected in the attenuation of muscle mass loss (Smith et al. 2005). There is also evidence that HMB might attenuate the muscle loss caused by aging (Vukovich et al. 2001), cancer cachexia (Zanchi et al. 2011), AIDS (Clark et al. 2000), and endotoxemia (Russell and Tisdale 2009; Kovarik et al. 2010). In addition, recent studies have demonstrated that dietary HMB supplementation attenuates dexamethasone-induced muscle wasting and might be used to prevent steroid myopathy (Noh et al. 2014). It is unknown whether HMB has a direct or indirect effect on the observed positive change in skeletal muscle mass.
Studies addressing the efficacy of the combination of supplementation of HMB with other nutrients, resulting in potentially increased strength and lean mass, are also of great importance. In this context, volunteers received dietary supplementation with HMB, creatine, a combination of both, or a placebo, and underwent strength training for 3 weeks. The authors found a higher gain in skeletal muscle strength and mass in the group supplemented with HMB and creatine, compared to the other groups, and a reduction in muscle damage and protein degradation in the group supplemented with HMB (Jowko et al. 2001).
Taken together, HMB has been evaluated alone or in combination with other nutrients, with or without exercise, as a supplement to augment LBM and to treat exercise, sepsis, and cancer-induced muscle damage. However, it is unclear whether these effects of HMB have negative effects on non-muscle tissues (such as liver and white adipose tissue). Bearing this question in mind, researchers carried out studies to evaluate the effects of HMB supplementation on skeletal muscle hypertrophy and the expression of proteins involved in insulin signaling. In this study, rats were treated with saline or HMB (320 mg/kg body weight) for 1 month (Pimentel et al. 2011). The authors found that HMB supplementation stimulated muscle hypertrophy in extensor digitorum longus (EDL) and soleus muscles, while enhancing serum insulin levels, the expression of the mammalian target of rapamycin, and phosphorylation of the 70 kDa ribosomal protein S6 kinase 1 in EDL muscle (Pimentel et al. 2011). Expression of the insulin receptor was enhanced only in liver. These observations indicate that HMB supplementation can be used to increase muscle mass without adverse health effects (Pimentel et al. 2011). Overall, HMB supplementation is safe and may potentially improve several markers of health. Additionally, the usual dose of 3 g/day may be routinely recommended to maintain or improve skeletal muscle mass and function in health and disease (Molfino et al. 2013).
Summary and perspectives
Our fundamental knowledge of the HMB regulation of skeletal muscle mitochondrial biogenesis and health has been greatly expanded over the past 20 years. Understanding the important role for HMB in mitochondrial biogenesis and muscle health may provide new strategies to improve human health and meat quality in livestock production. Importantly, HMB can be used as a nitrogen-free supplement to benefit the environment by reducing nitrogen excretion. Further studies are warranted to clearly define the effects of dietary HMB supplementation on in vivo skeletal muscle mitochondrial biogenesis in healthy subjects and farm animals.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- BCAA:
-
Branched-chain amino acid(s)
- BCAAem:
-
Branched-chain amino acid-enriched mixture
- CaMK:
-
Calmodulin-dependent protein kinase
- CaMKKβ:
-
Ca2+-activated kinase calmodulin-dependent kinase kinase-β
- EDL:
-
Extensor digitorum longus
- ERRα:
-
Estrogen-related receptor α
- HMB:
-
β-Hydroxy-β-methylbutyrate
- LBM:
-
Lean body mass
- LKB1:
-
Liver kinase B1
- Mef2:
-
Myocyte enhancer factor 2
- MHC:
-
Myosin heavy chain
- OXPHOS:
-
Oxidative phosphorylation
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma co-activator 1-alpha
- PPARα:
-
Peroxisome proliferator-activated receptor α
- ROS:
-
Reactive oxygen species
- SIRT1:
-
Silent information regulator transcript 1
- TFAM:
-
Mitochondrial transcription factor A
References
Ahn BH, Kim HS, Song SW et al (2008) A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA 105:14447–14452
