
Abstract
Since d-amino acids were identified in mammals, d-serine has been one of the most extensively studied “unnatural amino acids”. This brain-enriched transmitter-like molecule plays a pivotal role in the human central nervous system by modulating the activity of NMDA receptors. Physiological levels of d-serine are required for normal brain development and function; thus, any alterations in neuromodulator concentrations might result in NMDA receptor dysfunction, which is known to be involved in several pathological conditions, including neurodegeneration(s), epilepsy, schizophrenia, and bipolar disorder. In the brain, the concentration of d-serine stored in cells is defined by the activity of two enzymes: serine racemase (responsible for both the synthesis and degradation) and d-amino acid oxidase (which catalyzes d-serine degradation). Both enzymes emerged recently as new potential therapeutic targets for NMDA receptor-related diseases. In this review we have focused on human d-amino acid oxidase and provide an extensive overview of the biochemical and structural properties of this flavoprotein and their functional significance. Furthermore, we discuss the mechanisms involved in modulating enzyme activity and stability with the aim to substantiate the pivotal role of d-amino acid oxidase in brain d-serine metabolism in physiological and pathological conditions and to highlight its great significance for novel drug design/development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
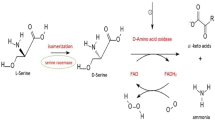
The flavoenzyme d-amino acid oxidase (DAAO, EC 1.4.3.3) is the only enzyme which has been proven to catalyze the strictly specific oxidative deamination of neutral d-amino acids. It was first discovered in the porcine kidney by Krebs (1935) and since then has been extensively studied as a model of FAD-dependent oxidase. DAAO has a broad substrate specificity and metabolizes a number of neutral, hydrophobic, polar, and basic d-amino acids (Pilone et al. 1989; Curti et al. 1992; Pilone 2000; Pollegioni et al. 2007a, b). The different substrates are oxidized through the concomitant reduction of the FAD cofactor to give the corresponding imino acids and hydrogen peroxide, produced upon flavin reoxidation on molecular oxygen; subsequently, the imino acids nonenzymatically hydrolyze to α-keto acids and ammonia (Fig. 1a).

a Scheme of the oxidative deamination of d-serine catalyzed by DAAO. The hydride transfer of the α-proton of d-serine to the oxidized FAD cofactor (DAAOox) yields imino-serine and reduced flavin (DAAOred). The imino acid is subsequently nonenzymatically hydrolyzed to hydroxypyruvate and ammonia, while the reduced flavin is reoxidized by molecular oxygen, producing hydrogen peroxide. b Role of hDAAO and pLG72 on d-serine bioavailability at glutamatergic synapses: pLG72 modulates the amount of active hDAAO acting on the stability of the holoenzyme. Under pathological conditions, the supposed lower expression level of pLG72 would result in an overactivation of hDAAO and a consequent decrease in d-serine cellular concentration
Putative DAAO genes have been identified in all kingdoms (plants representing the main exception) and DAAO from a variety of sources, ranging from microorganisms to mammals, have been purified and characterized (Pollegioni et al. 2007a). Several DAAO protein sequence regions are highly conserved across species, including residues that constitute the active site and those interacting with FAD, see below (Pollegioni et al. 2007b). All known DAAOs possess a C-terminal PTS1 sequence for peroxisomal targeting. Indeed, owing to this localization the cytotoxic product hydrogen peroxide can be efficiently removed.
DAAO plays diverse roles in different organisms: its main function in microorganisms is to make d-amino acids—via enzymatic oxidation—available for cell metabolism. However, several specific functions in nematodes, insects and lower vertebrates have also been proposed (Pollegioni et al. 2007a; Saitoh et al. 2012). In mammals DAAO is mainly present in kidney and liver (although it is not present in the mouse liver) (Konno et al. 1997) and, unexpectedly, it is enriched in the brain. Historically, endogenous d-amino acids were considered to be absent in higher animals. In fact, although DAAO enzyme activity was detected in mammal brains over 40 years ago (Neims et al. 1966; Arnold et al. 1979), its role remained enigmatic until d-amino acids (including d-alanine, d-serine, d-leucine and d-proline) were discovered in brain tissues. Among them d-serine is by far the most abundant (Nagata et al. 1992, 1994; Hashimoto et al. 1993; Hamase et al. 1997; Inoue et al. 2000; Morikawa et al. 2003; Song et al. 2008).
d-Serine: synthesis and role
d-Serine, an atypical signaling molecule, is synthesized from l-serine by the PLP-dependent enzyme serine racemase (EC 5.1.1.18, SR, also present in the brain) (Wolosker et al. 1999a, b; Conti et al. 2011) and is presumably degraded by both SR, which also catalyzes the α,β-elimination of water from l- and d-serine (Foltyn et al. 2005), and DAAO (Pollegioni and Sacchi 2010). d-Serine binds to the “strychnine-insensitive glycine modulatory site” of the N-methyl-d-aspartate type glutamate receptor (NMDAR) (Mothet et al. 2000); thus, it plays a key role as a coagonist of this excitatory receptor, which is critically involved in brain development, neuronal cell migration, and plasticity as well as in learning, memory, and excitotoxicity (Wolosker et al. 1999b; Foltyn et al. 2005; Mothet et al. 2000; Snyder and Kim 2000; Martineau et al. 2006; Wolosker et al. 2008). Interestingly, the distribution pattern of DAAO gene and protein expression in the central nervous system (CNS) is inversely correlated with d-serine content and distribution (Horiike et al. 1994). Brain DAAO activity can be detected and increases rapidly in the cerebellum, pons, and medulla oblongata of rodents around postnatal day 10 (Weimar and Neims 1977) when d-serine concentration starts to significantly decrease in the same areas/regions (Hashimoto et al. 1995). Based on these findings, it was proposed that DAAO might physiologically affect NMDAR function by modulating d-serine cellular storage/turnover and thus, indirectly, its availability at the synapses (Fig. 1b). Although the observation that, at the cellular level, exogenously applied yeast DAAO reduces NMDAR-mediated currents supports this notion (Mothet et al. 2000; Stevens et al. 2003; Panatier et al. 2006), the biological relevance of DAAO has occasionally been questioned because of the modest affinity of the mammalian flavoenzyme for its substrates (in the millimolar range, see below) (Molla et al. 2006a; Pollegioni and Sacchi 2010) and the low d-amino acids concentration in the brain. Indirect data from studies performed in transgenic animals strikingly substantiate the crucial physiological role of the flavoenzyme: in ddY/DAAO−/− mice DAAO activity is lost due to an inactivating point mutation (Gly181Arg) (Konno and Yasumura 1983; Sasaki et al. 1992) and a substantial increase (several fold) in the concentrations of d-serine and other d-amino acids is detected. ddY/DAAO−/− mice display behavioral phenotypes consistent with altered NMDAR signaling, including decreased stereotypy and ataxia elicited by MK-801 (Almond et al. 2006; Hashimoto et al. 2005) and enhanced spatial learning and long-term potentiation (LTP) in the hippocampus (Maekawa et al. 2005). Furthermore, oral or systemic administration of DAAO inhibitors to normal rodents elevated central d-serine levels (Adage et al. 2008; Duplantier et al. 2009) and increased NMDAR-mediated function (Verrall et al. 2010). These findings support a major role for DAAO in d-amino acid—in particular d-serine—metabolism. However, the recent observation that SR is predominantly expressed in neurons (which release d-serine upon membrane depolarization) (Kartvelishvily et al. 2006; Rosenberg et al. 2010) and poorly expressed in astrocytes (the cells containing the highest amount of d-serine in most brain regions) (Schell et al. 1997; Kartvelishvily et al. 2006; Puyal et al. 2006; Williams et al. 2006; Ding et al. 2011) argues against its role as the main d-serine-degrading enzyme, at least in the latter cell type.
