
Abstract
Sarcopenia, which is characterized by reduction in muscle mass and strength, contributes to several age-related conditions, including insulin resistance and frailty. Despite the importance of maintaining muscle mass for healthy aging, the mechanisms contributing to sarcopenia are not fully elucidated. Nevertheless, mitochondria appear to play a key role in the underlying condition, and importantly, respond robustly to exercise interventions. Mitochondria are intracellular organelles largely attributed to maintaining ATP concentrations, however, the importance of this organelle in overall cellular homeostasis has been expanded in the last decades to include redox signaling, calcium homeostasis, inflammation, and apoptosis. Several lines of evidence suggest that mitochondrial bioenergetics are altered in aged skeletal muscle, resulting in an increase in reactive oxygen species (ROS) production, while conversely genetic/pharmacological approaches that attenuate mitochondrial ROS promote healthy aging and maintenance of muscle mass. These observations suggest that increased free radicals are one of the bases of the aging process and related sarcopenia. Here, we reviewed the current knowledge regarding mitochondrial function and redox balance in aged human skeletal muscle, highlighting the implications of redox unbalance on skeletal muscle mass maintenance and muscle health. Additionally, we describe the benefits of exercise and nutrition interventions in the context of improving mitochondrial bioenergetics and functional outcomes regarding skeletal muscle mass and function.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Skeletal muscle comprises approximately 40% of total body mass, and therefore represents an important tissue in the context of healthy aging, as it is essential for locomotion, overall metabolism and glucose homeostasis [57]. One of the most significant changes with aging is a loss of muscle mass and strength, a process called sarcopenia. Sarcopenia is implicated in the reduction of functional capacity, mobility, increasing the risk of falls and resulting in frailty [11]. Therefore, a loss of muscle mass ultimately compromises the quality of life of older individuals and increases the risk of mortality. While the direct health-care costs associated with sarcopenia are estimated to be ~ 20 billion dollars [31] since decreased physical activity increases the risk of developing hypertension, heart disease, and diabetes, the overall consequence of a loss of muscle mass is likely much higher. In addition, in almost every country worldwide, the proportion of individuals over 60 years of age is estimated to triple by the year 2050, at which time these individuals will represent ~ 25% of the world’s population, placing an unprecedented burden on the health care system [69].
While the etiology of sarcopenia is poorly defined, and likely involves a complex interaction of a variety of mechanisms, the basic phenotypic changes within muscle have been well characterized. The current working model suggests that aging is associated with anabolic resistance and a preferential loss of motor units (MU) innervating type II muscle fibres. This process progressively results in a reduction of type II muscle fibres cross-sectional area (CSA) and a higher proportion of type I muscle fibres [49]. Since type I muscle fibres are smaller and produce less force, these basic changes compromise muscle function [24]. While the observation for deterioration in the neuromuscular junction with aging was reported over 50 years ago, the underlying mechanisms are only now being identified. In this context, alterations in mitochondrial function have been implicated as a cause of sarcopenia [19], as well as several co-morbidities associated with aging [30]. The mitochondrial participation in these processes has been highlight by studies showing that attenuating mitochondrial reactive oxygen species (ROS) prevents disuse-mediated atrophy [46].
Mitochondria are small organelles within almost every cell that are required for aerobic ATP production, and also play a central role in redox biology. Several lines of evidence suggest that mitochondria have a prominent role in the development of sarcopenia. For instance, aged skeletal muscle displays a loss of mitochondrial content [18, 21] and increases mitochondrial-mediated apoptosis [9, 52]. Moreover, transgenic models that decrease the capacity of mitochondria to produce ATP dismantle neuromuscular junctions and trigger distal degeneration of motor neurons [14]. In addition, genetic approaches that increase the prevalence of mitochondrial DNA (mtDNA) mutations and oxidative damage result in premature aging [68]. On the other hand, induction of mitochondrial biogenesis through the overexpression of PGC-1α attenuates aging-associated derangements [71]. Combined, these data strongly suggest that mitochondrial dysfunction contributes to pathological aging.
While a lot of research is currently being dedicated to understanding mitochondrial quality control with aging, including mechanisms influencing mitophagy and the removal of damaged mitochondria [18, 38, 50], this review will focus on the underlying mechanisms impairing mitochondrial function with aging, as well as how lifestyle modifications prevent age-associated skeletal muscle derangements. Additionally, we will discuss the mechanisms causing an increase in mitochondrial-derived ROS with aging which is of paramount importance, as increased ROS emission has been associated with MU loss and abnormal morphology [36], muscle fiber atrophy [34, 49], inflammation [32] and apoptosis [65]. Additionally, transgenic and pharmacological approaches that attenuate mitochondrial ROS have been shown to preserve mitochondrial content, increase muscle mass, and prolongs lifespan [39, 63], while conversely decreasing the antioxidant capacity of muscle compromises neuromuscular junction morphology as well as muscle mass and force [29]. Altogether, these data strongly implicate mitochondrial ROS as a cause of aging-related sarcopenia. However, we must acknowledge that physical inactivity similarly affects mitochondria and skeletal muscle physiology, and it is, therefore, difficult to distinguish the effects of aging from a sedentary lifestyle that usually manifests in older individuals. Rather than focusing on the limitations of our current knowledge, instead of these data further implicate the necessity of physical activity to promote healthy aging.
Role of Mitochondria on Redox Balance
The first description of “free radical” comes from 1954 [10]. By definition, one atom or molecule containing one or more unpaired electrons is called a free radical. These radicals can be produced from homolytic, heterolytic, or redox reactions. Free radicals can be nominated reactive oxygen species, however, the term reactive oxygen species (ROS) is a generic statement for oxygen-centered radicals, but also, for non-radical but still reactive derivatives of oxygen, such as hydrogen peroxide (H2O2) [48]. Excessive ROS production without suitable buffering is called oxidative stress [64]. This condition can damage biomolecules, leading to cell dysfunction and eventually cell death.