Aquilano K, Vigilanza P, Baldelli S et al (2010) Peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC-1 alpha) and sirtuin 1 (SIRT1) reside in mitochondria possible direct function in mitochondrial biogenesis. J Biol Chem 285:21590–21599
Arany Z, Novikov M, Chin S et al (2006) Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-γ coactivator 1α. Proc Natl Acad Sci USA 103:10086–10091
Aspnes LE, Lee CM, Weindruch R et al (1997) Caloric restriction reduces fiber loss and mitochondrial abnormalities in aged rat muscle. FASEB J 11:573–581
Bazer FW, Ying W, Wang XQ et al (2015) The many faces of interferon tau. Amino Acids 47:449–460
Berchtold MW, Brinkmeier H, Muntener M (2000) Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol Rev 80:1215–1265
Bruckbauer A, Zemel MB (2013) Synergistic effects of metformin, resveratrol, and hydroxymethylbutyrate on insulin sensitivity. Diabetes Metab Syndr Obes 6:93–102
Buler M, Aatsinki SM, Izzi V et al (2014) SIRT5 is under the control of PGC-1alpha and AMPK and is involved in regulation of mitochondrial energy metabolism. FASEB J 28:3225–3237
Calvo S, Venepally P, Cheng J et al (1999) Fiber-type-specific transcription of the troponin l slow gene is regulated by multiple elements. Mol Cell Biol 19:515–525
Calvo JA, Daniels TG, Wang X et al (2008) Muscle-specific expression of PPARγ coactivator-1α improves exercise performance and increases peak oxygen uptake. J Appl Physiol 104:1304–1312
Canto C, Gerhart-Hines Z, Feige JN et al (2009) AMPK regulates energy expenditure by modulating NAD(+) metabolism and SIRT1 activity. Nature 458:1056–1060
Canto C, Jiang LQ, Deshmukh AS et al (2010) Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 11:213–219
Chalkiadaki A, Igarashi M, Nasamu AS et al (2014) Muscle-specific SIRT1 gain-of-function increases slow-twitch fibers and ameliorates pathophysiology in a mouse model of duchenne muscular dystrophy. PLoS Genet 10:e1004490
Cheng W, Phillips B, Abumrad N (1997) Beta-hydroxy-beta-methyl butyrate increases fatty acid oxidation by muscle cells. FASEB J 11:A381
Cheng W, Phillips B, Abumrad N (1998) Effect of HMB on fuel utilization, membrane stability and creatine kinase content of cultured muscle cells. FASEB J 12:A950
Clark RH, Feleke G, Din M et al (2000) Nutritional treatment for acquired immunodeficiency virus-associated wasting using beta-hydroxy beta-methylbutyrate, glutamine, and arginine: a randomized, double-blind, placebo-controlled study. JPEN J Parenter Enteral Nutr 24:133–139
Columbus DA, Fiorotto ML, Davis TA (2015) Leucine is a major regulator of muscle protein synthesis in neonates. Amino Acids 47:259–270
Crabtree GR, Olson EN (2002) NFAT signaling: choreographing the social lives of cells. Cell 109(2):S67–S79
Dai ZL, Wu ZL, Yang Y et al (2013) Nitric oxide and energy metabolism in mammals. BioFactors 39:383–391
D’Antona G, Ragni M, Cardile A et al (2010) Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab 12:362–372
Davis TA, Suryawan A, Orellana RA et al (2010) Amino acids and insulin are regulators of muscle protein synthesis in neonatal pigs. Animal 4:1790–1796
de Moura MB, dos Santos LS, Van Houten B (2010) Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ Mol Mutagen 51:391–405
Demling RH (2009) Nutrition, anabolism, and the wound healing process: an overview. Eplasty 9:e9
Drey M (2011) Sarcopenia—pathophysiology and clinical relevance. Wien Med Wochenschr 161:402–408
Duan Y, Li F, Li Y et al (2015a) The role of leucine and its metabolites in protein and energy metabolism. Amino Acids. doi:10.1007/s00726-015-2067-1
Duan YH, Li FN, Liu HN et al (2015b) Nutritional and regulatory roles of leucine in muscle growth and fat reduction. Front Biosci Landmark 20:796–813
Duan YH, Li FN, Tan KR et al (2015c) Key mediators of intracellular amino acids signaling to mTORC1 activation. Amino Acids 47:857–867
Duchen MR (2004) Roles of mitochondria in health and disease. Diabetes 53:S96–S102
Eddinger TJ, Moss RL (1987) Mechanical-properties of skinned single fibers of identified types from rat diaphragm. Am J Physiol 253:C210–C218