Apart from NMDAR modulation, d-serine also antagonizes AMPA glutamate receptors (Gong et al. 2007), suggesting that this neuromodulator (and its degrading enzyme) could be involved in both positive and negative modulatory effects at glutamatergic synapses. d-Serine is also an endogenous ligand of the ionotropic glutamate-like receptor GluRδ2, which is important in cerebellar development and plasticity (Hansen et al. 2009). Moreover, d-serine may antagonize NR1/NR3A or NR1/NR3B receptors (which are insensitive to glutamate and activated by glycine) and thus modulate glycinergic transmission (Chatterton et al. 2002; Takarada et al. 2009). On the other hand, DAAO might be involved in the metabolism of both d-proline, which does not act at NMDARs but can activate glycine receptors (Hamasu et al. 2008), and d-leucine, a potent regulator of the blood–brain barrier enkephalin transport system (Banks and Kastin 1991).
DAAO brain localization
DAAO is widely present in the adult human CNS, but its distribution in terms of brain regions, cell types, and subcellular compartments is still under debate, in particular concerning the levels of enzyme expression and activity. Although a regional heterogeneity has been observed, DAAO is generally considered to be a hindbrain enzyme that is enriched in the cerebellum and in the brainstem (Horiike et al. 1994). In agreement with previous studies in rodents (Schell et al. 1997; Moreno et al. 1999; Wolosker et al. 2008), DAAO expression and activity were robustly detected in the cerebellum in human brain (Kapoor et al. 2006; Verrall et al. 2007), particularly in Bergmann glia (Verrall et al. 2007) (Fig. 2a–d). DAAO distribution in the human forebrain is less clear. Notwithstanding the barely measurable levels of enzyme activity, a consistent amount of DAAO mRNA and protein has been highlighted. Bendikov et al. (2007) detected robust levels of this flavoprotein in the parietal lobe, and Verrall et al. (2007) confirmed that DAAO protein is unequivocally expressed in human cerebral cortex and hippocampus, also showing that its mRNA is mainly distributed within neurons and to a small degree in glia. Since it was not feasible to detect the protein by Western blot analysis, they raised the intriguing issue of a regional heterogeneity in DAAO antigenicity, possibly due to differences in post-translational modifications (Verrall et al. 2007). Flavoenzyme expression was further confirmed in the prefrontal cortex by immunolocalization and immunoprecipitation studies performed in our laboratory (Sacchi et al. 2008); DAAO seems to be present essentially in glial cells. We hypothesized that DAAO activity could not be detected by the commonly used assay in the forebrain because some kind of negative effector (a small ligand or an inactivating protein) is present along with the enzyme, downregulating its—already low—activity (Molla et al. 2006a).

Human DAAO regional and cellular distribution. a In the prefrontal cortex, hDAAO immunoreactivity is localized essentially in the gray matter (gm), compared to the unstained white matter (wm); b in the same brain region the immunorecognition signal corresponding to the flavoprotein is mainly evident in pyramidal neurons, which are labeled throughout their cell bodies (asterisk); c in the cerebellum hDAAO is mainly distributed in the Purkinje cell layer (pcl) with moderate staining observed in the molecular layer (ml), granule cell layer (gcl), and white matter (wm); d in this region, Bergmann glia cells in the Purkinje cell layer (pcl) are strongly immunopositive (arrows), whereas Purkinje cells are unstained (asterisk); in the molecular layer (ml) moderate immunolabeling of Bergmann glia radial processes is also observed. Scale bars a 1 mm, c 0.5 mm, b, d 50 μm. Modified from (Verrall et al. 2007). e, f hDAAO and pLG72 subcellular localization. The subcellular distribution of the two proteins has been investigated in U87 glioblastoma cells stably expressing EYFP-hDAAO and pLG72-ECFP: e EYFP-hDAAO signal clearly overlaps with the stained peroxisomal protein PMP70; f pLG72-ECFP fluorescence distribution largely overlaps with the pattern of signal obtained by staining the cells with a mitochondrial marker (Sacchi et al. 2011). Scale bars 3 μm
Subcellular localization
DAAO subcellular distribution, in particular concerning the way by which it might exert its putative physiological function as a peroxisomal enzyme (Fig. 2e, f), represents an additional point for discussion. It is conceivable that d-serine could access the peroxisomes through a not yet identified membrane transport mechanism; otherwise, an active form of DAAO might be present in the cytosol, thus directly consuming d-serine from cytosolic storage. Interestingly, the presence of an extraperoxisomally active DAAO has been reported both in glial cells (Sacchi et al. 2008, 2011) and neurons (Popiolek et al. 2011). The localization of DAAO outside peroxisomes is mainly related to interaction with regulatory interaction proteins (see below).
