
Abstract
Vascular abnormalities predisposing to atherosclerosis are present in patients with rheumatoid arthritis (RA) and associate with excess cardiovascular risk. Symmetric dimethylarginine (SDMA), an endogenous inhibitor of nitric oxide (NO) synthase activity, has been recognised as novel risk factor for endothelial dysfunction and cardiovascular disease. We aimed to compare SDMA levels in RA patients and controls and to investigate whether they are influenced by demographic, inflammatory or metabolic factors. Serum SDMA levels were measured in 197 RA individuals [median age: 67 years (quartiles: 59–3), 153 (78 %) females] and 82 controls [median age: 44 years [quartiles: 33–55, 50 (61 %) females]. Routine biochemistry tests, lipid profile, glycemic profile [glucose, insulin, homeostasis model assessment (HOMA-IR), quantitative insulin sensitivity check index (QUICKI)], as well as inflammatory markers were measured in all patients. Paired analysis was employed for the comparison of SDMA in two groups and multivariable regression models were performed to identify predictors of SDMA in the RA cohort. SDMA was significantly lower in RA than control patients in both unpaired and paired analyses (P < 0.001 and P = 0.005, respectively), with the magnitude of the difference being similar in both models. QUICKI (P = 0.005) and disease activity score-28 (P = 0.007) were positively related to SDMA in the RA cohort, whilst a negative correlation with renal function (eGFR) was detected (P = 0.005). The molecular explanation of lower serum SDMA is unclear, but the established relationships with indices of disease activity and insulin resistance, may underline the pathogenetic role of the l-arginine/NO pathway dysregulation in the development of atherosclerosis in RA. The biological and clinical importance of SDMA in RA remains to be evaluated in clinical and experimental studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Accelerated atherosclerosis leading to excess cardiovascular (CV) risk has been recognised as a major cause of premature morbidity and mortality in patients with rheumatoid arthritis (RA) (Gonzalez-Gay et al. 2005). In addition to higher frequency of CV disease risk factors in RA compared to the general population (Chung et al. 2012), autoimmunity and systemic inflammation have a pivotal role in the development of vascular disease, as rheumatoid synovia and atherosclerotic plaques share a common inflammatory cellular and molecular signalling milieu (Abou-Raya A and Abou-Raya S 2006).
Endothelial dysfunction is a central event in the development of atherosclerosis and contributes to all stages of coronary artery disease, from the early formation to the rupture of atherosclerotic plaque. Balanced production of endothelium-derived vasoactive substances maintains the integrity of vascular endothelium, providing an atheroprotective environment within the vessel. Nitric oxide (NO) is considered the most important of these substances and factors affecting its bioavailability are involved in the pathophysiology of vascular damage and injury (Sandoo et al. 2010). In chronic high-grade inflammatory conditions such as RA, increased systemic expression of inflammatory mediators exerts detrimental effects on vascular haemostasis, mainly by affecting NO metabolism, resulting in functional and structural changes, such as dysregulation of vascular tone and disorganization of the vascular architecture that promote thrombosis and atherosclerosis (Desideri and Ferri 2005).
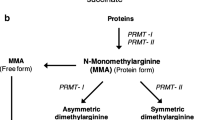
Dimethylarginines are endogenous guanidine-substituted analogues of l-arginine, the precursor of NO, which are naturally liberated in biological fluids following proteolysis. During endogenous protein turnover asymmetrically and symmetrically methylated l-arginine residues are released as asymmetric (ADMA) and symmetric (SDMA) dimethylarginine, respectively. Several lines of evidence support the adverse effect of these molecules on vascular health, as they hold a key homeostatic role in NO production by inhibiting NO synthase (Caplin and Leiper 2012). ADMA, the most studied dimethylarginine today, has been linked to vascular outcomes and overall mortality in different CV disease settings as well as in the general population (Böger et al. 2009; Leong et al. 2008). ADMA has also been considered as a potential CV disease risk factor in patients with rheumatic diseases, with reports demonstrating higher levels in patients with RA compared to controls (Sandoo et al. 2012a), as well as associations with morphological markers of premature atherosclerosis (Surdacki et al. 2007) and RA-related inflammatory burden (Sandoo et al. 2014).
Symmetric dimethylarginine (SDMA), the structural counterpart of ADMA, was initially considered biologically inert. However, SDMA is a weak inhibitor of neuronal NO synthase (Tsikas et al. 2000). Recent insights suggest that SDMA influences NO synthesis indirectly, by enhancing formation of reactive oxygen species and/or by inhibiting cellular transport of l-arginine and other amino acids (Schepers et al. 2009; Strobel et al. 2012). SDMA has emerged as an independent CV risk factor in the general population and some high-risk populations (Siegerink et al. 2013; Schulze et al. 2010; Schwedhelm et al. 2014). Coronary artery disease is the leading cause of death in patients with RA (Levy et al. 2008) and there is increasing interest in biomarkers that might better identify patients at higher risk for CV disease who may benefit from early preventive strategies and targeted therapeutic interventions for CV risk factors. Little attention has been paid in the role of SDMA and other arginine analogues such as homoarginine (Kayacelebi et al. 2014) in vascular pathology characterising RA.