Eley HL, Russell ST, Baxter JH et al (2007) Signaling pathways initiated by beta-hydroxy-beta-methylbutyrate to attenuate the depression of protein synthesis in skeletal muscle in response to cachectic stimuli. Am J Physiol Endocrinol Metab 293:E923–E931
Eley HL, Russell ST, Tisdale MJ (2008a) Mechanism of attenuation of muscle protein degradation induced by tumor necrosis factor-alpha and angiotensin II by beta-hydroxy-beta-methylbutyrate. Am J Physiol Endocrinol Metab 295:E1417–E1426
Eley HL, Russell ST, Tisdale MJ (2008b) Attenuation of depression of muscle protein synthesis induced by lipopolysaccharide, tumor necrosis factor, and angiotensin II by beta-hydroxy-betamethylbutyrate. Am J Physiol Endocrinol Metab 295:E1409–E1416
Ennion S, Pereira JS, Sargeant AJ et al (1995) Characterization of human skeletal-muscle fibers according to the myosin heavy-chains they express. J Muscle Res Cell Motil 16:35–43
Evans M, Rees A (2002) Effects of HMG-CoA reductase inhibitors on skeletal muscle: are all statins the same? Drug Saf 25:649–663
Feldman JL, Dittenhafer-Reed KE, Denu JM (2012) Sirtuin catalysis and regulation. J Biol Chem 287:42419–42427
Figueiredo PA, Powers SK, Ferreira RM et al (2009) Aging impairs skeletal muscle mitochondrial bioenergetic function. J Gerontol A Biol Sci Med Sci 64:21–33
Filhiol TM (2012) The effects of leucine on mitochondrial biogenesis and cell cycle in A-375 melanoma cells, Master Thesis. The University of Tennessee, Knoxville
Fillmore N, Jacobs DL, Mills DB et al (2010) Chronic AMP-activated protein kinase activation and a high-fat diet have an additive effect on mitochondria in rat skeletal muscle. J Appl Physiol 109:511–520
Fu WJ, Haynes TE, Kohli R et al (2005) Dietary l-arginine supplementation reduces fat mass in Zucker diabetic fatty rats. J Nutr 135:714–721
Gerhart-Hines Z, Rodgers JT, Bare O et al (2007) Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1 alpha. EMBO J 26:1913–1923
Gouspillou G, Sgarioto N, Norris B et al (2014) The relationship between muscle fiber type-specific PGC-1 alpha content and mitochondrial content varies between rodent models and humans. PLoS One 9:e103044
Handschin C, Chin S, Li P et al (2007) Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem 282:30014–30021
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13:251–262
Hirschey MD, Shimazu T, Goetzman E et al (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–125
Hock MB, Kralli A (2009) Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol 71:177–203
Horton MJ, Brandon CA, Morris TJ et al (2001) Abundant expression of myosin heavy-chain IIB RNA in a subset of human masseter muscle fibres. Arch Oral Biol 46:1039–1050
Horvath TL, Erion DM, Elsworth JD et al (2011) GPA protects the nigrostriatal dopamine system by enhancing mitochondrial function. Neurobiol Dis 43:152–162
Hou YQ, Wang L, Yi D et al (2015a) N-acetylcysteine and intestinal health: a focus on mechanisms of its actions. Front Biosci 20:872–891
Hou YQ, Yin YL, Wu G (2015b) Dietary essentiality of “nutritionally nonessential amino acids” for animals and humans. Exp Biol Med 240:997–1007
Huss JM, Torra IP, Staels B et al (2004) Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor at signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol 24:9079–9091
Jager S, Handschin C, St-Pierre J et al (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 104:12017–12022
Jobgen WS, Fried SK, Fu WJ et al (2006) Regulatory role for the arginine-nitric oxide pathway in metabolism of energy substrates. J Nutr Biochem 17:571–588
Johnston APW, De Lisio M, Parise G (2008) Resistance training, sarcopenia, and the mitochondrial theory of aging. Appl Physiol Nutr Metab 33:191–199
Joseph AM, Joanisse DR, Baillot RG et al (2012) Mitochondrial dysregulation in the pathogenesis of diabetes: potential for mitochondrial biogenesis-mediated interventions. Exp Diabetes Res. doi:10.1155/2012/642038
Jowko E, Ostaszewski P, Jank M et al (2001) Creatine and beta-hydroxy-beta-methylbutyrate (HMB) additively increase lean body mass and muscle strength during a weight-training program. Nutrition 17:558–566
Karagounis LG, Hawley JA (2010) Skeletal muscle: increasing the size of the locomotor cell. Int J Biochem Cell Biol 42:1376–1379
Kim JS, Wilson JM, Lee SR (2010) Dietary implications on mechanisms of sarcopenia: roles of protein, amino acids and antioxidants. J Nutr Biochem 21:1–13