Pathological aspects
Since d-serine-dependent, aberrant NMDAR activation is involved in a variety of neurological diseases, a possible imbalance in the levels or activities of the enzymes related to the metabolism of this neuromodulator might be also implicated. Multiple lines of evidence suggest that excessive production or release of d-serine is involved in acute and chronic degenerative disorders (stroke, epilepsy, amyotrophic lateral sclerosis (ALS), and Parkinson’s, Alzheimer’s, and Huntington’s disease), while an anomalous drop in the concentration of the neuromodulator can be observed in psychiatric disorders such as schizophrenia and bipolar disorders (Rossi et al. 2007; Labrie and Roder 2010; Mustafa et al. 2010; Fuchs et al. 2011; Lu et al. 2011). In particular, d-serine and SR levels are greatly increased in the spinal cord of patients affected by familial and sporadic ALS as well as in Alzheimer’s disease, while they are downregulated in schizophrenia (Wolosker et al. 2008; Billard 2008 and references therein). Furthermore, it has been reported that a unique mutation in the DAAO gene yielding an almost total loss of enzymatic activity is associated with the adult onset of familial ALS (Mitchell et al. 2010).
The molecular mechanisms thought to trigger NMDAR hypofunction in schizophrenic patients have been investigated. Genetic studies have identified several risk genes for schizophrenia that influence NMDAR activity (Coyle 2006; Ross et al. 2006): these include genes that encode proteins responsible for d-serine metabolism. Furthermore, reduced levels of d-serine have been reported in the serum and cerebrospinal fluid (CSF) of affected individuals (Bendikov et al. 2007; Hashimoto et al. 2003) and, when used as an adjuvant to both conventional neuroleptics (Tsai et al. 1998) and new atypical antipsychotics (Heresco-Levy et al. 2005), d-serine significantly improves the symptoms of the disease. At present, there is weak evidence that the d-serine-producing enzyme SR may be involved as a schizophrenia susceptibility gene (Yamada et al. 2005; Strohmaier et al. 2007) and that the protein expression levels might be altered in forebrain areas of these patients (Duplantier et al. 2009; Bendikov et al. 2007).
Since the landmark analysis of Chumakov et al. (2002) was published in 2002, several association studies have linked DAAO and its proposed regulatory interactor pLG72 with schizophrenia susceptibility. pLG72 is a small protein (153 amino acids, 18 kDa) with no recognizable motifs and a high α-helix content, which suggests a membrane localization (Fig. 3) (Molla et al. 2006b). It is encoded by the G72 gene located within the chromosome 13q14 region, which provides significant genetic evidence for linkage with the disease. G72 represents an exceptional case of a primate-specific gene encoding for a rapidly changing protein which is related to fast evolution of underlying brain functions (Chen and Li 2001); Chumakov et al. 2002). In brain, expression of pLG72 protein is maximal in the midbrain and prefrontal cortex (Chumakov et al. 2002; Korostishevsky et al. 2004), but the distribution pattern is still controversial (Benzel et al. 2008). Early studies on transiently transfected cells reported that pLG72 was localized in the endoplasmic reticulum/Golgi apparatus (Chumakov et al. 2002). More recently however, a mitochondrial localization has been proposed for this protein (Otte et al. 2009; Sacchi et al. 2011). By means of a yeast two-hybrid system, pLG72 was identified as a putative DAAO-interacting partner and it was suggested to modulate the flavoenzyme activity (Chumakov et al. 2002).

pLG72 properties deduced from its amino acidic sequence. Secondary structure prediction was performed using Jpred server (http://www.compbio.dundee.ac.uk/www-jpred/). Hydrophobicity plot (arbitrary units) was calculated using Expasy ProtScale tool (http://web.expasy.org/protscale/) and classical Kyte and Doolittle parameters with a nine-residue window. Probability of disorder plot (arbitrary units) was calculated using RONN software (http://www.strubi.ox.ac.uk/RONN). Gray shaded areas represents hydrophobic (top) or disordered (bottom) protein regions
Notably, altered DAAO expression levels and activity have been reported in schizophrenia patients. In particular, DAAO activity was determined in postmortem brain tissues from individuals who had been diagnosed as schizophrenia, bipolar disorder, or major depressive disorder (Madeira et al. 2008). Differently from the latter two conditions, an increase in DAAO activity was demonstrated for the first time in the cerebral cortex of schizophrenia patients, suggesting that the enzyme is active in the forebrain areas (regions particularly relevant for cognitive deficits) and may play a role in the pathophysiology of the disorder. Elevated DAAO activity in the cerebellum of affected individuals (Kapoor et al. 2006; Burnet et al. 2008) accompanied by an increase in gene expression was subsequently reported (Verrall et al. 2007; Burnet et al. 2008). In addition to the cerebellum, elevated DAAO immunoreactivity was also recently detected in the medulla oblongata and choroid plexus (CP) (Ono et al. 2009) of individuals affected by schizophrenia, suggesting that the onset of the disease is associated with abnormal DAAO levels in different brain areas. In particular, since CP plays an important role in the homeostasis of nutrients (including amino acids) in the CSF (Redzic and Segal 2004), the low d-serine levels in the CSF of schizophrenia patients (Hashimoto et al. 2003; Bendikov et al. 2007) might arise from increased DAAO expression in the CP (Ono et al. 2009).
Increased DAAO activity in schizophrenia provides the rationale to propose inhibition of d-serine-degrading enzyme(s) (eventually in combination with d-serine administration) as a therapeutic approach. Although no clinical trials based on DAAO inhibitors to treat schizophrenia have been performed to date, several preliminary preclinical studies showed that DAAO inactivation (either in ddY/DAAO−/− mice or after pharmacological inhibition of the enzyme in rodents) produces behavioral, electrophysiological, and neurochemical effects that suggest a procognitive profile (Duplantier et al. 2009). However, DAAO inhibitors to be used as drugs for therapeutic treatments must be carefully designed since the maintenance of the physiological balance between d-serine synthesis and degradation appears extremely important. In fact, the lack of degradation of the neuromodulator—as observed in ALS affected individuals (Sasabe et al. 2012)—has been shown to induce motoneuron degeneration.