Therefore, we aimed to compare serum SDMA levels in patients with RA and healthy controls and to explore the associations between SDMA and indices of disease activity and insulin resistance.
Materials and methods
Study population
One hundred and ninety-seven consecutive Caucasian RA patients were recruited from the rheumatology outpatient clinics of the Dudley Group NHS Foundation Trust, UK, between March 2011 and March 2013. All patients met the retrospective application of the 1987 revised RA criteria of the American College of Rheumatology (Arnett et al. 1988). Patients with previous confirmed acute coronary syndrome, evidence of chronic kidney disease or serious psychiatric disorders according to their medical records were excluded. Eighty-two healthy participants were recruited from hospital staff and their friends. The exclusion criteria were the same as for the RA patients. The study received approval from the Black Country Research Ethics Committee and all patients gave their written informed consent according to the Declaration of Helsinki.
Demographic data were recorded and all participants underwent a thorough assessment including a detailed review of their medical history, hospital records and a physical examination. Disease activity was assessed by the 28-joint disease activity score—ESR (DAS28-ESR) (Prevoo et al. 1995) and physical function using the anglicised version of the Health Assessment Questionnaire (HAQ) (Kirwan and Reeback 1986). All current medications including biologic and non-biologic disease-modifying drugs were recorded.
Insulin resistance was evaluated from fasting glucose and insulin using the Homeostasis Model Assessment of Insulin Resistance (HOMA-IR) and Quantitative Insulin Sensitivity Check Index (QUICKI) (Matthews et al. 1985). The formulas used for the calculation were as follows (Wallace et al. 2004; Chen et al. 2003):
.
Blood tests for biochemical and serological markers were carried out in the Research Laboratory at Russells Hall Hospital. Total cholesterol, high-density lipoprotein cholesterol, triglycerides and C-reactive protein (CRP) were measured using the Vitros 5.1chemistry system (www. orthoclinical.com). Estimated glomerular filtration rate (eGFR) was calculated using modification of diet in renal disease study equation (Levey et al. 1999). Levels of erythrocyte sedimentation rate (ESR) were determined using the Westergren method via a Starrsed Compact (Mechatronics BV, Netherlands). This is an immunoturbidimetric method.
Measurement of SDMA
SDMA was measured in the serum of the study participants. The SDMA assay is based on the method of competitive enzyme using a commercial ELISA kit (Immundiagnostik, Bensheim, Germany). The sample preparation includes the addition of a derivatization reagent for SDMA coupling. During the incubation period, the target SDMA in the sample competes with the SDMA derivative (tracer) immobilised on the wall of the microtiter plates for the binding of the polyclonal antibodies. The SDMA in the sample displaces the antibodies out of the binding to the tracer. Therefore, the concentration of the tracer-bound antibody is inversely proportional to the SDMA concentration in the sample. The absorbance is measured at 450 nm and patient samples are read from a standard curve. The intra-assay relative standard deviation was 7.5 % and inter-assay was 6 %. The lowest concentration detected was 0.05 µM.
Statistical analysis
Initially, SDMA levels, and other relevant factors, were compared between patients and controls using Mann–Whitney and Fisher’s exact tests. To account for the effects of potentially confounding factors such as age, renal function, inflammation, individuals within the groups were then matched, using a propensity score, and the resulting subset of paired individuals were compared using Wilcoxon’s signed rank and McNemar’s tests.
Within RA patients, associations between a range of variables and SDMA were then assessed using univariable regression models. Factors that were found to be significantly associated with the outcomes were then included in multivariable regression models, to account for potentially confounding factors. Prior to the analysis, the distributions of all of the variables were assessed by plotting histograms. Where skew was detected, log2-transformations were applied, after adding a constant, where applicable, to remove zero values (Bland and Altman 1996). All analyses were performed using IBM SPSS Statistics 22 (IBM Corp. Armonk, NY, USA), with P < 0.05 deemed to be indicative of statistical significance.
Results
Baseline characteristics
SDMA data were available for 197 RA and 82 healthy control patients. The demographic characteristics and basic biochemical evaluation of the study participants are summarized in Table 1. RA patients were significantly older than controls, with significantly greater predominance of females, as expected. The RA group was found to have significantly higher triglycerides and CRP levels and lower eGFR.
Comparison of SDMA between RA and controls
Initial comparisons between groups (Table 1) demonstrated that serum SDMA levels were significantly lower in the RA patients compared to controls with medians of 0.47 µM (quartiles: 0.41–0.53) and 0.52 (0.49–0.57) µM, respectively (P < 0.001).
In an attempt to remove the potential confounding effects of different distributions of age and biochemical variables between patients and controls, a propensity score was produced. This was based on a multivariable binary logistic regression model, with the patient cohort being the dependent variable (Supplementary Table S1). eGFR was not included in the propensity model, due to issues with multicollinearity, as it was highly correlated with both age and creatinine. CRP was converted into a categorical variable, since a large proportion of individuals had values <3 mg/l, which was a cutoff for truncation at the point of data collection.