Kong X, Wang R, Xue Y et al (2010) Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 5:e11707
Kovarik M, Muthny T, Sispera L et al (2010) Effects of beta-hydroxy-beta-methylbutyrate treatment in different types of skeletal muscle of intact and septic rats. J Physiol Biochem 66:311–319
Koves TR, Li P, An J et al (2005) Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem 280:33588–33598
Lage R, Dieguez C, Vidal-Puig A et al (2008) AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med 14:539–549
Leone TC, Lehman JJ, Finck BN et al (2005) PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3:e101
Li F, Yin Y, Tan B et al (2011) Leucine nutrition in animals and humans: mTOR signaling and beyond. Amino Acids 41:1185–1193
Li HL, Xu MJ, Lee J et al (2012) Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Am J Physiol Endocrinol Metab 303:E1234–E1244
Liang C, Curry BJ, Brown PL et al (2014) Leucine modulates mitochondrial biogenesis and SIRT1-AMPK signaling in C2C12 myotubes. J Nutr Metab 2014:239750
Lin J, Wu H, Tarr PT et al (2002) Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418:797–801
Lombard DB, Alt FW, Cheng HL et al (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27:8807–8814
McKnight JR, Satterfield MC, Jobgen WS et al (2010) Beneficial effects of l-arginine on reducing obesity: potential mechanisms and important implications for human health. Amino Acids 39:349–357
Molfino A, Gioia G, Rossi Fanelli F et al (2013) Beta-hydroxy-beta-methylbutyrate supplementation in health and disease: a systematic review of randomized trials. Amino Acids 45:1273–1292
Mootha VK, Bunkenborg J, Olsen JV et al (2003) Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115:629–640
Murgia M, Serrano AL, Calabria E et al (2000) Ras is involved in nerve-activity-dependent regulation of muscle genes. Nat Cell Biol 2:142–147
Narkar VA, Downes M, Yu RT et al (2008) AMPK and PPARdelta agonists are exercise mimetics. Cell 134:405–415
Naya FJ, Mercer B, Shelton J et al (2000) Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J Biol Chem 275:4545–4548
Nissen SL, Abumrad NN (1997) Nutritional role of the leucine metabolite β-hydroxy β-methylbutyrate (HMB). J Nutr Biochem 8:300–311
Nissen SL, Sharp RL (2003) Effect of dietary supplements on lean mass and strength gains with resistance exercise: a meta-analysis. J Appl Physiol 94:651–659
Nissen S, Sharp R, Ray M et al (1996) Effect of leucine metabolite beta-hydroxy-beta-methylbutyrate on muscle metabolism during resistance-exercise training. J Appl Physiol 81:2095–2104
Noh KK, Chung KW, Choi YJ et al (2014) Beta-hydroxy-beta-methylbutyrate improves dexamethasone-induced muscle atrophy by modulating the muscle degradation pathway in SD rat. PLoS One 9:e102947
Olson EN, Williams RS (2000) Remodeling muscles with calcineurin. BioEssays 22:510–519
Pagliarini DJ, Calvo SE, Chang B et al (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134:112–123
Pansarasa O, Flati V, Corsetti G et al (2008) Oral amino acid supplementation counteracts age-induced sarcopenia in elderly rats. Am J Cardiol 101:35e–41e
Panton LB, Rathmacher JA, Baier S et al (2000) Nutritional supplementation of the leucine metabolite beta-hydroxy-beta-methylbutyrate (HMB) during resistance training. Nutrition 16:734–739
Pette D, Hofer HW (1979) The constant proportion enzyme group concept in the selection of reference enzymes in metabolism. Ciba Found Symp 73:231–244
Pette D, Staron RS (2001) Transitions of muscle fiber phenotypic profiles. Histochem Cell Biol 115:359–372
Pimentel GD, Rosa JC, Lira FS et al (2011) Beta-hydroxy-beta-methylbutyrate (HMbeta) supplementation stimulates skeletal muscle hypertrophy in rats via the mTOR pathway. Nutr Metab (Lond) 8:11
Poyton RO, McEwen JE (1996) Crosstalk between nuclear and mitochondrial genomes. Annu Rev Biochem 65:563–607
Price NL, Gomes AP, Ling AJY et al (2012) SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 15:675–690
Prince FP, Hikida RS, Hagerman FC et al (1981) A morphometric analysis of human-muscle fibers with relation to fiber types and adaptations to exercise. J Neurol Sci 49:165–179