The equilibrium between the production and breakdown/buffering of ROS is referred to as the redox state within muscle and is maintained in a narrow range mainly by the action of antioxidant system. In humans, there are two broadly defined systems that work together to maintain the redox balance: namely enzymatic and non-enzymatic antioxidants (e.g. GSH, vitamin E, carotenoids, vitamin C, bilirubin) [55]. The main mechanism of action of the non-enzymatic antioxidants is the capacity to donate one electron to electron-unpaired molecules. This reduction mechanism decreases the reactivity of the molecules, preserving adjacent biomolecules. On the other hand, the enzymatic antioxidant system utilizes a sophisticated system composed of proteins, substrates, and cofactors to inactivate ROS. These enzymes are distributed in all cellular compartments and work together to keep the redox state in physiological ranges. Among these enzymes are superoxide dismutase (SOD), catalase, glutathione peroxidase system (GPX), peroxiredoxins (PRX), and thioredoxins (TRX). Although there is an apparent overlap between these enzymes as a result of them sharing the same substrate (H2O2, with exception of SOD), they have different roles in the cells because of the affinity with substrates, protein residues potentially oxidizable and cellular distribution [5]. Thus, the balance between mitochondrial ROS production and buffering by enzymatic and non-enzymatic systems keep superoxide/H2O2 in narrow concentrations. Transitory variations on the production of these molecules lead to adaptations of these antioxidant processes in order to maintain homeostasis, however, chronic unbalances may result in oxidative stress with damage of biomolecules.
All redox reactions involve the transfer of electrons between one donor and one acceptor [48], and in biological systems, oxidation–reduction (redox) reactions play a fundamental role in life. Indeed, oxidative stress has been suggested as one of the theories of aging, however, living cells have adapted to these reactive species by developing a sophisticated system of alternative pathways to create second messengers and less reactive compounds. Specifically, the single-electron reduction of O2 results in the formation of superoxide radical anion (O·−2) [48], however, superoxide can react with biomolecules or it can react with nitric oxide (NO) to form peroxynitrite (ONOO−), a highly reactive compound. Also, superoxide can be converted into hydrogen peroxide (H2O2), a less reactive compound, by SOD action (Mn-SOD and Cu/Zn-SOD for mitochondrial and cytosolic isoforms, respectively). Although not categorized as a free radical, H2O2 is considered a ROS, and can also be transformed into a hydroxyl radical (HO·) in the presence of transition metals, the predominant reactions are Fenton and Haber–Weiss reactions. On the other hand, H2O2 can be converted to inert H2O and O2, by the activity of catalase. Regardless of the complexity, the production of superoxide is considered paramount to understand redox biology, as these ROS species are directly required for the production of ONOO−, H2O2, and HO·.
Although there are many sites of superoxide production within the muscle, mitochondria have a primary role in redox balance. From a quantitative viewpoint, mitochondria are thought to be the largest contributors to intracellular oxidant production [5]. Mitochondria generate ATP in an oxygen-dependent process, called oxidative phosphorylation (OXPHOS). Briefly, OXPHOS is a process that harnesses the free energy released as electrons move through the respiratory chain to pump protons from the mitochondrial matrix, against a concentration gradient, into the intermembrane space. This process generates an electrochemical gradient (Δp), also called proton-motive force, between these two mitochondrial compartments. This proton-motive force is harnessed by F1F0 ATP synthases to produce ATP from ADP + Pi, resulting in the dissipation of Δp [47]. In general, an increase in the proton-motive force is correlated with increased ROS production while a decrease in the proton-motive force reduces ROS generation.
The first descriptions of mitochondrial superoxide radical anion (O·−2) production and hydrogen peroxide (H2O2) come from the 1970 s [41]. Initially, free radical mitochondrial production was thought to be a result of inefficiency of electron transfer within the electron transfer system (ETS) and was harmful to cell function. However, the notion that redox biology is complicated and intricately regulated, has gained attention in recent years, as it is now widely recognized that ROS serves as an important signaling molecule, in addition to the classically considered harmful compounds attributed to the aging process [22]. Within mitochondria, the major sites of superoxide production are believed to be within complex I and complex III of the electron transport chain (ETC). Complex I ROS production works based on NADH/NAD+ ratio, with redox potential (Eh) equal to − 28 mV, furthermore other ROS production sites work based in function of ubiquinol/ubiquinone (QH2/Q) ratio, which has an Eh of +20 mV. Additionally, other mitochondrial sources of ROS have been described, such as 2-oxoglutarate dehydrogenases (OGDH), pyruvate dehydrogenases (PDH), and complex II (subunit IIF) [5]. The exact location of ROS generation is important because it determinates the rates of superoxide/H2O2 production, and potentially the biological effects. Additionally, it is noteworthy that complex I can produce superoxide only on the matrix side, whilst complex III is able to produce superoxide on both sides of the inner mitochondrial membrane. These vector differences between mitochondrial complexes may have profound effects on mitochondrial function and cellular signaling. For instance, ROS production to the mitochondrial matrix can react with mtDNA and other mitochondrial proteins, impairing mitochondrial function; however, ROS emission within the intermembrane space may have a more pronounced role as a signaling molecule, since it is easier for these molecules to diffuse to the cytosol and react with cytosolic molecules. The topology of ROS production is not totally understood; however, some important data has emerged which can impact the aging process. For instance, we have shown that increased ROS production may be specific for individual sites in skeletal muscle mitochondria of aged people [26] as while H2O2 production was the same when mitochondria were stimulated with succinate (a classical substrate for mitochondrial complex II), on the other hand, a substrate for mitochondrial complex I (i.e. pyruvate) increased ROS emission in older people compared to young. This information may be particularly important since the FMN site on complex I only produces ROS on the mitochondrial matrix side [5], and therefore may preferentially damage mtDNA. This ROS-induced mtDNA alteration has been proposed as one of the theories of aging, which results in a vicious circle based on mitochondrial oxidative stress [23].