Individuals from the two cohorts were then paired on the resulting propensity score. A nearest neighbour approach was used, using a match tolerance of 0.05, and with matches selected at random in the case of ties. Matches could be found for a total of 44 individuals from each group, which are compared in Table 2. No significant differences between the groups were detected for any of the variables from the propensity score on which the patients were matched. However, the difference in SDMA levels between the groups remained significant (P = 0.005), and of a similar magnitude to the unpaired analysis, with medians of 0.46 µM (quartiles: 0.40–0.54) in the RA group, and 0.53 µM (0.49–0.56) in controls.
Associations between SDMA with disease-related and metabolic factors
A second analysis was performed within the RA cohort. Initial review of the distributions of serum SDMA concentrations showed that they were highly skewed, and so log2-transformations were applied. The resulting variables closely followed a normal distribution, with the exception of 11 (5.6 %) patients with serum SDMA levels >1 µM, who did not fit into the distribution of the remainder (Supplementary Figure S1). These outliers (Supplementary Table S2) were excluded from subsequent analysis, meaning that the remaining data met the normality assumptions, allowing parametric analyses to be used. This left 186 patients, with a median age of 66 years (range 32–89), of whom 144 (77.4 %) were females.
Univariable regression models identified three factors that were significant predictors of SDMA (Table 3). Both DAS28 (P = 0.010) and QUICKI (P = 0.032) showed significant positive associations, with SDMA, whilst eGFR was significantly negatively associated (P = 0.024) with SDMA. In multivariable analysis, the three factors associated with SDMA remained significant (Table 4). A one-unit increase of DAS28 was found to be associated with 3.7 % higher SDMA serum levels (95 % CI 1.1 %, 6.3 %; P = 0.005). An increase of 0.1 unit in QUICKI was associated with a 10.8 % higher SDMA levels (95 % CI 2.8 %, 19.5 %; P = 0.007). For eGFR, a one-unit increase was associated with 0.3 % lower SDMA (95 % CI 0.1 %, 0.6 %; P = 0.005).
A sensitivity analysis was then performed, to test whether removing the outliers had introduced bias into the analysis. SDMA values were correlated with the three factors found to be significant, using Spearman’s rho on all 197 patients, and Pearson’s r on the 186 with outliers excluded. Coefficients for DAS28 (rho = 0.218, r = 0.157), eGFR (rho = −0.206, r = −0.194) and QUICKI (rho = 0.108, r = 0.153) were similar in both analyses, hence it was concluded that there was no evidence to suggest that excluding the outliers had adversely affected the conclusions of the analysis.
Discussion
The main finding of our study is that serum SDMA levels are lower in patients with RA compared to healthy controls, and are associated with the QUICKI index, a commonly used measure of insulin sensitivity. We also found positive correlations between SDMA and disease activity score.
Unlike ADMA, SDMA may affect vascular haemostasis through other mechanisms, such as the stimulation of reactive oxygen species by modulating calcium channels in monocytic leukocytes (Schepers et al. 2009). Such proinflammatory effects have not been described with ADMA which, on the other hand, upregulates endothelial adhesion molecules and contributes to oxidative stress by inhibiting NO isoenzymes in various tissues via different mechanisms (Karbach et al. 2014; Kielstein et al. 2007). These observations suggest that these NO synthase inhibitors may promote endothelial insult via the involvement of different cell types and mechanisms (Lüneburg et al. 2012). Until recently, the role of SDMA in clinical studies assessing the relationship between dimethylarginines and CV disease was underestimated but, over the last few years, numerous reports suggest that SDMA may be a predictor of CV mortality and morbidity in a wide range of cardio- and cerebro-vascular disease (Meinitzer et al. 2011; Riccioni et al. 2014; Siegerink et al. 2013; Schulze et al. 2010). It is worth noting that ADMA and SDMA had similar predictive values for cardiovascular risk in a population-based cohort of older individuals (Kiechl et al. 2009) and higher SDMA levels have been linked with increased overall mortality in the general population (Gore et al. 2013; Schwedhelm et al. 2014).
To the best of our knowledge, this is the first study reporting on SDMA concentrations in RA patients and controls as well as on associations between SDMA insulin sensitivity and inflammation. A similar trend of decreased SDMA in RA was found in a small study including 29 RA patients and 26 controls, but the results did not reach statistical significance (Klimek et al. 2014). Given that RA is associated with enhanced CV risk, and SDMA has been proposed as a potential marker of atherosclerosis and vascular dysfunction, the results of our study look at first glance fundamentally counterintuitive. However, reduced SDMA levels have been reported in insulin-resistant patients (Zsuga et al. 2007) and a strong negative relationship between SDMA and insulin resistance has been described in Caucasians (Schutte et al. 2010). In concordance with these data, we demonstrated a positive relationship between SDMA and insulin sensitivity assessed by QUICKI in individuals with RA. A similar insignificant tendency to a negative correlation between SDMA and HOMA-IR has been reported to 80 non-diabetic men with stable angina in whom ADMA levels predicted deterioration of glucose tolerance (Surdacki et al. 2013).