Human DAAO biochemical properties
Over the past few decades, DAAO has become the paradigm for the dehydrogenase/oxidase class of flavoproteins: several aspects of the structural/functional relationships have been extensively investigated using the protein purified from pig kidney and the recombinant DAAOs from microbial sources, mostly yeasts (Pollegioni et al. 1993, 2007a; Pilone 2000). The expression of human DAAO (hDAAO) in E. coli was first reported in 2000 (Raibekas et al. 2000), but this procedure yielded a low amount of purified protein (0.5 mg l−1 culture medium). Most recently, a robust and reliable process at the laboratory bioreactor scale was established (Romano et al. 2009): this procedure achieved ~100 mg of hDAAO/liter culture medium. Recombinant hDAAO was purified as a stable and active holoenzyme (347 residues, ~40 kDa), showing the typical absorbance spectrum and properties of the FAD-containing flavoenzymes (Molla et al. 2006a): (1) hDAAO contains a single molecule of noncovalently bound FAD per protein monomer; (2) hDAAO binds the classical competitive inhibitors benzoate and anthranilate (with K d values in the μM range, see below); (3) the oxidized flavin covalently binds sulfite; (4) the oxidized cofactor is fully converted to the reduced species upon the addition of a substrate under anaerobic conditions and then quickly reacts with dioxygen; (5) the enzyme stabilizes the anionic semiquinone form of the cofactor. The reaction catalyzed by hDAAO follows a sequential kinetic mechanism in which the rate-limiting step is represented by the product release from the reoxidized enzyme, analogously to porcine DAAO, although the rate of flavin reduction is slower for the human enzyme (Molla et al. 2006a).
The human flavoprotein possesses a number of peculiar properties. Since the binding of the flavin cofactor is the weakest among the purified DAAOs (K d = 8 × 10−6 M, 40- and 400-fold higher than for the porcine and yeast enzymes, respectively), the recombinant hDAAO exists in solution as an equilibrium between the active holoenzyme and the inactive apoprotein (Fig. 4). Indeed, a 20-fold tighter interaction with FAD is observed when a ligand (e.g., benzoate) is present in the active site of the enzyme (Molla et al. 2006a). In the reconstitution process that leads to the active holoenzyme, first the flavin cofactor binds to the apoprotein (thus acquiring catalytic activity) and then a slower secondary conformational change follows. The latter is assumed to definitively stabilize the protein (Caldinelli et al. 2009). hDAAO is a dimeric holoenzyme in the 1- to 24-mg ml−1 protein concentration range which retains this oligomeric state in the apoprotein form (Molla et al. 2006a), whereas all known DAAO apoproteins are stable 40-kDa monomers. Holo- and apoprotein forms of hDAAO show substantial differences in conformation while they share highly similar secondary structural elements.

Scheme depicting the different forms of hDAAO observed in vitro. Apoprotein (colorless in solution) is shown in gray; holoenzyme in yellow, and apoprotein–CPZ complex in pale yellow. Substrate d-serine and inhibitor 3-hydroxyquinolin-2(1H)-one (Duplantier et al. 2009) are shown in purple. Remarkably, hDAAO is able to bind the substrate d-serine even in the apoprotein form (K d = 3.0 mM, not shown) (Caldinelli et al. 2010)
Likewise to other previously characterized DAAOs, the human enzyme oxidizes small, uncharged d-amino acids. It shows a low catalytic efficiency and substrate affinity on the physiological substrate d-serine, whereas a higher activity is apparent on d-alanine (Table 1). Interestingly, the human enzyme is also active on two additional signaling molecules present at relevant concentrations in the brain, i.e., the alternative NMDAR coagonist glycine and the agonist of the receptor d-aspartate; however, the relative affinity for these substrates is very low (Table 1). In contrast, no activity is evident on NMDA as substrate. hDAAO shows a higher catalytic efficiency for bulky and hydrophobic substrates, such as d-tryptophan, d-phenylalanine, d-tyrosine, and d-proline, and catalyzes the oxidation of d-3,4-dihydroxy-phenylalanine (d-DOPA) into dihydroxyphenylpyruvic acid (Kawazoe et al. 2007). It has been proposed that d-DOPA might be pharmacologically relevant since its l-stereoisomer is commonly used in the treatment of Parkinson’s disease (human cells synthesize dopamine from it). Interestingly, at low concentration d-DOPA is metabolized more efficiently by hDAAO than d-tyrosine and d-phenylalanine (stereoisomer of the dopamine precursors) or any previously identified substrate (Table 1); nevertheless, a substrate inhibition effect is evident at high concentration (as also observed with d-tyrosine and d-phenylalanine) (Kawazoe et al. 2007). As l-DOPA is quickly converted to dopamine, the most effective symptomatic treatment for Parkinson’s disease consists in administering it in combination with a peripheral DOPA decarboxylase inhibitor (Fernandez and Odin 2011). The slower utilization of d-DOPA and the longer retention of its metabolites in the brain indicate that the combination of the two isomers might represent a more efficacious therapeutic strategy (Kawazoe et al. 2007).
More recently, DAAO was also reported to catalyze the conversion of d-kynurenine (d-KYN) to kynurenic acid (KYNA), a neuroactive metabolite of the kynurenine pathway of tryptophan degradation with antioxidant properties (Lugo-Huitron et al. 2011). Although it was originally described as a broad spectrum antagonist of excitatory amino acids receptors (Perkins and Stone 1982), KYNA is known to inhibit the NMDAR (Ganong et al. 1983) through a preferential interaction with the d-Ser/Gly coagonist site (Kessler et al. 1989). In addition, it acts as an antagonist of the α7 nicotinic acetylcholine receptor and also stimulates the orphan G-protein-coupled receptor GPR-35. Recent investigations provided evidence that DAAO may contribute to KYNA production by the oxidative deamination of d-KYN in human and rat brain (Fukushima et al. 2009; Ishii et al. 2010). In particular, (in vivo) focal application of d-KYN by reverse microdialysis induced a significant increase in extracellular KYNA levels in the rat striatum and a concomitant reduction in extracellular dopamine (Pérez-de la Cruz et al. 2012): preadministration of a selective DAAO inhibitor markedly suppressed the increase in extracellular KYNA (Fukushima et al. 2009). Elevated KYN levels in mammalian tissues might be a consequence of microbial infections. d-KYN and DAAO may, therefore, be at least in part responsible for the significant increases in brain KYNA levels seen under inflammatory conditions (Holtze et al. 2008; Pérez-de la Cruz et al. 2012).