Mitochondrial Control of ROS Production: Implications to Aging-Related Sarcopenia
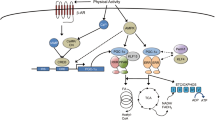
Age-associated sarcopenia develops as a result of anabolic resistance (under feeding or exercise stimulation) or increased protein breakdown (during fasting and fed conditions) [6]. While protein synthesis is an energetically demanding process, a direct link between compromised mitochondrial bioenergetics and anabolic resistance/attenuated protein synthesis remains to be defined. Alternatively, increased mitochondrial ROS emission has been implicated in the aging-related atrophy process. The causal relationship between ROS unbalance and atrophy has been shown by genetic deletion of the muscle antioxidant enzyme Cu/Zn-SOD, which results in muscle oxidative damage, myofiber atrophy, and neuromuscular junction degeneration [59]. These cellular effects may be direct through oxidation of cysteine residues on specific protein-targets, or indirect, as is the case for ROS-induced apoptosis [65]. It is well-established that mitochondrial play an important role in skeletal muscle homeostasis by energy supply, ROS signaling, and apoptosis [7, 22, 26]. For this reason, it has been speculated that mitochondrial ROS emission may have a role in apoptosis and aging-related muscle loss. Mitochondria can trigger apoptosis by several mechanisms, one of them is the opening of mitochondrial permeability transition pores (mPTP). The complete regulation of mPTP opening is not fully elucidated, however, mitochondrial ROS production seems to be involved in this process. In fact, mitochondria from older people show a reduction in calcium retention capacity, suggesting an increase sensitization for mPTP opening [20]. While rodent data has generated a strong link between mitochondrial ROS unbalance, apoptosis and atrophy, the mechanisms behind these events need to be investigated. In addition, although elegant rodent models provide compelling evidence to link mitochondrial ROS with age-associated pathologies, the data in humans remain ambiguous, in vitro assessments of mitochondrial ROS emission do not increase in association with redox stress [3, 7, 13, 26]. These contradictory findings suggest that either mitochondria are not responsible for the increased oxidative stress with human aging or, alternatively, contemporary in vitro assessment of mitochondrial ROS emission do not accurately reflect in vivo responses. The most important factor involved in the regulation of in vivo mitochondrial ROS production is ATP production [48]. Mitochondrial membrane potential is modulated by the rate of ATP production, which is dependent on ADP availability. In this respect, rates of ROS emission have almost exclusively been determined in the absence of ADP (usually with oligomycin), and therefore have examined the capacity for mitochondrial ROS production under artificial conditions, which likely does not reflect the in vivo situation. In fact, resting skeletal muscle has ~ 25–100 μM free ADP [51], and from a physiological standpoint, it is, therefore, more appropriate to examine mitochondrial bioenergetics (respiration and ROS emission) in the presence of submaximal concentrations of ADP. As a result, previous data from human skeletal muscle may have underestimated the importance of mitochondrial ROS in the aging process. In support of this supposition, our recent data has shown that while maximal ROS (absence of ADP) is not altered with aging, in the presence of physiological ADP concentrations, ROS emission in older subjects is higher. Additionally, our work demonstrates a reduction in mitochondrial ADP sensitivity with aging [26] which can contribute to this observed higher mitochondrial ROS emission under low ADP concentrations. Since mitochondrial ADP transport can influence several mechanisms associated with aging (e.g. ROS, autophagy, and motor unit retention), the regulation of the key mitochondrial membrane ADP transporter ANT may represent a nexus point for numerous processes that influence aging. Therefore, a molecular understanding of ANT, and ultimately mitochondrial ADP sensitivity, may have a broad impact on our understanding of aging.
Adenine nucleotide translocase (ANT) appears to be modulated by aging [12]. Indeed, ANT can be regulated through several post-translational modifications, such as glutathionylation, acetylation, and oxidation [15, 45, 73]. Since the reduction of ADP sensitivity results in increased mitochondrial ROS production, oxidation has been indicated to be one candidate to modulate ANT regulation in aging, and although this remains to be tested in humans, redox modification of ANT has been identified with aging in rodents and housefly muscles [73]. While further work is necessary to elucidate this relationship in humans, hyperacetylation of ANT is a possible alternative mechanism to consider [45] since SIRT-deacetylase activity has been shown reduced with aging [74]. Regardless of the underlying mechanism, a decrease in ANT function can increase mitochondrial ROS production rates, activating NF-κB signaling and nuclear sequestration of FoxO. Under NF-κB activation, FoxO regulates the ubiquitin proteolysis system and mitophagy, forming a vicious cycle over mitochondrial dynamics and quality control [60].
In addition to ANT function, mitochondrial content is thought to indirectly affect mitochondrial ADP responsiveness. In this respect, increasing mitochondrial content may be preventative in age-associated redox stress. Indeed, the overexpression of PGC-1α, a transcription co-activator implicated in the regulation of mitochondrial biogenesis, increased mitochondrial content, muscle cross-sectional area, and longevity in the aged mouse [71]. Additionally, improving mitochondrial dynamics and mitophagy (removal of damaged or dysfunction mitochondrial by autophagosomes) can similarly improve the quality control of mitochondria, ultimately increasing mitochondrial bioenergetics in the absence of stimulating PGC-1α [38]. Several studies show that the preservation of mitochondrial quality control improves muscle health, sarcopenia and insulin-resistance aging-associated. For instance, 4 weeks of Parkin overexpression, a protein involved in mitochondrial quality control, increases muscle force-production and attenuates sarcopenia in aging muscle [38]. Importantly, Parkin overexpression was associated with a reduction on oxidative stress, measured by 4-HNE content, which reinforces the crosstalk between mitochondrial ROS production, mitophagy, and sarcopenia.