RA and insulin resistance share several atherogenic processes, including vascular injury, acute phase response and aberrant lipid metabolism which might play a role in the increased CV risk observed in these patients (Dessein et al. 2003). Insulin resistance is the central mechanism in the development of metabolic syndrome, which is highly prevalent in RA, and is considered an additional aggravating factor for heightened CV morbidity and mortality in this population (da Cunha et al. 2012). In individuals with RA, insulin resistance maintains a chronic low grade inflammatory activity irrespective of effective—or not—control of joint disease, whereas systemic high-grade RA-related inflammation has adverse effects on vascular pathology precipitating endothelial dysfunction caused by components of the metabolic syndrome (Ferraccioli and Gremese. 2011). This reciprocal relationship is reflected in studies clearly demonstrating that the magnitude of CV risk in RA is virtually identical to that of diabetes mellitus, the prototypic disease carrying high CV risk burden (Lindhardsen et al. 2011; Stamatelopoulos et al. 2009). The lower SDMA concentrations in RA and insulin-resistant patients, as well as previous reports of associations between ADMA and insulin resistance in RA (Dimitroulas et al. 2013) suggest that common pathogenic mechanisms driven by abnormal regulation of l-arginine/NO pathway are involved in the development of accelerated atherosclerosis in both conditions.
The mechanisms related to low SDMA are incompletely understood; however decreased insulin sensitivity results in hyperinsulinemia that directly increases overexpression of y+ transporters, the main route of intracellular migration of endogenous dimethylarginines (Simmons et al. 1996; González et al. 2004). Taking into account that SDMA competitively inhibits ADMA for entry into the cells via y+ transporters, it is tempting to speculate that increased SDMA cellular uptake contributes to raised serum ADMA levels, the subsequent inhibition of NO synthesis and the evolution of vascular abnormalities leading to atherosclerosis. Recent reports have shed more light in the mechanisms governing SDMA clearance and degradation, indicating alanine-glyoxylate aminotransferase2 (AGXT2) as an alternative enzymatic cleavage route in healthy controls (Seppälä et al. 2014) and diabetic patients (Anderssohn et al. 2014). AGXT2 strongest mRNA expression was identified in renal and liver tissue (Lüneburg et al. 2014) suggesting that, apart from renal excretion, other organs may also contribute to SDMA clearance. Whether metabolic activation of hepatocytes triggered by proinflammatory cytokines in RA upregulates hepatic metabolic pathways of SDMA remains to be determined. It is worth noting that dimethylarginine dimethylaminohydrolase (DDAH) enzymatic action is not involved in SDMA degradation, despite its leading role in ADMA clearance (Caplin and Leiper 2012). High DDAH activity has been reported in RA and spondyloarthropathies (Chobanyan-Jürgens et al. 2011) but we did not find any relationship between DDAH gene variants and ADMA levels in our RA population (Dimitroulas et al. 2014).
The multivariable analysis confirmed previous studies in other populations, showing the close relationship between SDMA and eGFR which have led to the view that SDMA is a marker for renal disease (Kielstein et al. 2006). As a result, it has been suggested that SDMA may constitute a more reliable indicator of CV disease in patients with renal insufficiency, while ADMA may be more important in the presence of normal renal function (Schulze et al. 2010). Given that renal dysfunction is common in patients with RA, and is associated with classic CV risk factors (Daoussis et al. 2010), the potential of SDMA as a surrogate marker of CV disease in this population is promising but warrants further in-depth studies.
Systemic inflammatory burden has been considered one of the main drivers of vascular damage and derangement of endothelial haemostasis in RA. In the present study, and despite the lack of association between SDMA and acute phase response, the positive relationship with clinical markers of disease activity reinforces the hypothesis that ADMA and SDMA may mediate endothelial dysfunction not only by directly diminishing NO bioavailability, but also by exhibiting proinflammatory effects on the vascular wall. In that respect, ADMA and SDMA may affect vascular haemostasis in RA by other NO-independent mechanisms in keeping with observations in a German population (Willers and Hahn 2012). The decreased serum SDMA levels found in our study may reflect a mechanism associated specifically with RA similarly to what occurs in diabetes mellitus where intriguing interactions between ADMA and CV events have been described (Anderssohn et al. 2010). It has been reported that interstitial ADMA seems to play a minor role in NO-dependent regulation of adipose tissue blood flow and metabolism in obese and type 2 diabetic patients (May et al. 2014), suggesting that other NO-independent mechanisms may be involved in endothelial dysfunction in insulin-resistant patients. The complexity of CV disease in RA with multilevel interrelations between traditional, novel and disease-related risk factors pose significant difficulties in the establishment of biochemical markers able to encompass such a variety of pathophysiologic processes. Dimethylarginines can capture different elements of this puzzle and improve diagnostic capabilities in CV risk stratification, but translating these assumptions into clinical practice requires replication in multiple settings, ideally with large prospective studies to prove the predictive value of ADMA and SDMA in the RA population.
