
Abstract
Octreotide as a synthetic cyclic octapeptide is a somatostatin analog with longer half-life and more selectivity for inhibition of the growth hormone. The acetate salt of octreotide is currently used for medical treatment of somatostatin-related disorders such as endocrine and carcinoid tumors, acromegaly, and gigantism. Octreotide contains both cysteine and tryptophan residues which make the hydrolysis part of its amino acid analysis procedure very challenging. The current paper introduces a fast and additive-free method which preserves tryptophan and cysteine residues during the hydrolysis. Using only 6 M HCl, this hydrolysis process is completed in 30 min at 150 °C. This fast hydrolysis method followed by pre-column derivatization of the released amino acids with 4-N,N-dimethylaminoazobenzene-4ʹ-sulfonyl chloride (DABS-Cl) which takes only 20 min, makes it possible to do the complete amino acid analysis of an octreotide sample in a few hours. The highly stable-colored DABS-Cl derivatives can be detected in 436 nm in a reversed phase chromatographic system, which eliminates spectral interferences to a great extent. The amino acid analysis of octreotide acetate including hydrolysis, derivatization, and reversed phase HPLC determination was validated according to International Conference of Harmonization (ICH) guidelines.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
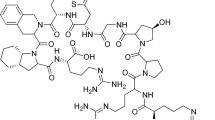
The peptide hormone somatostatin by inhibitory effect on the growth hormone regulates the endocrine system (Brazeau et al. 1973; Reichlin et al. 1983a, b). It also affects cell profiltration and secretion of a range of hormones including insulin and glucagon (Astruc et al. 2005). The limitations in the application of somatostatin for medical purposes according to its short half-life in vivo (a few min), has led to the synthesis of its more stable analogs. Octreotide is a synthetic cyclic octapeptide which in comparison to somatostatin exhibits a longer half-life (60–90 min in humans) and more selectivity for inhibition of the growth hormone (Bauer et al. 1982). The acetate salt of octreotide has been approved for the treatment of endocrine and carcinoid tumors and treatment of other somatostatin-related disorders such as acromegaly and gigantism (Woltering et al. 2005, 2008; Ludlam et al. 2011). The labeled octreotide has been used by imaging techniques to visualize tumors expressing somatostatin receptors (Albert et al. 1998; Hsieh et al. 1999). The structure of the octreotide acetate and its consisting amino acids can be seen in Fig. 1.

The amino acid composition and structure of octreotide
One of the most important quality control items for pharmaceutical grade peptides such as octreotide is the determination of their amino acid content. The amino acid analysis of peptides and protein samples typically includes two main steps, hydrolysis of the sample to obtain the free amino acids and then using an appropriate analytical technique to determine the amino acid content (Rutherfurd et al. 2009). The most widely used hydrolysis method is the conventional acid hydrolysis with 6 M HCl for 24 h at 110 °C (Moore et al. 1951). One of the drawbacks of this method is the partial destruction of some amino acid residues such as glutamine, asparagine, and cysteine and their conversion to glutamate, aspartate, and cystine, respectively (Fountoulakis et al. 1998). The main problem with conventional acid hydrolysis is almost complete destruction and loss of the tryptophan residue (Silvestre 1997). Octreotide contains both cysteine and tryptophan residues which make the hydrolysis part of the analysis very challenging.
To overcome these limitations some new methods and also modifications to the conventional acid hydrolysis method has been introduced. The enzymatic hydrolysis is one of the methods that has been suggested for this purpose (Church et al. 1984). However, due to relative specificity of the proteases for certain residues, to achieve complete enzymatic digestion the sample should be incubated with several proteases and the reactions usually last long. Therefore, the application of this method is limited (Fountoulakis et al. 1998). The alkaline hydrolysis is another method which is specially designed to recover the tryptophan residues which are stable under basic conditions (Molnár-Perl 1997). The major drawback of the method is that serine, threonine, arginine, and cysteine are destroyed and the other amino acids are racemized (Greenstein et al. 1961). The conventional acid hydrolysis has also been modified either by changing the type of the acid (methanesulfonic acid, mercaptoethanesulfonic acid, or p-toluenesulfonic acid), or using some chemical additives to protect/convert certain amino acids to achieve better results (Rutherfurd et al. 2009; Fountoulakis et al. 1998; Dai et al. 2014). Almost all of the methods mentioned above require multiple and often tedious preparation steps specially in the case of chemical additives.