It is important to recognize some inconsistent data in the literature. For instance, mitochondria from older people have been reported to display mild uncoupling, which would decrease ROS rates [1, 12]. However, in our recent study, there were no differences in various indexes of mitochondrial uncoupling with aging [26]. Also, decreased ADP responsiveness through the genetic ablation of ANT has been associated with an attenuation in ROS emission, however, our data suggest a reduction of ADP/ATP transport increases ROS production. It is likely that these differences reflect compensatory adaptations in a genetic animal to maintain cellular survival, as ablation of ANT would be expected to dramatically increase ROS, while the observed uncoupling may represent a feedback-loop to prevent apoptosis and cellular death. Similarly, in our study, a reduction in ADP transport may represent a mechanism to stimulate mitochondrial biogenesis, as the expected minor increase in cytosolic ADP and induction of mitochondrial ROS have both been linked to PGC-1α mediated gene transcription. Regardless of these possible benefits, it appears that an age-associated increase in ROS favors a detrimental phenotype, and therefore the potential feed-back-loops (uncoupling and gene transcription) are not sufficient.
Lifestyle Modifications
Since one of the hallmarks of muscle aging is the presence of oxidative stress in aged skeletal muscle [7, 26], additional stimuli resulting in attenuated redox stress may be required to mitigate aging-related declines on skeletal muscle mass and function. In this context, physical inactive and poor dietary habits are known to be risk factors for the development of many chronic diseases, including diabetes, cancer, hypertension, and musculoskeletal disorders. In fact, it has been reported that the amount of time spent being physically active decreases with age, which can potentiate/cause aging-related conditions [31]. Since lifestyle modifications can promote health, well-being, and functional improvements to minimize the appearance and/or progression of aging-related diseases, exercise and nutrition interventions may be particularly beneficial in these conditions [8, 42].
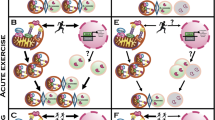
Although elderly individuals typically present with an unbalanced redox state, it is unclear if the observed increase in redox stress associated with aging is indirectly influenced by a lack of physical activity, as exercise training improves the antioxidant capacity of muscle, mitochondrial ADP responsiveness and ultimately redox control [26] (Please, see Fig. 1 for a summary of aging and physical activity on mitochondrial function and redox balance). Additionally, exercise training improves cardiovascular function, muscle mass, strength, bone density, vascularization among others in adults and elderly people, and therefore represents a comprehensive preventative strategy. Therefore, while physically inactivity likely contributes to pathological aging, importantly, regardless of age, individuals can benefit from the regular practice of physical exercise. The link between the effects of exercise on mitochondrial function and aging can best be exemplified by the observation that exercise prevents severe mitochondrial dysfunction, aging, and prolongs life expectancy in a genetically modified mouse prone to mtDNA deletions (PolG mutant), while also preserving muscle mass, increasing mitochondrial content and reducing mtDNA damage [58]. Altogether, these data highlight the beneficial effects of exercise training with respect to mitigating age-associated changes within skeletal muscle. In addition to these intracellular mechanisms, redox unbalance has been linked to compromised neuromuscular junction morphology [29, 50], a mechanism believed to contribute to age-associated sarcopenia [24]. While not tested in PolG mice, exercise training has been shown to prevent the loss of neuromuscular innervation, as master athletes display preserved motor unit number and neuromuscular stability [54]. Therefore, it appears that exercise training in both humans and rodents improves muscle function, muscle mass, and inflammation, which may be associated mitochondrial bioenergetics [21, 26, 33, 36, 58]. In the next sections we describe the positive effects of exercise and nutritional lifestyle interventions within the elderly population. Additionally, we highlight how these interventions can target mitochondrial function and redox balance in order to support the beneficial outcomes of these strategies.

Aging and physical activity on mitochondrial function and redox balance
Exercise
Exercise is a front-line intervention for the prevention and treatment of a large range of chronic diseases, including, type-2 diabetes mellitus (T2DM), metabolic syndrome, neurological disease, osteoporosis, and cardiovascular disease. Exercise training can be classically distinguished as endurance or resistance in nature, although all weight-bearing exercise should be considered on a continuum of power and duration. Endurance “type” exercise is composed from minutes up to several hours at various intensities, incorporating repetitive, low-resistance load contractions. On the other hand, resistance focussed training consists of short-duration activity at high intensity, with relatively few repetitions. The divergent adaptations as a result of these two kinds of exercise modalities can easily be observed, as in general terms, but not exclusively, endurance training improves cardiac function and aerobic performance while resistance training is known to increase muscle mass and strength. Older individuals benefit from both kinds of exercise training, and therefore a multicomponent plan of physical training, based on exercises to improve force-production, aerobic fitness, balance, and motor coordination, has been suggested for the elderly population.
Aerobic training is well established as a strategy to induce mitochondrial biogenesis and improvements on mitochondrial function in skeletal muscle. These parameters are inversely associated with age-related chronic diseases. Indeed, trained older individuals can present with mitochondrial ATP production and whole-body health status at the same level than young sedentary people. This demonstrates the independent positive effects of exercise over skeletal muscle mitochondrial function and total body health [62]. Specifically, aerobic training increases gene expression related to mitochondrial function (COX4 and ND4) and mitochondrial biogenesis (PGC-1α, NRF-1, and TFAM) [44]. Indeed, 12 weeks of endurance exercise training increase cardiorespiratory fitness in older people. These improvements are associated with increments in skeletal muscle mitochondrial content, such as cardiolipin and mtDNA. Additionally, aerobic training increases mitochondrial NADH oxidase and succinate oxidase activity [44]. Furthermore, aerobic training increases catalase expression, which can improve the antioxidant defense in the skeletal muscle of older people [18].