The findings of our study should be considered against the background of its strengths and limitations. We acknowledge that the RA and control groups were not matched for age, gender and renal function but we took particular care to attenuate the impact of these differences on the outcomes with the propensity matched paired analysis. The lack of variation in the magnitude of SDMA difference between the two groups in unpaired and paired analysis is reassuring regarding the validity of our results. In the regression analyses, we excluded SDMA outliers from the data, to achieve a normal distribution, to maximise the fit and reliability of the final models. However, a sensitivity analysis found that conclusions would have been similar if these patients were included. Patients in our cohort had, on average, moderate to low disease activity which may have contributed to the lower SDMA levels observed in these RA individuals; therefore our findings should not be extrapolated to all RA populations, particularly to patients with active uncontrolled disease, without confirmation in further studies. Despite the cross-sectional design, our study is the first to assess SDMA in RA patients and controls. In fact very few studies assessing SDMA in other conditions characterised by increased CV risk have utilised a control group, either in a cross-sectional or prospective manner (Zsuga et al. 2007). Another strength of the present work is the inclusion of a large cohort of real-life RA patients on a variety of disease-modifying drugs and steroids. We took particular care to consider the influence of antirheumatic medications on SDMA levels in our analysis, none of which were found to be significantly predictive of SDMA in univariate analysis. In addition, biologic and non-biologic disease-modifying drugs do not appear to affect ADMA levels in previous cohorts of RA patients (Turiel et al. 2010; Sandoo et al. 2012b). Last but not least our RA population was almost exclusively Caucasian, diminishing the effect of ethnic differences previously reported for dimethylarginines (Schutte et al. 2010).
Conclusion
We showed that serum SDMA is lower in RA patients and that it is inversely correlated with insulin resistance. Preliminary data indicate SDMA as a useful tool for determining CV risk, but there is still inadequate understanding of its role in promoting endothelial dysfunction by altering NO metabolism. The link between dimethylarginines and parameters of disease activity and insulin sensitivity supports the notion that ADMA and SDMA induce vascular damage in chronic high-grade inflammatory conditions, such as RA, but further longitudinal studies are warranted to elucidate the importance of l-arginine/NO pathway in the development of atherosclerosis and CV risk in patients with RA.
Abbreviations
- ADMA:
-
Asymmetric dimethylarginine
- AGXT2:
-
Alanine-glyoxylate aminotranaferase2
- CRP:
-
C-reactive protein
- CV:
-
Cardiovascular
- DAS 28:
-
Disease activity score 28
- DDAH:
-
Dimethylamine dimethylaminohydrolase
- eGFR:
-
Estimated glomerular filtration rate
- ESR:
-
Estimated sedimentation rate
- HAQ:
-
Health assessment questionnaire
- HOMA-IR:
-
Homeostasis model assessment of insulin resistance
- NO:
-
Nitric oxide
- QUICKI:
-
Quantitative insulin sensitivity check index
- RA:
-
Rheumatoid arthritis
- SDMA:
-
Symmetric dimethylarginine
References
Abou-Raya A, Abou-Raya S (2006) Inflammation: a pivotal link between autoimmune diseases and atherosclerosis. Autoimmun Rev 5:331–337
Anderssohn M, Schwedhelm E, Lüneburg N, Vasan RS, Böger RH (2010) Asymmetric dimethylarginine as a mediator of vascular dysfunction and a marker of cardiovascular disease and mortality: an intriguing interaction with diabetes mellitus. Diab Vasc Dis Res 7:105–118
Anderssohn M, McLachlan S, Lüneburg N, Robertson C, Schwedhelm E, Williamson RM, Strachan MW, Ajjan R, Grant PJ, Böger RH, Price JF (2014) Genetic and environmental determinants of dimethylarginines and association with cardiovascular disease in patients with type 2 diabetes. Diabetes Care 37:846–854
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31:315–324
Bland JM, Altman DG (1996) Transformations, means, and confidence intervals. BMJ Br Med J 312:1079
Böger RH, Sullivan LM, Schwedhelm E, Wang TJ, Maas R, Benjamin EJ, Schulze F, Xanthakis V, Benndorf RA, Vasan RS (2009) Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation 119:1592–1600