Structural details of human DAAO (and functional significance)
The overall three-dimensional structure of DAAO has been largely conserved during evolution from microorganisms (yeasts) to mammals (Pollegioni et al. 2002, 2007a, b); in fact, the RMSD (root-mean-square deviation) between the human and yeast (Rhodotorula gracilis) protein is only 1.48 Å (when superimposing the α-C of 228 residues), the main differences being concentrated in a long C-terminal loop. As expected, the structural similarity between porcine (pkDAAO) and human enzyme is even higher, with a RMSD of 0.40 Å when superimposing the α-C of 308 residues. DAAO tertiary structure can be divided in two large domains formed by several noncontiguous sequence regions (Fig. 5a): a FAD-binding domain possessing the classical dinucleotide binding fold and an interface (dimerization) domain characterized by a large twisted antiparallel β-sheet formed by eight β-strands (Kawazoe et al. 2006). Notwithstanding the conservation of the general flavo-oxidase fold, relevant and specific structural and functional differences are observed in the enzyme isolated from different species (Pollegioni et al. 2007a).

Ribbon representation of hDAAO in complex with 3-hydroxyquinolin-2(1H)-one (PDB code 3g3e) (Duplantier et al. 2009). a Mode of hDAAO dimerization. The monomer on the left is represented using a color scaling for temperature factors (or B-factor, a parameter used to fit a potential structure to the electron density map obtained from X-ray crystallography which measures the disorder of the atoms in the structure): blue corresponds to a low and magenta to a high B-factor values. b Detail of hDAAO active site. The inhibitor is represented in brown and FAD cofactor in yellow. Hydrophobic interactions are shown in pale yellow, H-bonds and polar interactions are represented by black dotted lines, and π–π interactions are shown by green arrows
Dimerization
The quaternary structure of hDAAO in solution is always dimeric, with a head-to-head mode of monomer–monomer interaction (Fig. 5a). This is different from other mammalian DAAOs, which are either monomeric (e.g., DAAO from rat, rDAAO) or show a concentration-dependent oligomerization state (e.g., pkDAAO) (Pollegioni et al. 2007a; Frattini et al. 2011). The dimerization surface of hDAAO is located at the top of the apical interface domain and is formed by 36 residues: its composition was proposed as the main determinant responsible for the different quaternary structure observed in mammalian DAAOs. This region is a hot spot for mutations with a substitution frequency (33 %, 12 out of 36 residues) twofold higher than for the remainder of the protein (13 %, 41 out of 311 residues). This results in a completely different distribution of charges at the interface between the human and the porcine enzyme: in pkDAAO, the dimerization interface is mostly positively charged (Mattevi et al. 1996) while in the human enzyme it is mainly negatively charged (Kawazoe et al. 2006). Comparison of hDAAO with the closely related monomeric rDAAO (80 % sequence identity) (Frattini et al. 2011) shows that substitution of two of the few positively charged residues at the hDAAO monomer–monomer interface totally abolished the protein’s ability to dimerize. These residues are His78 and, in particular, Arg120, which is involved in electrostatic interactions with the carboxylic group of Asp73 and hydrogen-bonded to the backbone C = O groups of Asp109, Thr110, and Val111 (all belonging to the facing monomer). Interestingly, hDAAO retains the dimeric state in the apoprotein form, indicating that a large portion of the dimerization interface is conserved even after FAD dissociation (Caldinelli et al. 2009).
FAD binding
The FAD cofactor is bound in an elongated conformation, completely buried in the protein core (Fig. 5); the isoalloxazine ring is the sole part of the cofactor that is accessible from bulk solvent. hDAAO binds FAD cofactor mainly through the Rossmann fold of the FAD-binding domain (N-terminal region of the protein). The complex network of noncovalent contacts formed by the cofactor with the protein moiety increases the stability of the protein against chemical and thermal denaturation (see below) (Caldinelli et al. 2009). Interestingly, comparative analysis of the contact network between FAD and the surrounding residues (within 6 Å from the cofactor) did not reveal remarkable differences between human and porcine DAAO, with the exception of a short hydrophobic stretch (47-VAAGL-51) located close to the si-face of the flavin isoalloxazine ring. This stretch is classified as a structurally ambivalent peptide (SAP), i.e., a peptide that assumes different conformations depending on the structural context. In particular, the lower affinity for FAD to hDAAO could arise from the loss of the H-bond between the backbone nitrogen atom of Ala49 of SAP and flavin N(5): the distance between these two atoms, which is 3.7 Å in pkDAAO, is increased up to 4.9 Å in hDAAO. The loss of the binding energy of a H-bond (~2 kcal mol−1) could account for the 40-fold lower cofactor affinity (Gao and Kelly 2008). In addition, the absence of the aforementioned H-bond also correlated with the slower rate of flavin reduction in hDAAO (180 s−1) as compared with the porcine enzyme (4000 s−1) (Porter et al. 1977; Kawazoe et al. 2006; Molla et al. 2006a).
The isoalloxazine ring, located at the interface between the FAD binding and the interface domain, forms the “floor” of the active site (Fig. 5b). The protein residues surrounding the isoalloxazine are fundamental for modulating the functional properties of the cofactor (i.e., the redox potentials). Two major interactions are observed in this region: the first is an electrostatic interaction between the flavin N(1) and the partial positive charge of a dipole, generated by helix F5, which is assumed to stabilize the negative charge of the reduced flavin during catalysis. The second is a H-bond between FAD O(2) and Thr317 hydroxyl. This latter interaction is important both for cofactor binding and function.