On the other hand, strength training is well recognized as one of the most effective strategies to reduce age-related skeletal muscle loss. In older individuals, the age-related loss of skeletal muscle mass is primarily caused by a reduction in muscle fiber cross-sectional area, which predominately occurs in type II fibres [70]. Type II fibres are distinguished from type I fibres by a greater force- and power- producing capacity. The atrophy of type II fibres in elderly subjects is most likely related to a decrease in physical activity, muscle denervation and/or redox unbalance [26, 49]. Resistance-type exercise training has been shown to increase skeletal muscle mass and strength in a variety of older and more compromised populations [35, 56]. More specifically, various studies have demonstrated that 12–24 weeks structured resistance-type exercise program can lead to a 1–2 kg gain in whole-body lean mass and a substantial (20–50%) increase in muscle strength in the older population [27, 40]. On the muscle fiber level, the hypertrophic response to resistance exercise training predominantly occurs in type II muscle fibres [56]. Heavy resistance exercise training has been demonstrated to effectively increase muscle mass and strength in women over the age of 90 years [16]. These findings indicate that the anabolic properties of resistance-type exercise training on skeletal muscle mass and strength are not restricted by an individual’s age. While endurance training is known to induce mitochondrial adaptations, resistance ‘type’ exercise training is also capable of inducing functional and mitochondrial improvements in elderly subjects. In a recent study, we demonstrated an increase in maximal oxidative phosphorylation (OXPHOS) after 12-week of resistance exercise training in elderly subjects. Also, after the training period, mitochondrial ADP sensitivity was increased in the skeletal muscle of the participants, and the ability of ADP to decrease mitochondrial ROS emission was increased [26]. These data demonstrate the ability of exercise to improve mitochondrial bioenergetics in the skeletal muscle of older individuals.
High-intensity interval training (HIIT) has been proposed as an alternative training based on extremely low-volume of sessions. Older adults who performed 12-week of HIIT on cycle ergometer showed an increase in several markers of mitochondrial content in skeletal muscle [72]. Additionally, when comparing different models of exercise training, 12-week of HIIT increased mitochondrial respiration while there was no effect following resistance training. However, when the data from HIIT training participants were normalized by mitochondrial protein content, there was no effect of HIIT on mitochondrial respiration [72]. These data reinforce the importance of mitochondrial biogenesis in response to exercise training, however, the influence of HIIT on redox biology within aging remains to be determined. Since a multicomponent plan for exercise training has been suggested for older people, a mix of endurance and resistance training could be an alternative to improve the cardiovascular system, mitochondrial function, and skeletal muscle strength. Indeed, a combination of exercise training, composed of endurance and resistance training was able to increase mitochondrial oxidative phosphorylation and body composition in older people [28].
Nutrition
In addition to exercise, nutrition interventions can be advantageous to increase muscle health, antioxidant capacity, and mitochondrial bioenergetics. Currently, there are several nutritional experiments aiming to improve quality of life and longevity in humans. Polyphenols compounds and polyunsaturated fatty acids are among the most interventions studied. Here, we highlight how these dietetic manipulations can impact mitochondrial function, redox status, and inflammation in older people.
Aging is linked with low-grade chronic inflammation, which has been implicated in skeletal muscle health and sarcopenia [2]. Omega-3 (n-3) polyunsaturated fatty acids (PUFAs) are receiving increasing attention due to their anti-inflammatory action in several conditions, such as cancer, diabetes, and metabolic syndrome. The main biological effects come from the presence of docosahexaenoic (DHA) and eicosapentaenoic acid (EPA), which can be incorporated in the sarcolemmal and mitochondrial phospholipidic membranes [25, 43], increasing fluidity due to the presence of unsaturated binding in its structural composition. Importantly, omega-3 PUFA supplementation increases mitochondrial ADP sensitivity without alterations in mitochondrial content or oxidative stress markers in healthy humans [25]. Recently, the effects of n-3 PUFA supplementation were tested in skeletal muscle mitochondria from older men. Sixteen weeks of n-3 PUFA supplementation was not able to change maximal mitochondrial oxygen consumption, however, there was a decrease in mitochondrial ROS emission in the presence of several substrates [37]. These findings raise the positive biological effects of n-3 PUFA on mitochondrial function and redox state, which can lower inflammation and improve muscle function in elderly.
Omega-3 PUFA can present positive effects on muscle protein synthesis (MPS) in younger and older adults [66]. Supplementation with n-3 PUFA increased MPS rate and attenuates muscle mass loss during immobilization [43]. In older humans, 6 months of n-3 PUFA supplementation attenuated muscle mass loss and muscle strength [67]. Also, these effects of n-3 PUFA supplementation in older people were associated with alterations on several genes set pathways related to mitochondrial function [75]. These results from n-3 PUFA supplementation and muscle mass maintenance reinforce the role of mitochondrial function in redox balance and inflammation status of older people.