Caplin B, Leiper J (2012) Endogenous nitric oxide synthase inhibitors in the biology of disease: markers, mediators, and regulators? Arterioscler Thromb Vasc Biol 32:1343–1353
Chen H, Sullivan G, Yue LQ, Katz A, Quon MJ (2003) QUICKI is a useful index of insulin sensitivity in subjects with hypertension. Am J Physiol Endocrinol Metab 284:E804–E812
Chobanyan-Jürgens K, Pham VV, Stichtenoth DO, Tsikas D (2011) Elevated dimethylarginine dimethylaminohydrolase (DDAH) activity in rheumatoid arthritis and spondyloarthritis. Nitric Oxide 25:436–438
Chung C, Giles J, Petri M, Szklo M, Post W, Blumenthal RS, Gelber AC, Ouyang P, Jenny NS, Bathon JM (2012) Prevalence of traditional modifiable cardiovascular risk factors in patients with rheumatoid arthritis: comparison with control subjects from the multi-ethnic study of atherosclerosis. Semin Arthritis Rheum 41:535–544
da Cunha VR, Brenol CV, Brenol JC, Fuchs SC, Arlindo EM, Melo IM, Machado CA, de Castro Chaves H, Jr Xavier RM (2012) Metabolic syndrome prevalence is increased in rheumatoid arthritis patients and is associated with disease activity. Scand J Rheumatol 41:186–191
Daoussis D, Panoulas VF, Antonopoulos I, John H, Toms TE, Wong P, Nightingale P, Douglas KM, Kitas GD (2010) Cardiovascular risk factors and not disease activity, severity or therapy associate with renal dysfunction in patients with rheumatoid arthritis. Ann Rheum Dis 69:517–521
Desideri G, Ferri C (2005) Endothelial activation. Sliding door to atherosclerosis. Curr Pharm Des 11:2163–2175
Dessein PH, Joffe BI, Stanwix AE (2003) Inflammation, insulin resistance, and aberrant lipid metabolism as cardiovascular risk factors in rheumatoid arthritis. J Rheumatol 30:1403–1405
Dimitroulas T, Sandoo A, Veldhuijzen van Zanten JJ, Smith JP, Hodson J, Metsios GS, Stavropoulos-Kalinoglou A, Kitas GD (2013) Predictors of asymmetric dimethylarginine levels in patients with rheumatoid arthritis: the role of insulin resistance. Scand J Rheumatol 42:176–181
Dimitroulas T, Sandoo A, Hodson J, Smith J, Panoulas VF, Kitas GD (2014) Relationship between dimethylarginine dimethylaminohydrolase gene variants and asymmetric dimethylarginine in patients with rheumatoid arthritis. Atherosclerosis 237:38–44
Ferraccioli G, Gremese E (2011) Adiposity, joint and systemic inflammation: the additional risk of having a metabolic syndrome in rheumatoid arthritis. Swiss Med Wkly 141:w13211
González M, Flores C, Pearson JD, Casanello P, Sobrevia L (2004) Cell signalling-mediating insulin increase of mRNA expression for cationic amino acid transporters-1 and -2 and membrane hyperpolarization in human umbilical vein endothelial cells. Pflugers Arch 448:383–394
Gonzalez-Gay MA, Gonzalez-Juanatey C, Martin J (2005) Rheumatoid arthritis: a disease associated with accelerated atherogenesis. Semin Arthritis Rheum 35:8–17
Gore MO, Lüneburg N, Schwedhelm E, Ayers CR, Anderssohn M, Khera A, Atzler D, de Lemos JA, Grant PJ, McGuire DK, Böger RH (2013) Symmetrical dimethylarginine predicts mortality in the general population: observations from the Dallas heart study. Arterioscler Thromb Vasc Biol 33:2682–2688
Karbach S, Wenzel P, Waisman A, Münzel T, Daiber A (2014) eNOS uncoupling in cardiovascular diseases—the role of oxidative stress and inflammation. Curr Pharm Des 20:3579–3594
Kayacelebi AA, Pham VV, Willers J, Hahn A, Stichtenoth DO, Jordan J, Tsikas D (2014) Plasma homoarginine (hArg) and asymmetric dimethylarginine (ADMA) in patients with rheumatoid arthritis: is homoarginine a cardiovascular corrective in rheumatoid arthritis, an anti-ADMA? Int J Cardiol 176:1129–1131
Kiechl S, Lee T, Santer P, Thompson G, Tsimikas S, Egger G, Holt DW, Willeit J, Xu Q, Mayr M (2009) Asymmetric and symmetric dimethylarginines are of similar predictive value for cardiovascular risk in the general population. Atherosclerosis 205:261–265
Kielstein A, Tsikas D, Galloway GP, Mendelson JE (2007) Asymmetric dimethylarginine (ADMA)—a modulator of nociception in opiate tolerance and addiction? Nitric Oxide 17:55–59
Kielstein JT, Salpeter SR, Bode-Boeger SM, Cooke JP, Fliser D (2006) Symmetric dimethylarginine (SDMA) as endogenous marker of renal function—a meta-analysis. Nephrol Dial Transplant 21:2446–2451
Kirwan JR, Reeback JS (1986) Stanford Health Assessment Questionnaire modified to assess disability in British patients with rheumatoid arthritis. Br J Rheumatol 25:206–209