Very few publications with focus on the amino acid analysis of the octreotide can be found in the literature. The hydrolysis with 6 M HCl at 115 °C for 24 h by Albert et al. (1998) resulted in partial destruction of tryptophan and cysteine. They reported molar ratios of 0.78 and 0.20 for cysteine and tryptophan, respectively. Because in the structure of octreotide there are two cysteine residues and only one tryptophan residue, the expected molar ratios for them are 2 and 1, respectively. Hsieh et al. (1999) used 6 M HCl:trifluoroacetic acid (4:1, v/v) at 130 °C for 3 h for the hydrolysis step and almost recovered cysteine (molar ratio = 1.94) but they lost the tryptophan residue due to degradation in harsh hydrolysis medium. In another study the elemental analysis of the DOTA-(Tyr3)-octreotide gave the molar ratio of 1.2 for cysteine while in case of tryptophan only its presence was reported (Reubi et al. 2000).
For quantitative analysis of the free amino acids several analysis techniques can be used, among which high-performance liquid chromatography (HPLC), gas chromatography (GC), capillary electrophoresis (CE) can be mentioned (Kaspar et al. 2008, 2009; Zhang et al. 2012; Harder et al. 2011; Veledo et al. 2005). For simultaneous analysis of the amino acids—which are rather different in terms of polarity, volatility, and electrostatic characteristics—often derivatization techniques are employed (Kaspar et al. 2009). There are instrumental techniques which can be used for the determination of unmodified amino acids like CE or HPLC assisted by an electrochemical detector but they suffer from low sensitivity and incompatibility by the gradient elution, respectively (Yang et al. 2003; Casella et al. 2003). Tandem mass spectroscopy coupled with HPLC can be used for sensitive detection of unmodified amino acids (Qu et al. 2002). However, this technique is very expensive and the instrument cannot be found in many laboratories. As a result, pre- or post-column derivatizations are still the most common methods which make it possible to detect and quantify the free amino acids after their separation in a liquid chromatographic system equipped by a simple UV–visible or fluorescence detector (Molnár-Perl 2003). Although post-column dervatization is a reliable method, but it is costly and needs special apparatus. The reversed phase HPLC for the separation and quantitation of pre-column derivatized amino acids is the preferred routine in many studies.
DABS-Cl (4-N,N-dimethylaminoazobenzene-4ʹ-sulfonyl chloride) as a reagent for pre-column derivatization has attracted a considerable attention for amino acid analysis (Krause et al. 1995; Lin et al. 1980; Schneider 1989; Sethuraman et al. 2004; Pinho et al. 2001). DABS-Cl has advantages over the other pre-column derivatization reagents such as o-phthalaldehyde (OPA) and phenyl isothiocyanate (PITC). DABS-Cl reacts with both primary and secondary amino acid, and produces derivatives which are stable for weeks at room temperature. The colored derivatives can easily be detected at picomolar concentrations with high sensitivity in the visible region of the spectrum within 430–460 nm. Working in the visible region increases the specificity of the detection of the amino acids as it eliminates baseline noise and interferences from the other biological components in the system which might be seen at lower (UV) wavelengths.
The current paper compares the conventional method of acid hydrolysis with a fast and additive-free method presented here for the hydrolysis and further analysis of octreotide acetate. This novel method uses only 6 M HCl and hydrolysis process is completed in 30 min at 150 °C without a significant loss of tryptophan and cysteine. This fast hydrolysis method followed by derivatization of the resulted free amino acids with DABS-Cl which takes only 20 min makes it possible to do the complete amino acid analysis of an octreotide sample in a few hours. The whole procedure for amino acid analysis of the octreotide acetate including hydrolysis, derivatization by DABS-Cl, and the final determination by reversed phase HPLC was validated according to ICH guidelines (International Conference of Harmonization 1996).