Substrate binding
The DAAO active site is striking in its simplicity, see Fig. 5b (Umhau et al. 2000). DAAO catalyzes the transfer of a hydride ion from the substrate to the N(5) atom: direct participation of active-site residues is not required for the chemical step of catalysis, e.g., there is no need for an active-site base (Harris et al. 1999; Molla et al. 2000). The main requirement favoring an efficient hydride transfer is a very precise binding of the substrate relative to the FAD cofactor (Umhau et al. 2000); in particular, the α-C of the substrate must be at ~3 Å from the N(5) of FAD with the α-H pointing toward that atom. This condition is achieved by formation of a tight network of H-bonds between the substrate and the active-site residues. At the physiological pH of the peroxisome (hDAAO main subcellular localization), the substrate amino acids are largely present in the zwitterionic form (Harris et al. 2001). Hence, the negatively charged oxygen atoms of the α-carboxylic group of the substrate interact with the positively charged Arg283 and with the hydroxyl oxygen of Tyr228. The H-bonds between the α-amino group of the substrate and FAD O(4) and the Gly313 main chain C = O complete the bond network. The substrate side chain points toward the inner/upper part of the active site that is formed by an array of bulky hydrophobic residues (mainly Leu51, Leu215, Tyr228, and Ile230). The van der Waals and hydrophobic interactions formed in this region of the active site contribute significantly to the overall binding energy of the substrate and predispose the substrate specificity of the enzyme to hydrophobic amino acids, Table 1. Remarkably, Leu215 (Ile215 in pkDAAO) corresponds to Met213 of yeast DAAO that was demonstrated to be the most important residue for determining enzyme substrate specificity (Sacchi et al. 2002).
The volume of the human enzyme active site is ~220 A3, which is slightly larger than the pkDAAO active site, whose volume is ~160 A3 and thus suitable to accommodate d-amino acids composed of up to nine heavy atoms (e.g., d-valine possesses a volume of ~170 A3) (Mattevi et al. 1996). The roof of the active site of hDAAO is formed by the side chain of Tyr224 (Fig. 5b), which is not necessary for catalysis (Mattevi et al. 1996; Pollegioni et al. 2007a). This residue belongs to loop 216–228, which is thought to switch from an open to a closed conformation upon substrate binding. In yeast DAAO, this loop is replaced by a single side chain (Tyr238) (Pollegioni et al. 2002). The conformational movement of such an active-site loop limits the turnover number of mammalian DAAOs as compared to the yeast counterparts (14.7 and 350 s−1, respectively) and increases the hydrophobicity of the active site, thus improving the efficiency of the hydride transfer reaction. The flexibility of loop 216–228, and in particular of the side chain of Tyr224, allows hDAAO to bind substrates possessing large side chains, such as d-Phe, d-Tyr, or even d-DOPA, as compared to its putative physiological substrate d-serine (Table 1) (Molla et al. 2006a; Kawazoe et al. 2007).
Inhibitor binding
Classical DAAO inhibitors, such as benzoate, have been known since the 1960s, see below (Fonda and Anderson 1968), but recently the discovery that DAAO and d-amino acids are implicated in several physiological (and pathological) processes in mammalian CNS prompted a quest for more effective hDAAO inhibitors that potentially could be employed as drugs. The combined use of structural bioinformatics (in particular virtual screening procedures based on automated docking analysis), high-throughput screening, and structural biochemistry studies discovered several high-affinity DAAO inhibitors and experimentally determined the structure of six different hDAAO-inhibitor complexes (Table 2). These compounds can be subdivided into three distinct chemotypes (Lange et al. 2011): (1) the hetero(bi)cyclic carboxylic acids, as 5-methylpyrazole-3-carboxylic acid (AS057278) (IC50 = 910 nM) (Adage et al. 2008), 4H-furo[3,2-b]pyrrole-5-carboxylic acid (IC50 = 141 nM) (Sparey et al. 2008), and 4H-thieno[3,2-b]pyrrole-5-carboxylic acid (IC50 = 145 nM) (Smith et al. 2009); (2) a series of compounds based on benzo[d]isoxazol-3-ol, the most potent of which was 6-chlorobenzo[d]isoxazol-3-ol (CBIO) (IC50 = 188 nM) (Ferraris et al. 2008); and (3) the very potent hDAAO inhibitor 3-hydroxyquinoline-2-(1H)-one (IC50 = 4 nM) and its analogs (Table 2) (Duplantier et al. 2009). Their main properties and pharmacokinetics have been reviewed recently (Sacchi et al. 2012).
From the huge amount of structural data, we were able to elucidate in detail the binding mode of inhibitors at the hDAAO active site (Table 2; Fig. 5b). hDAAO inhibitors mimic d-amino acid binding behavior, i.e., the ligand molecule can be divided into two functional regions: a first region comprising polar or charged groups (e.g., a carboxylic acid, an hydroxyl, or an isoxazole ring) able to form H-bonds with polar residues of the active site (Arg283, Tyr228 and Gly313 main chain C = O, see above) and a second hydrophobic region (usually formed by a six- or five-atom aryl ring) which forms close contacts with hydrophobic residues of the active site (Fig. 5b). These hydrophobic interactions are of utmost importance since the inhibitors (the only exception being the classical ligand o-aminobenzoate) usually lack the substrate α-amino group. In addition, the aromatic ring of the inhibitor is stacked between the aryl ring of Tyr224 and the isoalloxazine ring of FAD (Ferraris et al. 2008), establishing strong π–π interactions (Fig. 5b). In silico simulations demonstrated that the best π–π interaction (in terms of binding energy) is achieved when the aromatic rings are not perfectly stacked (parallel-displaced configuration), with an estimated value of 2.8 kcal mol−1 for each π-stacking (Sinnokrot et al. 2002). This is true for the hDAAO inhibitors showing the highest affinities (i.e., 3-hydroxyquinoline-2(1H)-one or inhibitors with a fused pyrrole carboxylic acid) as compared to benzoate or o-aminobenzoate where an almost perfect superimposition between Tyr244 and the aromatic ring of the inhibitor is observed (Sparey et al. 2008; Duplantier et al. 2009). In some cases, both the overall molecular volume and the hydrophobicity of the inhibitor are enhanced by adding a halogen substituent to the aromatic ring, e.g., chlorine in CBIO (Ferraris et al. 2008) or chlorine or fluorine in compounds listed in Duplantier et al. (2009). Unfortunately, the results of such additions are unpredictable since, in several cases, large decreases in affinity were observed, probably because of steric clashes between the halogen substituent and the protein moiety (Duplantier et al. 2009).