Klimek E, Skalska A, Kwaśny-Krochin B, Surdacki A, Sulicka J, Korkosz M, Fedak D, Kierzkowska I, Wizner B, Grodzicki TK (2014) Differential associations of inflammatory and endothelial biomarkers with disease activity in rheumatoid arthritis of short duration. Mediators Inflamm 2014:681635
Leong T, Zylberstein D, Graham I, Lissner L, Lissner L, Ward D, Fogarty J, Bengtsson C, Björkelund C, Thelle D, Collaboration Swedish-Irish-Norwegian (2008) Asymmetric dimethylarginine independently predicts fatal and nonfatal myocardial infarction and stroke in women: 24-year follow-up of the population study of women in Gothenburg. Arterioscler Thromb Vasc Biol 28:961–967
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D (1999) A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of diet in Renal Disease Study Group. Ann Intern Med 130:461–470
Levy L, Fautrel B, Barnetche T, Schaeverbeke T (2008) Incidence and risk of fatal myocardial infarction and stroke events in rheumatoid arthritis patients. A systematic review of the literature. Clin Exp Rheumatol 14:673–679
Lindhardsen J, Ahlehoff O, Gislason GH, Madsen OR, Olesen JB, Torp-Pedersen C, Hansen PR (2011) The risk of myocardial infarction in rheumatoid arthritis and diabetes mellitus: a Danish nationwide cohort study. Ann Rheum Dis 70:929–934
Lüneburg N, von Holten RA, Töpper RF, Schwedhelm E, Maas R, Böger RH (2012) Symmetric dimethylarginine is a marker of detrimental outcome in the acute phase after ischaemic stroke: role of renal function. Clin Sci (Lond) 122:105–111
Lüneburg N, Lieb W, Zeller T, Chen MH, Maas R, Carter AM, Xanthakis V, Glazer NL, Schwedhelm E, Seshadri S, Ikram MA, Longstreth WT Jr, Fornage M, König IR, Loley C, Ojeda FM, Schillert A, Wang TJ, Sticht H, Kittel A, König J, Benjamin EJ, Sullivan LM, Bernges I, Anderssohn M, Ziegler A, Gieger C, Illig T, Meisinger C, Wichmann HE, Wild PS, Schunkert H, Psaty BM, Wiggins KL, Heckbert SR, Smith N, Lackner K, Lunetta KL, Blankenberg S, Erdmann J, Munzel T, Grant PJ, Vasan RS, Böger RH (2014) Genome-Wide Association Study of l-Arginine and Dimethylarginines Reveals Novel Metabolic Pathway for Symmetric Dimethylarginine. Circ Cardiovasc Genet. (Epub ahead of print)
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
May M, Batkai S, Zörner AA, Tsikas D, Jordan J, Engeli S (2014) Clinical evaluation of extracellular ADMA concentrations in human blood and adipose tissue. Int J Mol Sci 15:1189–1200
Meinitzer A, Kielstein JT, Pilz S, Drechsler C, Ritz E, Boehm BO, Winkelmann BR, März W (2011) Symmetrical and asymmetrical dimethylarginine as predictors for mortality in patients referred for coronary angiography: the Ludwigshafen Risk and Cardiovascular Health study. Clin Chem 57:112–121
Prevoo ML, Van ‘t Hof MA, Kuper HH, Van Leeuwen MA, van de Putte LB, Van Riel PL (1995) Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 38:44–48
Riccioni G, Scotti L, D’Orazio N, Gallina S, Speziale G, Speranza L, Bucciarelli T (2014) ADMA/SDMA in elderly subjects with asymptomatic carotid atherosclerosis: values and site-specific association. Int J Mol Sci 15:6391–6398
Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD (2010) The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J 4:302–312
Sandoo A, Dimitroulas T, Veldhuijzen van Zanten JJ, Smith JP, Metsios GS, Nightingale P, Stavropoulos-Kalinoglou A, Kitas GD (2012a) Lack of association between asymmetric dimethylarginine and in vivo microvascular and macrovascular endothelial function in patients with rheumatoid arthritis. Clin Exp Rheumatol 30:388–396
Sandoo A, Dimitroulas T, Toms TE, Hodson J, Veldhuijzen van Zanten JJ, Smith JP, Kitas GD (2012b) Clinical remission following treatment with tumour necrosis factor-alpha antagonists is not accompanied by changes in asymmetric dimethylarginine in patients with rheumatoid arthritis. Clin Biochem 45:1399–1403
Sandoo A, Dimitroulas T, Hodson J, Smith JP, Douglas KM, Kitas GD (2014) Cumulative inflammation associates with asymmetric dimethylarginine in rheumatoid arthritis: a 6 year follow-up study. Rheumatology (Oxford) Sep 3. pii: keu349 (Epub ahead of print)
Schepers E, Glorieux G, Dhondt A, Leybaert L, Vanholder R (2009) Role of symmetric dimethylarginine in vascular damage by increasing ROS via store-operated calcium influx in monocytes. Nephrol Dial Transplant 24:1429–1435
Schulze F, Carter AM, Schwedhelm E, Ajjan R, Maas R, von Holten RA, Atzler D, Grant PJ, Böger RH (2010) Symmetric dimethylarginine predicts all-cause mortality following ischemic stroke. Atherosclerosis 208:518–523