Experimental
Chemicals and reagents
All amino acids as well as DABS-Cl (4-N,N-dimethylaminoazobenzene-4′-sulfonyl chloride) were purchased from Sigma-Aldrich Chemical Co. HPLC-grade acetonitrile, ethanol, and dimethylformamide were purchased from Merck. Hydrochloric acid, sodium acetate, sodium bicarbonate, and disodium hydrogen phosphate were also purchased from Merck. Ultrapure water used to prepare all aqueous solutions was obtained from Milli-Q water purification system (Millipore). Glass vials for octreotide hydrolysis were heated at 300 °C for 2 h to remove any organic contaminants.
Preparation of derivatization solutions
To prepare the stock solution of DABS-Cl as derivatization reagent (10 mM), approximately 65 mg DABS-Cl was dissolved in 20 ml acetonitrile and stirred for 5 min at room temperature. Sample buffer (50 mM NaHCO3, pH 8.4) was prepared in Milli-Q water and the pH adjusted to 8.4. Diluent solution was prepared by slow addition of 25 ml ethanol to 25 ml of 50 mM Na2HPO4 solution (pH 7.0).
Accelerated and conventional hydrolysis methods
In the presented accelerated method, the hydrolysis at 150 °C was performed at different durations i.e., 15, 30, 45, and 60 min. In the initial stages of the study, for each experiment 1 mg of the octreotide standard sample was accurately weighed in a clean glass vial and 1 ml of 6 M HCl was added and the vial was sealed under the vacuum. After hydrolyses for the desired duration, the vials were cooled down to the room temperature and then the mixtures were dried under the vacuum. For the method validation experiments, the standard octreotide samples (1 mg) were hydrolyzed at 150 °C for 30 min. In traditional hydrolyses method, the same amount of the sample was weighed in a clean vial and after the addition of 1 ml 6 M HCl and vacuum seal, it was hydrolyzed for 24 h at 110 °C. Afterward, the hydrolyzed sample was dried under vacuum. In both accelerated and traditional hydrolysis methods no other chemicals were added to the hydrolysis mixtures.
DABS-Cl derivatization of free amino acids
1000 µL of sample buffer was added to each hydrolyzed and dried sample and the vial was vortexed for 1 min for the complete dissolution of the hydrolysates. In case of unclear solutions, they were centrifuged at 12,000 rpm for 5 min at room temperature to remove the solid particles. For the initial studies, 100 µL from each dissolved sample was transferred to a separate clean vial. To prepare the calibration solutions at the method validation step, the aliquots of 20, 40, 60, 80, 100, 120, and 140 µL of the dissolved standard sample hydrolyzed at 150 °C for 30 min was added to seven clean vials. To prepare a derivatized solution not included in the calibration step for method validation studies, in another vial 90 µL from the same standard solution was added. In the next step to all of the above-mentioned vials, appropriate amounts of the sample buffer were added in such a way that the total volume of the mixture at each vial became exactly 200 µL. The amount of the derivatization solution that was then added to every vial was 300 µL. At this step the vials were vortexed thoroughly for 1 min and then sealed and heated for 20 min at 70 °C. After placing the vials in oven, they were briefly shaken at 5 and 15 min. The vials were then cooled down in an ice bath for 2 min to stop the reaction. To dilute all the mixtures to the final volume of 1000 µL, 500 µL of the dilution solution was added to every vial and mixed well.
Chromatographic conditions
HPLC analysis was performed on a Shimadzu Prominence system (Shimadzu Corporation, Kyoto, Japan) composed of a LC-20AD pump, an on-line DGU-20A degassing system, a CTO-20A column oven, and a SPD-20A UV–visible detector. The separation was achieved using a Hector C18-M column (250 × 4.6 mm, 5 μm particle size). Chromatograms were recorded and processed with the LabSolutions software version 5.51 (Shimadzu). Column temperature was kept constant at 40 °C and the flow rate of 1.0 mL/min was used in the gradient mode of the elution. The injection volume was 10 μL. UV detector wavelength was set at 436 nm. Sodium acetate buffer (25 mM, pH = 6.5) containing 4 vol % DMF was used as mobile phase A and acetonitrile was used as mobile phase B. The mobile phase A was prepared daily and filtered through a 0.45-μm membrane. The derivatized samples were completely clear and injected to the column by the auto sampler without any further filtration. Gradient elution program (time/ %B) was as follow: 0.01/15, 20/40, 32/70, 34/70, 35/15, 45/15.