Concerning the size of the inhibitor, hDAAO possesses a large active-site plasticity: the best inhibitors are composed of more than nine atoms and the presence of the aromatic ring renders them quite compact. In fact, the best inhibitor, 3-hydroxyquinoline-2(1H)-one, is formed by 12 atoms (and a volume of 149 A3) and it fits easily into the active site of the enzyme (Table 2).
Modulation of hDAAO structural–functional properties
In vitro studies
The conformation, function, and stability of hDAAO are affected by interactions with several partners: small-size molecules (FAD, substrates, and inhibitors) and interacting proteins.
FAD cofactor, mandatory for enzyme activity, is only weakly bound to hDAAO apoprotein (K d = 8 μM) (Molla et al. 2006a). By associating FAD to the apoprotein moiety, a holoenzyme is generated that possesses a more “compact” tertiary structure, decreased exposure of hydrophobic surfaces, and lower sensitivity to proteolysis and thermal denaturation (Table 3) (Caldinelli et al. 2009).
The classical competitive inhibitor benzoate (K d = 7 μM, K i = 7 μM) (Molla et al. 2006a; Kawazoe et al. 2006) binds to the hDAAO active site, yielding a ~20-fold increase in affinity for the cofactor (K d = 0.3 μM, Fig. 4) (Molla et al. 2006a). This raises the amount of the holoenzyme formed in solution and protects hDAAO against proteolysis and thermal denaturation (a T m increase of ~5 °C was apparent in the presence of benzoate, Table 3) (Caldinelli et al. 2010). Similar effects were observed in the presence of the pseudosubstrate CF3-d-alanine (K i = 3 μM), which also binds to hDAAO apoprotein and increases the K d for FAD binding (Caldinelli et al. 2010).
Another well-known hDAAO inhibitor is chlorpromazine (CPZ), a drug widely employed for the treatment of schizophrenia. CPZ competes with FAD for the binding to hDAAO apoprotein (K d = 5 μM; K i = 0.7 mM, Table 2) (Sacchi et al. 2008; Iwana et al. 2008). When in complex with CPZ, hDAAO is supposed to assume a conformation resembling that of the apoprotein since it is more sensitive to proteolysis and thermal denaturation than the native holoenzyme (Table 3; Fig. 4) (Caldinelli et al. 2010).
Concerning the modulation of hDAAO function by protein partners, pLG72 was identified by a yeast two-hybrid approach as a putative interacting partner of mammalian DAAOs (Chumakov et al. 2002). Subsequently, in slices from human brain cortex, we identified hDAAO and pLG72 in the same astrocytes and reported on the coimmunoprecipitation of the two proteins from human cortex extracts. We also demonstrated that pLG72 binding to both the holo- and apoprotein forms of hDAAO yields an ~200-kDa complex constituted by two hDAAO homodimers (2 × 80 kDa) and two pLG72 molecules (2 × 20 kDa) (Sacchi et al. 2008). Real-time analysis by the SPR method of the interaction between the holoenzyme or the apoprotein species of hDAAO to immobilized pLG72 showed similar apparent affinities (K d values were 5.3 vs. 3.1 × 10−7 M with and without free FAD, respectively) (Caldinelli et al. 2010). By using hDAAO-immobilized sensors and flushing different concentrations of pLG72, the association rate constants were higher than those with immobilized pLG72 (and traces were not affected by the presence of FAD): K d value was 0.8 × 10−7 M.
pLG72 binding does not affect the kinetic parameters of the reaction catalyzed by hDAAO on d-serine, the affinity for the cofactor, and the rate constant of FAD binding to the apoprotein: the main effect is a faster time course of hDAAO inactivation (Sacchi et al. 2008). We demonstrated that pLG72 binding modifies hDAAO tertiary structure and produces a decrease in the holoenzyme (active) form. In fact, the amount of FAD cofactor bound to the enzyme that can be reduced by the substrate decreases after the pLG72-hDAAO complex is formed. Visible absorbance and near-UV CD spectroscopy also show that pLG72 binding altered the tertiary structure of hDAAO. Indeed, limited proteolysis studies show that the presence of a stoichiometric amount of pLG72 increases the rate of hDAAO holoenzyme degradation (Fig. 6a), a destabilization most apparent at increasing pLG72 concentrations, see (Caldinelli et al. 2010). In contrast, the time course of proteolysis of the apoprotein form is not affected by adding pLG72 (Fig. 6a) (Caldinelli et al. 2010). The destabilization effect of pLG72 interaction on hDAAO half-life was also apparent at the cellular level (see below). Taken together, we propose that pLG72 binding switches off hDAAO activity and, under normal conditions, prevents glial d-serine from being fully degraded (Fig. 1b).

pLG72 modulates hDAAO stability/activity and cellular d-serine levels. a The time course of trypsin digestion of hDAAO (holo- and apo-form) is compared to that obtained for the hDAAO-pLG72 complex. As observed by monitoring the 40-kDa band corresponding to the intact monomer (top panel) by SDS-PAGE and densitometric analysis (bottom panel), binding of pLG72 to the holoenzyme produces an increase in hDAAO sensitivity to proteolytic degradation; no significant change in protein stability is apparent for the apoprotein form. The intensity of the 40-kDa band of the sample before adding trypsin (sample 0, top panel) is indicated as 100 %. Open square in bottom panels represent hDAAO-pLG72 complex, while closed circle represent the free hDAAO form. b d-Serine/l-serine concentration ratio determined by HPLC analysis on cell extracts from U87 and U87 pLG72-ECFP cells transiently transfected with EYFP-hDAAO. The measured values are higher in the cells stably expressing pLG72, thus confirming the negative effect exerted by pLG72 on hDAAO stability/activity. Errors bars represent the standard error; *p < 0.001, **p < 0.0001 (Sacchi et al. 2011)
None of the small ligands tested (FAD, benzoate, d-serine, or CPZ) modified the hDAAO-pLG72 complex formation, as judged by SPR and gel permeation analyses. Binding of FAD, benzoate, and CF3-d-alanine counteracts the destabilizing effect of pLG72, while CPZ binding further increases susceptibility to proteolysis (Caldinelli et al. 2010).