Several other nutritional compounds possess some antioxidant activity or impact mitochondrial function and have therefore been applied to balance redox state in older people and slow the impact of oxidative stress. Among them, much attention has been given to the therapeutic effect of dietary polyphenols, which have been shown positive effects on age-related oxidative stress, inflammation and toxin accumulation [61]. Resveratrol is a non-flavonoid belonging to the stilbene class of compounds found abundantly is peanuts, blueberries, pine nuts, and grapes. Resveratrol is believed to modulate the expression and activity of different molecular targets, such as AMP-regulated kinase (AMPK) and NAD-dependent deacetylase Sirt-1 [4]. Mitochondrial content and function are regulated by these molecular pathways, and both appear to be altered in aging [74]. Therefore, resveratrol has been studied to restore these molecular pathways and redox equilibrium in senescent cells. Although several studies in cell culture or laboratory animals’ models show positive effects of resveratrol on antioxidant defense, inflammation, and lifespan [4, 53], human studies are not so consistent. Resveratrol supplementation for 6 weeks in older adults increases the expression of several mitochondrial genes, however, it is noteworthy that mitochondrial function or redox state was not directly addressed in this study [53]. In addition, resveratrol supplementation seems to blunt exercise training-induced adaptations, such as improvements on VO2max and angiogenesis, in aged humans [17]. Due to antioxidant effects of resveratrol, it may be speculated that exercise-induced cellular signals (mainly ROS production) could be blunted by resveratrol supplementation. It is noteworthy that other polyphenols, such as curcumin and quercetin, have been pointed out as aging regulator molecules due to their antioxidant’s proprieties. However, regarding inflammation and oxidative stress aged-related, the effects of these antioxidants are not clear yet. Also, it is still unknown how these molecules can act in the presence of exercise-induced adaptations on mitochondrial function and redox status.
In summary, nutrition interventions and exercise training may be modulated to target mitochondrial function in skeletal muscle of aged people. These interventions can restore mitochondrial function, content and redox state, which could support the positive effects on inflammation, oxidative stress, and sarcopenia in this population. However, it remains to be demonstrated the combined effects of exercise training and nutrition interventions on mitochondrial function. Also, these interventions need to be more elucidated in several situations that are common for elderly people, such as immobility, physical inactivity or metabolic diseases.
Conclusion
One hallmark of aging is a decrease in mitochondrial content and redox unbalance. Although historically ROS production was seen as harmful to cell function, currently ROS must be interpreted as signaling molecules, controlling inflammation, skeletal muscle mass, atrophy, and mitochondrial quality, by mitophagy, fusion, and fission. This new vision of ROS requires future studies examining mitochondrial ROS emission/buffering under physiological conditions as well as following different stresses (i.e. immobility, exercise, and/or nutrition interventions) to fully delineate a mechanistic relationship between mitochondrial bioenergetics, anabolic resistance, atrophy and ultimately sarcopenia. Additionally, a detailed topology of ROS emission during aging is necessary for a better understanding of the role of free radicals in mitochondrial damage and retrograde cell signaling. Nevertheless, despite the absence of a clear optimal exercise prescription, exercise training has consistently demonstrated a protective effect on pathological aging, and therefore should be advocated as an affordable and reliable intervention.
References
Amara CE, Shankland EG, Sharon AJ, Marcinek DJ, Kushmerick MJ, Conley KE. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc Natl Acad Sci USA. 2007;104(3):1057–62.
Arnold P, Njemini R, Vantieghem S, Gorus E, Pool-Goudzwaard A, Buyl R, Bautmans I. Reaction time in healthy elderly is associated with chronic low-grade inflammation and advanced glycation end product. Exp Gerontol. 2018;108(15):118–24.
Barrientos A, Casademont J, Cardellach F, Ardite E, Estivill X, Urbano-Márquez A, Fernández-Checa JC, Nunes V. Qualitative and quantitative changes in skeletal muscle mtDNA and expression of mitochondrial-encoded genes in the human aging process. Biochem Mol Med. 1997;62(2):165–71.
Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high calorie diet. Nature. 2006;444(7117):337–42.
Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. 2016;100(11):14–31.
Breen L, Phillips SM. Interactions between exercise and nutrition to prevent muscle waste during ageing. Br J Clin Pharmacol. 2012;75(3):708–15.
Capel F, Rimbert V, Lioger D, Diot A, Rousset P, Mirand PP, Boirie Y, Morio B, Mosoni L. Due to reverse electron transfer, mitochondrial H2O2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mech Ageing Dev. 2005;126(4):505–11.
Craig BW, Everhart J, Brown R. The influence of high-resistance training on glucose tolerance in young and elderly subjects. Mech Ageing Dev. 1989;49(2):147–57.
Chabi B, Ljubicic V, Menzies KJ, Huang JH, Sallem A, Hood DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008;7(1):2–12.
Commoner B, Townsend J, Pake GE. Free radicals in biological materials. Nature. 1954;9(174):689–91.
Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyère O, Cederholm T, Cooper C, Landi F, Rolland Y, Sayer AA, Schneider SM, Sieber CC, Topinkova E, Vandewoude M, Visser M, Zamboni M. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing. 2019;48(1):16–31.
Diolez P, Bourdel-Marchasson I, Calmettes G, Pasdois P, Detaille D, Rouland R, Gouspillou G. Hypothesis on skeletal muscle aging: mitochondrial adenine nucleotide translocator decreases reactive oxygen species production while preserving coupling efficiency. Front Physiol. 2015;6(369):1–9.
Distefano G, Standley RA, Dubé JJ, Carnero EA, Ritov VA, Stefanovic-Racic M, Toledo FG, Piva SR, Goodpaster BH, Coen PM. Chronological age does not influence ex vivo mitochondrial respiration and quality control in skeletal muscle. J Gerontol A Biol Sci Med Sci. 2017;72(4):535–42.
Dupuis L, Aguilar JLG, Echaniz-Laguna A, Eschbach J, Rene F, Oudart H, Halter B, Huze C, Schaeffer L, Bouillaud F, Loeffler JP. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS One. 2009;4(4):5390.
Feng J, Zhu M, Schaub MC, Gehrig P, Roschitzki B, Lucchinetti E, Zaugg M. Phosphoproteome analysis of isoflurane-protected heart mitochondria: phosphorylation of adenine nucleotide translocator-1 on Tyr194 regulates mitochondrial function. Cardiovasc Res. 2008;80(1):20–9.