Results and discussion
Traditional and accelerated hydrolysis methods
Hydrolysis is an extremely important step in amino acid analysis as it affects both qualitative and quantitative results obtained from peptide and protein samples. In the initial part of the study to digest the octreotide acetate sample, the traditional method of hydrolysis i.e., 24 h of incubation of the sample with 6 M hydrochloric acid at 110 °C, was tried. After hydrolysis, the mixture turned to rather a dark solution suggesting the presence of oxidative degradation products of the less stable residues. After sample preparation and derivatization with DABS-Cl, the derivatized samples were injected to the chromatographic system. The obtained chromatogram, in the time scale in which the amino acid peaks appeared, is shown in Fig. 2a. The peaks were identified using derivatized reference standard materials of the amino acids. As can be seen, tryptophan is almost completely destroyed and cannot be found in its expected position. Partial destruction for other residues especially for cysteine and threonine-ol is also noticeable.

Chromatograms obtained from hydrolyzed octreotide acetate standard samples with 6 M HCl for a 24 h, 110 °C, b 60 min, 150 °C, c 45 min, 150 °C, d 30 min, 150 °C, e 15 min, 150 °C. All samples had the same weight (1 mg) and were dried under vacuum before derivatization with DABS-Cl
To check if reducing the duration of the hydrolysis down to 60 min or less at a higher temperature could be helpful in saving the less stable residues from destruction, a series of experiments were designed and carried out. Four octreotide samples, each about 1 mg, were hydrolyzed at 150 °C for 15, 30, 45, and 60 min. After hydrolysis, the sample solutions especially in case of 30 and 15 min hydrolysis were clear with a light yellow color, indicating less oxidative products compared to traditional method of hydrolysis. After pretreatment and functionalization, the samples were injected to the chromatographic system. The obtained chromatograms for samples hydrolyzed for 60, 45, 30, and 15 min can be observed in Fig. 2b–e, respectively. Although compared to the conventional hydrolysis, the cysteine peak has grown significantly for the 60 and 45 min hydrolysis experiments, the tryptophan peak is still missing. Reducing the time of hydrolysis down to 30 min had an improving effect on all amino acid peaks. The main event in this hydrolysis was the appearance of tryptophan, with a detectable and well-resolved peak. To check whether the intensity of the peaks can be further improved, 15 min of hydrolysis was also tried. As obvious from Fig. 2e, this new condition not only did not improve the tryptophan and cysteine peak, but also had a decreasing effect on the other peaks due to incomplete hydrolysis of the octreotide sample.
The obtained results emphasize that conventional method of hydrolysis may not be a good choice for octreotide acetate amino acid analysis; as no peak can be observed for tryptophan and the other peaks such as cysteine and threonine-ol are not reliable. The other point that should be noted is that, the long duration of exposing a small peptide such as octreotide in harsh hydrolysis medium may result in unwanted side reactions which complicate the interpretation of the results. It seems that based on findings of this study, hydrolysis at 150 °C for 30 min is a good alternative for conventional acid hydrolysis method in the amino acid analysis of octreotide acetate samples. The point that should be emphasized is that this fast hydrolysis has been performed without application of any other chemical additives. The accelerated hydrolysis method (with some probable duration adjustments) in combination with DABS-Cl functionalization could be a promising method for the amino acid analysis of other peptide or protein samples. In the next sections, the proposed method of hydrolysis (30 min, 150 °C) and subsequent DABS-Cl functionalization are validated for the analysis of a standard octreotide acetate sample.
Method validation
Specificity
To validate the specificity of the m ethod, five different concentrations of hydrolyzed and derivatized octreotide standard sample was injected to the chromatographic system and the chromatograms were obtained (Table 1). The values of t R for different amino acids were used as a measure of specificity. As can be seen in this table, the t R values for each amino acid after hydrolyses are quite close together. This indicates that t R values are not significantly influenced by the concentration of amino acids, the presence of other amino acids, the DABS-Cl peaks as the derivatization reagent, or by the medium. In the blank chromatogram, except the DABS-Cl peaks which did not interfere with the amino acid peaks, no interfering peaks were observed. The values of the relative standard deviation (RSD) of the retention times for amino acids were in the range of 0.07–0.13 % which allows highly specific peak identification. It should be noted that a ±3 % variation in the retention times is considered acceptable for the same amino acids present in the hydrolyzed sample (International Conference of Harmonization 1996).