Rasbach KA, Gupta RK, Ruas JL et al (2010) PGC-1alpha regulates a HIF2alpha-dependent switch in skeletal muscle fiber types. Proc Natl Acad Sci USA 107:21866–21871
Rivero JLL, Talmadge RJ, Edgerton VR (1998) Fibre size and metabolic properties of myosin heavy chain-based fibre types in rat skeletal muscle. J Muscle Res Cell Motil 19:733–742
Rodgers JT, Lerin C, Haas W et al (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118
Rodgers JT, Lerin C, Gerhart-Hines Z et al (2008) Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett 582:46–53
Russell ST, Tisdale MJ (2009) Mechanism of attenuation by beta-hydroxy-beta-methylbutyrate of muscle protein degradation induced by lipopolysaccharide. Mol Cell Biochem 330:171–179
Russell AP, Foletta VC, Snow RJ et al (2014) Skeletal muscle mitochondria: a major player in exercise, health and disease. Biochim Biophys Acta 1840:1276–1284
Sahin E, Colla S, Liesa M et al (2011) Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470:359–365
Scarpulla RC (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88:611–638
Scarpulla RC (2011) Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Bba-Mol Cell Res 1813:1269–1278
Scheffler TL, Scheffler JM, Park S et al (2014) Fiber hypertrophy and increased oxidative capacity can occur simultaneously in pig glycolytic skeletal muscle. Am J Physiol Cell Physiol 306:C354–C363
Shaw RJ, Kosmatka M, Bardeesy N et al (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA 101:3329–3335
Slater GJ, Jenkins D (2000) Beta-hydroxy-beta-methylbutyrate (HMB) supplementation and the promotion of muscle growth and strength. Sports Med 30:105–116
Smerdu V, Karschmizrachi I, Campione M et al (1994) Type-iix myosin heavy-chain transcripts are expressed in type iib fibers of human skeletal-muscle. Am J Physiol Cell Physiol 267:C1723–C1728
Smith HJ, Wyke SM, Tisdale MJ (2004) Mechanism of the attenuation of proteolysis-inducing factor stimulated protein degradation in muscle by β-hydroxy-β-methylbutyrate. Cancer Res 64:8731–8735
Smith HJ, Mukerji P, Tisdale MJ (2005) Attenuation of proteasome-induced proteolysis in skeletal muscle by β-hydroxy-β-methylbutyrate in cancer-induced muscle loss. Cancer Res 65:277–283
Solerte SB, Fioravanti M, Locatelli E et al (2008) Improvement of blood glucose control and insulin sensitivity during a long-term (60 weeks) randomized study with amino acid dietary supplements in elderly subjects with type 2 diabetes mellitus. Am J Cardiol 101:82e–88e
Stancliffe RA (2012) Role of beta-hydroxy-beta-methylbutyrate (HMB) in leucine stimulation of mitochondrial biogenesis and fatty acid oxidation, Master Thesis. The University of Tennessee, Knoxville
Stefano GB, Kim C, Mantione K et al (2012) Targeting mitochondrial biogenesis for promoting health. Med Sci Monit 18:Sc1-Sc3
Sun X, Zemel MB (2009) Leucine modulation of mitochondrial mass and oxygen consumption in skeletal muscle cells and adipocytes. Nutr Metab (Lond) 6:26
Sun YL, Wu ZL, Li W et al (2015) Dietary l-leucine supplementation enhances intestinal development in suckling piglets. Amino Acids 47:1517–1525
Tekwe CD, Lei J, Yao K et al (2013) Oral administration of interferon tau enhances oxidation of energy substrates and reduces adiposity in Zucker diabetic fatty rats. BioFactors 39:552–563
Termin A, Staron RS, Pette D (1989) Myosin heavy chain isoforms in histochemically defined fiber types of rat muscle. Histochemistry 92:453–457
Valerio A, D’Antona G, Nisoli E (2011) Branched-chain amino acids, mitochondrial biogenesis, and healthspan: an evolutionary perspective. Aging-Us 3:464–478
Vaughan RA, Garcia-Smith R, Gannon NP et al (2013) Leucine treatment enhances oxidative capacity through complete carbohydrate oxidation and increased mitochondrial density in skeletal muscle cells. Amino Acids 45:901–911
Venhoff N, Lebrecht D, Pfeifer D et al (2012) Muscle-fiber transdifferentiation in an experimental model of respiratory chain myopathy. Arthritis Res Ther 14:1–11
Verdijk LB, Snijders T, Beelen M et al (2010) Characteristics of muscle fiber type are predictive of skeletal muscle mass and strength in elderly men. J Am Geriatr Soc 58:2069–2075
Virbasius CA, Virbasius JV, Scarpulla RC (1993) NRF-1, an activator involved in nuclearmitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev 7:2431–2445