Fiatarone MA, Marks EC, Ryan ND, Meredith CN, Lipsitz LA, Evans WJ. High-intensity strength training in nonagenarians. Effects on skeletal muscle. JAMA. 1990;263(22):3029–34.
Gliemann L, Schmidt JF, Olesen J, Biensø RS, Peronard SL, Grandjean SU, Mortensen SP, Nyberg M, Bangsbo J, Pilegaard H, Hellsten Y. Resveratrol blunts the positive effects of exercise training on cardiovascular health in aged men. J Physiol. 2013;591(20):5047–59.
Gouspillou G, Bourdel-Marchasson I, Rouland R, Calmettes G, Biran M, Deschodt-Arsac V, Miraux S, Thiaudiere E, Pasdois P, Detaille D, Franconi JM, Babot M, Trézéguet V, Arsac L, Diolez P. Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell. 2014;13(1):39–48.
Gouspillou G, Hepple RT. Editorial: mitochondria in skeletal muscle health, aging and diseases. Front Physiol. 2016;7(1):446.
Gouspillou G, Sgarioto N, Kapchinsky S, Purves-Smith F, Norris B, Pion CH, Barbat-Artigas S, Lemieux F, Taivassalo T, Morais JA, Aubertin-Leheudre M, Hepple RT. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014;28(4):1621–33.
Gram M, Vigelso A, Yokota T, Helge JW, Dela F, Hey-Mogensen M. Skeletal muscle mitochondrial H2O2 emission increases with immobilization and decreases after aerobic training in young and older men. J Physiol. 2015;593(17):4011–27.
Hamanaka R, Navdeep RC. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2011;35(9):505–13.
Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1955;11(3):1.
Hepple RT, Rice CL. Innervation and neuromuscular control in ageing skeletal muscle. J Physiol. 2016;594(8):1965–78.
Herbst EAF, Paglialunga S, Gerling C, Whitfield J, Mukai K, Chabowski A, Heigenhauser GJ, Spriet LL, Holloway GP. Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J Physiol. 2014;592(6):1341–52.
Holloway GP, Holwerda AM, Miotto PM, Dirks ML, Verdijk LB, van Loon LJC. Age-associated impairments in mitochondrial ADP sensitivity contribute to redox stress in senescent human skeletal muscle. Cell Rep. 2018;22(11):2837–48.
Holwerda AM, Overkamp M, Paulussen KJM, Smeets JSJ, van Kranenburg J, Backx EMP, Gijsen AP, Goessens JPB, Verdijk LB, van Loon LJC. Protein supplementation after exercise and before sleep does not further augment muscle mass and strength gains during resistance exercise training in active older men. J Nutr. 2018;148(11):1723–32.
Irving BA, Lanza IR, Henderson CG, Rao RR, Spiegelman CM, Nair KS. Combined training enhances skeletal muscle mitochondrial oxidative capacity independent of age. J Clin Endocrinol Metab. 2015;100(4):1654–63.
Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A, Van Remmen H. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010;24(5):1376–90.
Janssen I, Heymsfield SB, Ross R. Low relative skeletal muscle mass (sarcopenia) in older persons is associated with functional impairment and physical disability. J Am Geriatr Soc. 2002;50(5):889–96.
Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc. 2004;52(1):80–5.
Khodr B, Khalil Z. Modulation of inflammation by reactive oxygen species: implications for aging and tissue repair. Free Radic Biol Med. 2001;30(1):1–8.
Kim Y, Triolo M, Hood DA. Impact of aging and exercise on mitochondrial quality control in skeletal muscle. Oxid Med Cell Longev. 2017;2017:3165396.
Kondo H, Miura M, Itokawa Y. Oxidative stress in skeletal muscle atrophied by immobilization. Acta Physiol Scand. 1991;142(4):527–8.
Kosek DJ, Kim JS, Petrella JK, Cross JM, Bamman MM. Efficacy of 3 days/wk resistance training on myofiber hypertrophy and myogenic mechanisms in young vs. older adults. J Appl Physiol. 2006;101(2):531–44.
Kuwahara H, Horie T, Ishikawa S, Tsuda C, Kawakami S, Noda Y, Kaneko T, Tahara S, Tachibana T, Okabe M, Melki J, Takano R, Toda T, Morikawa D, Nojiri H, Kurosawa H, Shirasawa T, Shimizu T. Oxidative stress in skeletal muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radic Biol Med. 2010;48(9):1252–62.
Lalia AZ, Dasari S, Robinson MM, Abid H, Morse DM, Klaus KA, Lanza IR. Influence of omega-3 fatty acids on skeletal muscle protein metabolism and mitochondrial bioenergetics in older adults. Aging. 2017;9(4):1096–115.
Leduc-Gaudet JP, Reynaud O, Hussain SN, Gouspillou G. Parkin overexpression protects from aging-related loss of muscle mass and strength. J Physiol. 2019;597(7):1975–91.
Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12(6):668–74.
Leenders M, Verdijk LB, van der Hoeven L, van Kranenburg J, Nilwik R, van Loon LJ. Elderly men and women benefit equally from prolonged resistance-type exercise training. J Gerontol A Biol Sci Med Sci. 2013;68(7):769–79.
Loschen G. Respiratory chain linked H2O2 production in pigeon heart mitochondria. FEBS Lett. 1971;18(2):1–3.
Macaluso A, Vito G. Muscle strength, power and adaptations to resistance training in older people. Eur J Appl Physiol. 2004;91(4):450–72.
McGlory C, Gorissen SHM, Kamal M, Bahniwal R, Hector AJ, Baker SK, Chabowski A, Phillips SM. Omega-3 fatty acid supplementation attenuates skeletal muscle disuse atrophy during two weeks of unilateral leg immobilization in healthy young women. FASEB J. 2019;33(3):4586–97.
Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE, Goodpaster BH. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J Gerontol A Biol Sci Med Sci. 2006;61(6):534–40.