Although there is always a risk of contamination of the sample by other amino acids, their interferences in the current method seems to be unlikely. The pre-column DABS-Cl derivatization followed by RP-HPLC analysis of amino acids is a well-studied method and its high specificity for almost all amino acids is confirmed (Krause et al. 1995; Lin et al. 1980; Schneider 1989; Sethuraman et al. 2004; Pinho et al. 2001).
System suitability
System suitability parameters are used to verify the chromatographic system and to check if it works adequately and is not a source of systematic error in the overall analysis. Number of theoretical plates, resolution, and also tailing factor were the studied parameters which were extracted from the chromatograms of a standard octreotide sample (137.6 μM) after hydrolysis and derivatization (Table 2). The number of theoretical plates for all amino acid peaks was much more than 2000. The minimum number of theoretical plates was observed for threonine (151841) and the maximum number of 443,486 theoretical plates was achieved for lysine. Tailing factor for all amino acid peaks was below 2.0 and the resolution values for all the peaks were larger than 2.0 which are satisfactory results.
Linearity
In the linear range of the calibration curve, the area under the amino acid peaks are proportional to their concentration, i.e., it obeys the y = ax + b equation. Verifying the linearity is important, as the concentration of the sample selected for accuracy test should be within the linear range of the calibration plot. As explained in the experimental section, the calibration samples were obtained by dilution of a reference hydrolyzed octreotide sample. Every derivatized calibration sample was injected three times to the chromatographic system and the peak areas for the amino acids were determined. The three peak areas for each amino acid were then averaged. The calibration curves were obtained by treatment of the data of average peak area (n = 3) vs. the concentration of amino acids by linear least squares analysis (Fig. 3). The concentration of the standard solutions were in the range of 19.7–137.6 μM for threonine, tryptophan, threonine-ol, and lysine and 39.3–275.2 μM for phenylalanine and cysteine. The values of the slope, intercept, and the regression coefficients for amino acids are given in Table 3. The high value of the regression coefficient (R 2) which was more than 0.995 for all amino acids indicates the acceptable linearity of the calibration curves.

Calibration curves. Every point of the calibration curve is the average of three replicate measurements (n = 3). The error bars show the standard deviation of the calibration points
Accuracy
This validation parameter was estimated by analyzing a hydrolyzed reference material of octreotide acetate with concentration of 88.5 μM. The analysis was repeated 5 times by 5 injections of the derivatized sample. After the analysis, the molar ratio and the percent recovery was calculated for every amino acid (Table 4). As can be seen in Table 4, the average percent recoveries are all well within the acceptable range of 90–110 %, which confirms the good accuracy of the presented amino acid analysis (Reason 2003).
Precision
In this work, the precision was estimated by measuring the repeatability and intermediate precision. The data obtained from 5 injections of the accuracy test presented in the previous section, was used to calculate the RSD of the obtained molar ratios for each amino acid. The obtained RSD values shown in Table 5 were less than 5 % (0.17–0.63 %) which confirms the acceptable repeatability of the measurements.
Intermediate precision was also estimated by repeating the whole procedure on three different days (Table 6). The results shown for each day are averages of three replicate measurements. Although the RSD values for intermediate precisions are acceptable but they are not as satisfactory as those obtained in the repeatability measurements. It can be contributed to the sensitivity of the hydrolysis step to the deviations in thermal conditions in each individual experiment. These include the variations in reproducibility of the oven temperature and also the differences in the material and uniformity of hydrolysis tubes from run to run. A point that should be noted here is that the quantification of the obtained peak areas for each experiment have been done using the calibration equations reported earlier in this paper. The intermediate precision can be improved to a great extent if the reference and unknown octreotide samples experience the same thermal conditions during the hydrolysis, i.e., to have a separate calibration curve for each experiment.