Schutte AE, Schutte R, Huisman HW, van Rooyen JM, Fourie CM, Malan L, Malan NT, Schwedhelm E, Strimbeanu S, Anderssohn M, Böger RH (2010) Dimethylarginines: their vascular and metabolic roles in Africans and Caucasians. Eur J Endocrinol 162:525–533
Schwedhelm E, Wallaschofski H, Atzler D, Dörr M, Nauck M, Völker U, Kroemer HK, Völzke H, Böger RH, Friedrich N (2014) Incidence of all-cause and cardiovascular mortality predicted by symmetric dimethylarginine in the population-based study of health in pomerania. PLoS One 9:e96875
Seppälä I, Kleber ME, Lyytikäinen LP, Hernesniemi JA, Mäkelä KM, Oksala N, Laaksonen R, Pilz S, Tomaschitz A, Silbernagel G, Boehm BO, Grammer TB, Koskinen T, Koskinen T, Juonala M, Hutri-Kähönen N, Alfthan G, Viikari JS, Kähonen M, Raitakari OT, März W, Meinitzer A, Lehtimäki T (2014) Genome-wide association study on dimethylarginines reveals novel AGXT2 variants associated with heart rate variability but not with overall mortality. Eur Heart J 35:524–531
Siegerink B, Maas R, Vossen CY, Schwedhelm E, Koenig W, Böger R, Rothenbacher D, Brenner H, Breitling LP (2013) Asymmetric and symmetric dimethylarginine and risk of secondary cardiovascular disease events and mortality in patients with stable coronary heart disease: the KAROLA follow-up study. Clin Res Cardiol 102:193–202
Simmons WW, Closs EI, Cunningham JM, Smith TW, Kelly RA (1996) Cytokines and insulin induce cationic amino acid transporter (CAT) expression in cardiac myocytes. Regulation of l-arginine transport and no production by CAT-1, CAT-2A, and CAT-2B. J Biol Chem 271:11694–11702
Stamatelopoulos KS, Kitas GD, Papamichael CM, Chryssohoou E, Kyrkou K, Georgiopoulos G, Protogerou A, Panoulas VF, Sandoo A, Tentolouris N, Mavrikakis M, Sfikakis PP (2009) Atherosclerosis in rheumatoid arthritis versus diabetes: a comparative study. Arterioscler Thromb Vasc Biol 29:1702–1708
Strobel J, Mieth M, Endress B, Auge D, König J, Fromm MF, Maas R (2012) Interaction of the cardiovascular risk marker asymmetric dimethylarginine (ADMA) with the human cationic amino acid transporter 1 (CAT1). J Mol Cell Cardiol 53:392–400
Surdacki A, Martens-Lobenhoffer J, Wloch A, Marewicz E, Rakowski T, Wieczorek-Surdacka E, Dubiel JS, Pryjma J, Bode-Böger SM (2007) Elevated plasma asymmetric dimethyl-l-arginine levels are linked to endothelial progenitor cell depletion and carotid atherosclerosis in rheumatoid arthritis. Arthritis Rheum 56:809–819
Surdacki A, Kruszelnicka O, Rakowski T, Jaźwińska-Kozuba A, Dubiel JS (2013) Asymmetric dimethylarginine predicts decline of glucose tolerance in men with stable coronary artery disease: a 4.5-year follow-up study. Cardiovasc Diabetol 12:64
Tsikas D, Sandmann J, Savva A, Luessen P, Böger RH, Gutzki FM, Mayer B, Frölich JC (2000) Assessment of nitric oxide synthase activity in vitro and in vivo by gas chromatography-mass spectrometry. J Chromatogr B Biomed Sci Appl 26(742):143–153
Turiel M, Tomasoni L, Sitia S, Cicala S, Gianturco L, Ricci C, Atzeni F (2010) De Gennaro Colonna V, Longhi M, Sarzi-Puttini P (2010) Effects of long-term disease-modifying antirheumatic drugs on endothelial function in patients with early rheumatoid arthritis. Cardiovasc Ther 28:e53–e64
Wallace TM, Levy JC, Matthews DR (2004) Use and abuse of HOMA modelling. Diabetes Care 27:1487–1495
Willers J, Hahn A (2012) Cardiovascular risk in patients with rheumatoid arthritis: assessment of several traditional risk parameters and of a German risk score model. Rheumatol Int 32:3741–3749
Zsuga J, Török J, Magyar MT, Valikovics A, Gesztelyi R, Lenkei A, Csiba L, Kéki S, Zsuga M, Bereczki D (2007) Dimethylarginines at the crossroad of insulin resistance and atherosclerosis. Metabolism 56:394–399
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants
The study received approval from the Black Country Research Ethics Committee. All procedures performed in the study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study according to the Declaration of Helsinki.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dimitroulas, T., Hodson, J., Sandoo, A. et al. Symmetric dimethylarginine (SDMA) serum levels in rheumatoid arthritis: correlations with insulin resistance and disease activity scores. Amino Acids 47, 1995–2004 (2015). https://doi.org/10.1007/s00726-015-1953-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-015-1953-x