Mielke C, Lefort N, McLean CG, Cordova JM, Langlais PR, Bordner AJ, Te JA, Ozkan SB, Willis WT, Mandarino LJ. Adenine nucleotide translocase is acetylated in vivo in human muscle: modeling predicts a decreased ADP affinity and altered control of oxidative phosphorylation. Biochemistry. 2014;53(23):3817–29.
Min K, Smuder AJ, Kwon OS, Kavazis NA, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization induced muscle atrophy. J Appl Physiol (1985). 2011;111(5):1459–66.
Mitchel P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–8.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
Nilwik R, Snijders T, Leenders M, Groen BB, van Kranenburg J, Verdijk LB, van Loon LJ. The decline in skeletal muscle mass with aging is mainly attributed o a reduction in type II muscle fiber size. Exp Gerontol. 2013;48(5):492–8.
O’Leary MF, Vainshtein A, Iqbal S, Ostojic O, Hood DA. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am J Physiol Cell Physiol. 2013;304(5):422–30.
Perry CGR, Kane DA, Herbst EA, Mukai K, Lark DS, Wright DC, Heigenhauser GJ, Neufer PD, Spriet LL, Holloway GP. Mitochondrial creatine kinase activity and phosphate shuttling are acutely regulated by exercise in human skeletal muscle. J Physiol. 2012;590(21):5475–86.
Picard M, Ritchie D, Thomas MM, Wright KJ, Hepple RT. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell. 2011;10(6):1047–55.
Pollack RM, Barzilai N, Anghel V, Kulkarni AS, Golden A, O’Broin P, Sinclair DA, Bonkowski MS, Coleville AJ, Powell D, Kim S, Moaddel R, Stein D, Zhang K, Hawkins M, Crandall JP. Resveratrol improves vascular function and mitochondrial number but not glucose metabolism in older adults. J Gerontol A Biol Sci Med Sci. 2017;72(12):1703–9.
Power GA, Allen MD, Gilmore KJ, Stashuk DW, Doherty TJ, Hepple RT, Taivassalo T, Rice CL. Motor unit number and transmission stability in octogenarian world class athletes: can age-related deficits be outrun? J Appl Physiol (1985). 2016;121(4):1013–20.
Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88(4):1243–76.
Robinson MM, Dasari S, Konopka AR, Johnson ML, Manjunatha S, Esponda RR, Carter RE, Lanza IR, Nair KS. Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans. Cell Metab. 2017;25(3):581–92.
Rosenberg IH. Sarcopenia: origins and clinical relevance. Clin Geriatr Med. 2011;27(5):337–9.
Safdar A, Bourgeois JM, Obgorn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci USA. 2011;108(10):4135–40.
Sakellariou GK, McDonagh B, Porter H, Giakoumaki II, Earl KE, Nye GA, Vasilaki A, Brooks SV, Richardson A, Van Remmen H, McArdle A, Jackson MJ. Comparison of whole body SOD1 knockout with muscle-specific SOD1 knockout mice reveals a role for nerve redox signaling in regulation of degenerative pathways in skeletal muscle. Antioxid Redox Signal. 2018;28(4):275–95.
Sanchez AMJ, Candau RB, Bernardi H. FoxO transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell Mol Life Sci. 2014;71(9):1657–71.
Serino A, Salazar G. Protective role of polyphenols against vascular inflammation, aging and cardiovascular disease. Nutrients. 2019;11(1):53.
Short KR, Vittone JL, Bigelow ML, Proctor DN, Rizza RA, Coenen-Schimke JM, Nair KS. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes. 2003;52(8):1888–96.
Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS, Marcinek DJ. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell. 2013;12(5):763–71.
Sies H. Oxidative stress: introductory remarks. London: Academic Press; 1985. p. 1–8.
Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5(5):415–8.
Smith GI, Atherton P, Reeds DN, Mohammed BS, Rankin D, Rennie MJ, Mittendorfer B. Omega-3 polyunsaturated fatty acids augment the muscle protein anabolic response to hyperaminoacidemia–hyperinsulinemia in healthy young and middle-aged men and women. Clin Sci (Lond). 2011;121(6):267–78.
Smith GI, Julliand S, Reeds DN, Sinacore DR, Klein S, Mittendorfer B. Fish oil-derived n-3 PUFA therapy increases muscle mass and function in healthy older adults. Am J Clin Nutr. 2015;102(1):115–22.
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–23.
UN. World population ageing. New York: UN; 2009.
Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab. 2007;292(1):151–7.
Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA. 2009;106(48):20405–10.
Wyckelsma VL, Levinger I, McKenna MJ, Formosa LE, Ryan MT, Petersen AC, Anderson MJ, Murphy RM. Preservation of skeletal muscle mitochondrial content in older adults: relationship between mitochondria, fibre type and high-intensity exercise training. J Physiol. 2017;595(11):3345–59.
Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci USA. 1998;95(22):12896–901.
Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24(8):464–71.
Yoshino J, Smith GI, Kelly SC, Julliand S, Reeds DN, Mittendorfer B. Effect of dietary n-3 PUFA supplementation on the muscle transcriptome in older adults. Physiol Rep. 2016;4(11):e12785.
Acknowledgements
Henver S. Brunetta scholarship was funded by the Coordination for the improvement of Higher Education Personnel (CAPES) – Brazil. Graham P. Holloway is funded by the Natural Sciences and Engineering Research Council of Canada (NSERC).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The author(s) declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Brunetta, H.S., Holwerda, A.M., van Loon, L.J.C. et al. Mitochondrial ROS and Aging: Understanding Exercise as a Preventive Tool. J. of SCI. IN SPORT AND EXERCISE 2, 15–24 (2020). https://doi.org/10.1007/s42978-019-00037-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42978-019-00037-1