Limit of detection and limit of quantification
The limit of detection (LOD) and the limit of quantification (LOQ) were determined based on the standard deviation of the intercept and the slope of the calibration curves as described in the related ICH guideline (International Conference of Harmonization 1996). The LOD is calculated as 3.3α/b and LOQ as 10α/b, where α is the standard deviation of the intercept and b is the slope of the calibration curve. The LOD and LOQ values calculated for the amino acids of octreotide are presented in Table 7.
Conclusions
The achievement of this study is a reliable method for the hydrolysis and quantification of amino acids in the octreotide acetate samples. The method is based on pre-column derivatization of amino acids in octreotide by DABS-Cl, which guarantees highly stable derivatized amino acids, easily detectable in visible range. The method appeared to be specific, accurate, precise, and linear across the analytical range. The LOD and LOQ values were in the range of 1.7–25.6 μM and 5.2–77.5 μM, respectively. The acid hydrolysis with 6 M HCl at 150 °C for 30 min, not only shortened the whole analysis time to a few hours, but also resulted in the chromatograms that were significantly more appropriate for the quantification of less stable amino acids like tryptophan and cysteine in octreotide samples. The possibility of application of the presented accelerated hydrolysis method for peptide samples other than octreotide acetate is under study in our group. The obtained promising results offer that the method could easily be adopted for the analysis of other short length peptides.
References
Albert R, Smith-Jones P, Stolz B, Simeon C, Knecht H, Bruns C, Pless J (1998) Direct synthesis of [DOTA-DPhe1]-octreotide and [DOTA-DPhe1, Tyr3]-octreotide (SMT487): Two conjugates for systemic delivery of radiotherapeutical nuclides to somatostatin receptor positive tumors in man. Bioorg Med Chem Lett 8:1207–1210
Astruc B, Marbach P, Bouterfa H, Denot C, Safari M, Vitaliti A, Sheppard M (2005) Long-acting octreotide and prolonged-release lanreotide forumulation have different pharmacokinetic profiles. J Clin Pharmacol 45:836–844
Bauer W, Briner U, Doepfner W, Haller R, Huguenin R, Marbach P, Petcher TJ (1982) A very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sci 31:1133–1140
Brazeau P, Vale W, Burgus R, Ling N, Butcher M, Rivier J, Guillemin R (1973) Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 179:77–79
Casella IG, Contursi M (2003) Isocratic ion chromatographic determination of underivatized amino acids by electrochemical detection. Anal Chim Acta 478:179–189
Church FC, Swaisgood HE, Catignani GL (1984) Compositional analysis of proteins following hydrolysis by immobilized proteases. J Appl Biochem 6:205–211
Dai Z, Wu Z, Jia S, Wu G (2014) Analysis of amino acid composition in proteins of animal tissues and foods as pre-column o-phthaldialdehyde derivatives by HPLC with fluorescence detection. J Chromatogr B 964:116–127
Fountoulakis M, Lahm HW (1998) Hydrolysis and amino acid composition analysis of proteins. J Chromatogr A 826:109–134
Greenstein JP, Winitz M (1961) Chemistry of amino acids. John Wiley, New York
Harder U, Koletzko B, Peissner W (2011) Quantification of 22 plasma amino acids combining derivatization and ion-pair LC–MS/MS. J Chromatogr B 879:495–504
Hsieh HP, Wu YT, Chen ST, Wang KT (1999) Direct solid-phase synthesis of octreotide conjugates: precursors for use as tumor-targeted radiopharmaceuticals. Bioorg Med Chem Lett 7:1797–1803
International Conference of Harmonisation (1996) Harmon-ised tripartite guideline–validation of analytical procedures. Methodology
Kaspar H, Dettmer K, Gronwald W, Oefner PJ (2008) Automated GC–MS analysis of free amino acids in biological fluids. J Chromatogr B 870:222–232
Kaspar H, Dettmer K, Gronwald W, Oefner PJ (2009) Advances in amino acid analysis. Anal Bioanal Chem 393:445–452