Vukovich MD, Stubbs NB, Bohlken RM (2001) Body composition in 70-year-old adults responds to dietary beta-hydroxy-beta-methylbutyrate similarly to that of young adults. J Nutr 131:2049–2052
Wallace DC (2010) Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen 51:440–450
Wang YX, Zhang CL, Yu RT et al (2004) Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol 2:e294
Weber TA, Reichert AS (2010) Impaired quality control of mitochondria: aging from a new perspective. Exp Gerontol 45:503–511
Wenz T, Rossi SG, Rotundo RL et al (2009) Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA 106:20405–20410
Wilson GJ, Wilson JM, Manninen AH (2008) Effects of beta-hydroxy-beta-methylbutyrate (HMB) on exercise performance and body composition across varying levels of age, sex, and training experience: a review. Nutr Metab (Lond) 5:1
Wolfe RR (2006) The underappreciated role of muscle in health and disease. Am J Clin Nutr 84:475–482
Wu G (2013) Amino acids: biochemistry and nutrition. CRC Press, Boca Raton
Wu ZD, Puigserver P, Andersson U et al (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124
Wu H, Naya FJ, Mckinsey TA et al (2000) MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J 19:1963–1973
Wu H, Rothermel B, Kanatous S et al (2001) Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO J 20:6414–6423
Wu H, Kanatous SB, Thurmond FA et al (2002) Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 296:349–352
Wu G, Bazer FW, Dai ZL, Li DF, Wang JJ, Wu ZL (2014) Amino acid nutrition in animals: protein synthesis and beyond. Annu Rev Anim Biosci 2:387–417
Yan Z, Okutsu M, Akhtar YN et al (2011) Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J Appl Physiol 110:264–274
Yang Y, Wu ZL, Meininger CJ et al (2015) L-Leucine and NO-mediated cardiovascular function. Amino Acids 47:435–447
Yi D, Hou YQ, Wang L et al (2015) L-Glutamine enhances enterocyte growth via activation of the mTOR signaling pathway independently of AMPK. Amino Acids 47:65–78
Yin YL, Yao K, Liu ZJ et al (2010) Supplementing l-leucine to a low-protein diet increases tissue protein synthesis in weanling pigs. Amino Acids 39:1477–1486
Zanchi NE, Gerlinger-Romero F, Guimaraes-Ferreira L et al (2011) HMB supplementation: clinical and athletic performance-related effects and mechanisms of action. Amino Acids 40:1015–1025
Zhang D, Mott JL, Farrar P et al (2003) Mitochondrial DNA mutations activate the mitochondrial apoptotic pathway and cause dilated cardiomyopathy. Cardiovasc Res 57:147–157
Zierath JR, Hawley JA (2004) Skeletal muscle fiber type: influence on contractile and metabolic properties. PLoS Biol 2:1523–1527
Acknowledgments
This study was jointly supported by the National Natural Science Foundation of China (31330075;31110103909, 31572416, and 31372319), Special Fund for Agro-scientific Research in the Public Interest (201403047), Innovation Research Team Development Program of MOE of China (IRT0945), The Chinese Academy of Science STS Project (KFJ-EW-STS-063), Key Projects in the National Science and Technology Pillar Program (2013BAD21B04), Hunan Key Project (2015NK1002), Changsha Lvye Biotechnology Limited Company Academician Expert Workstation, Guangdong Wangda Group Academician Workstation for Clean Feed Technology Research and Development in Swine, Guangdong Hinapharm Group Academician Workstation for Biological Feed and Feed Additives and Animal Intestinal Health, Hunan New Wellful Co. Ltd, Academician Workstation, Hubei Provincial Key Project for Scientific and Technical Innovation (2014ABA022), Hubei Hundred Talent program, and the Natural Science Foundation of Hubei Province (2013CFA097, 2013CFB325, 2012FFB04805, and 2011CDA131).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have declared no conflict of interest.
Additional information
X. He and Y. Duan contributed equally to this study.
Rights and permissions
About this article

Cite this article
He, X., Duan, Y., Yao, K. et al. β-Hydroxy-β-methylbutyrate, mitochondrial biogenesis, and skeletal muscle health. Amino Acids 48, 653–664 (2016). https://doi.org/10.1007/s00726-015-2126-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-015-2126-7