Krause I, Bockhardt A, Neckermann H, Henle T, Klostermeyer H (1995) Simultaneous determination of amino acids and biogenic amines by reversed-phase high-performance liquid chromatography of the dabsyl derivatives. J Chromatogr A 715:67–79
Lin JK, Wang CH (1980) Determination of urinary amino acids by liquid chromatography with “dabsyl chloride”. Clin Chem 26:579–583
Ludlam WH, Anthony L (2011) Safety review: dose optimization of somatostatin analogs in patients with acromegaly and neuroendocrine tumors. Adv Ther 28:825–841
Molnár-Perl I (1997) Tryptophan analysis in peptides and proteins, mainly by liquid chromatography. J Chromatogr A 763:1–10
Molnár-Perl I (2003) Quantitation of amino acids and amines in the same matrix by high-performance liquid chromatography, either simultaneously or separately. J Chromatogr A 987:291–309
Moore S, Stein WH (1951) Chromatography of amino acids on sulfonated polystyrene resins. J Biol Chem 192:663–681
Pinho O, Ferreira IM, Mendes E, Oliveira BM, Ferreira M (2001) Effect of temperature on evolution of free amino acid and biogenic amine contents during storage of Azeitão cheese. Food Chem 75:287–291
Qu J, Wang Y, Luo G, Wu Z, Yang C (2002) Validated quantitation of underivatized amino acids in human blood samples by volatile ion-pair reversed-phase liquid chromatography coupled to isotope dilution tandem mass spectrometry. Anal Chem 74:2034–2040
Reason AJ (2003) Validation of amino acid analysis methods. In: Smith BJ (ed) Protein sequencing protocols, Methods in molecular biology, vol 211. Humana Press, Totowa
Reichlin S (1983a) Somatostatin I. New Engl J Med 309:1495–1501
Reichlin S (1983b) Somatostatin II. New Engl J Med 309:1556–1563
Reubi JC, Schär JC, Waser B, Wenger S, Heppeler A, Schmitt JS, Mäcke HR (2000) Affinity profiles for human somatostatin receptor subtypes SST1–SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur J Nucl Med 27:273–282
Rutherfurd SM, Gilani GS (2009) Amino acid analysis. Current protocols in protein science, Wiley Interscience
Schneider HJ (1989) Amino acid analysis using DABS-CL. Chromatographia 28:45–48
Sethuraman R, Lee TL, Tachibana S (2004) Simple quantitative HPLC method for measuring physiologic amino acids in cerebrospinal fluid without pretreatment. Clin Chem 50:665–669
Silvestre MPC (1997) Review of methods for the analysis of protein hydrolysates. Food Chem 60:263–271
Veledo MT, de Frutos M, Diez-Masa JC (2005) Amino acids determination using capillary electrophoresis with on-capillary derivatization and laser-induced fluorescence detection. J Chromatogr A 1079:335–343
Woltering EA, Mamikunian VAPM, Zietz S, Krutzik SR, Go VL, Vinik AL, Vinik E, Dorisio TMO, Mamikunian G (2005) Effect of octreotide LAR dose and weight on octreotide blood levels in patients with neuroendocrine tumors. Pancreas 31:392–400
Woltering EA, Salvo VA, O’Dorisio TM, Lyons J III, Li G, Zhou Y (2008) Clinical value of monitoring plasma octreotide levels during chronic octreotide long-acting repeatable therapy in carcinoid patients. Pancreas 37:94–100
Yang W, Zhang Z, Deng W (2003) A capillary electrophoresis detection scheme for underivatized amino acids based on luminol–BrO− chemiluminescence system. Talanta 59:951–958
Zhang X, Zhao T, Cheng T, Liu X, Zhang H (2012) Rapid resolution liquid chromatography (RRLC) analysis of amino acids using pre-column derivatization. J Chromatogr B 906:91–95
Acknowledgments
Authors would like to thank Tofigh Daru Co. for financial and technical support of this project. Authors are also grateful to Ms. Hedie Ghaffari for editing the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: D. Tsikas.
Rights and permissions
About this article

Cite this article
Akhlaghi, Y., Ghaffari, S., Attar, H. et al. A rapid hydrolysis method and DABS-Cl derivatization for complete amino acid analysis of octreotide acetate by reversed phase HPLC. Amino Acids 47, 2255–2263 (2015). https://doi.org/10.1007/s00726-015-1999-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-015-1999-9