Cellular studies
We recently tried to resolve the apparent discrepancy in subcellular localization of hDAAO and pLG72. In fact, DAAO is a well-known peroxisomal enzyme (see above and Fig. 2e) while pLG72 has been reported to be a mitochondrial protein for COS7-transfected cells, glioma U251, and primary neurons (Benzel et al. 2008; Kvajo et al. 2008). However, in human cultured astrocytes it showed a perinuclear and tubular distribution (Sacchi et al. 2008). Since only 5 % of the latter cells displayed (cytosolic) overlapping signals for pLG72 and hDAAO, with a mean average of 8 % colocalization, we proposed that pLG72 interaction with hDAAO is probably driven by specific, unknown spatiotemporal stimuli (Sacchi et al. 2008). Recently, by means of immunohistochemistry and organelle immunoprecipitation, we have confirmed that pLG72 has a mitochondrial localization in U87 glioblastoma cells (Fig. 2f) (Sacchi et al. 2011). By using U87 cells stably expressing pLG72-ECFP and transiently transfected with EYFP-hDAAO, we demonstrated that colocalization is most apparent at 24 h after hDAAO transfection, when the fluorescence signal corresponding to the flavoenzyme is largely diffused in cytosol and the associated FRET signal shows a distribution pattern resembling the one typically associated with mitochondria (and pLG72). Indeed, at 24 h from transfection the amount of hDAAO in the membrane-bound fraction is higher in cells overexpressing pLG72 than in U87 control cells, suggesting sequestration of newly synthesized hDAAO on the surface of mitochondria owing to the interaction with pLG72. The pLG72-hDAAO colocalization, as well as the FRET signal, was progressively lost for longer periods, which was paralleled by the appearance of a punctuate distribution of hDAAO, indicating that it was correctly targeted to peroxisomes. Indeed, subcellular fractionation analysis demonstrated that cytosolic hDAAO is active and its specific activity resembles that of the peroxisomal enzyme (Sacchi et al. 2011). Binding of hDAAO to pLG72—probably located on the cytosolic side of mitochondrial membranes—and ensuing enzyme inactivation and faster degradation plays a protective role against DAAO-induced depletion of cytosolic d-serine and also helps to prevent cellular oxidative stress.
The relationship between pLG72, hDAAO, and the cellular concentration of d-serine was assessed using human glioblastoma U87 cells transfected with pEGFP-C3 expression vectors encoding for EGFP-hDAAO and/or EGFP-pLG72 proteins and HPLC analyses to measure d- and l-serine concentrations (Sacchi et al. 2008). The d-/l-serine ratio determined on control cells decreased in the cells transfected with the plasmid encoding for hDAAO but did not show a statistically significant change in those transfected with the plasmid for pLG72 and in the cells cotransfected with the two plasmids. Similarly, hDAAO activity increased in U87 cells transfected with the pEGFP-hDAAO plasmid (Sacchi et al. 2008). The time course of d-serine concentration following hDAAO transient expression is different in U87 controls versus U87 pLG72-ECFP cells, as is the d-serine concentration at 24 h (Fig. 6b): the binding of pLG72 to newly synthesized hDAAO switches off the flavooxidase activity and limits the decrease in d-serine concentration.
Concerning the effect of small-size ligands, both benzoate and CPZ yield a statistically significant increase in d/(d + l)-serine concentration ratio in U87 cells stably transfected with hDAAO as compared to untreated controls (Caldinelli et al. 2010). Furthermore, CPZ treatment yields a statistically significant decrease in hDAAO cellular levels 48 h later. When the same experiment was carried out using U87 glioblastoma cells stably expressing EGFP-hDAAO and transiently expressing pLG72, CPZ treatment significantly decreased the amount of active hDAAO while benzoate slightly increased the cellular hDAAO in cotransfected cells. Both compounds increased the cellular d-serine concentration (because of hDAAO inhibition) but, intriguingly, using a different strategy for hDAAO inhibition (substrate vs. cofactor competition) might affect the in vivo concentration of d-serine to a different extent because protein stability is modulated dissimilarly (Caldinelli et al. 2010).
Recently, 24 putative DAAO-interacting proteins were identified by immunoprecipitation from rat cerebellar lysates using an anti-rDAAO antibody (Popiolek et al. 2011). Interestingly, the most robust interaction occurred with bassoon (BSN) and piccolo (PCLO), two components of the presynaptic active zone (Garner et al. 2000), a specialized subcellular domain where synaptic vesicles are docked and primed before they fuse to the membrane and release their neurotransmitter content (Owald and Sigrist 2009). Since the d-serine/d-alanine transporter Asc-1 is also enriched at the presynaptic terminus, the presence of extraperoxisomal DAAO suggests that some of the readsorbed d-serine/d-alanine may be metabolized, further fine-tuning their concentration at the synapse and consequently affecting NMDAR activation (Popiolek et al. 2011). Furthermore, BSN overexpression (HEK293 cell line stably expressing rDAAO and transiently transfected for BSN) decreased DAAO enzymatic activity by 30 %.
Evolution of human DAAO properties
DAAO represents a clear example of promiscuity, i.e. the enzyme shows the ability to act on different substrates and, for this reason, to be involved in different biochemical pathways. Owing to this property DAAO can play different physiological roles (Pollegioni et al. 2007a, see “Introduction”). The high versatility of this enzyme implies that specific structural features (confined to restricted regions since they do not modify the overall folding of the protein) related to the different specific roles have been acquired/altered during evolution. In particular, the role of DAAO in human brain requires a different substrate specificity and slower turnover than yeast DAAO. These features were gained because of the presence of the large lid loop covering the active-site entrance in mammalian DAAOs. In this step, the loop of hDAAO (which function is partially played by the side chain of Tyr238 in yeast DAAO) (Pollegioni et al. 2002) limits the rate of product release during catalysis and slows d-serine oxidation (Molla et al. 2006a). In addition, a second mechanism to control the enzyme activity evolved to prevent excessive degradation of local d-serine. In yeast DAAO, FAD cofactor is bound with a high affinity, it is necessary for protein dimerization, and it plays a fundamental role in stabilizing the enzyme against chemical and thermal denaturation. On the other hand, FAD interaction with the hDAAO apoprotein moiety is weak and its binding does not alter the dimeric quaternary structure of the protein (Pollegioni et al. 2007a; Caldinelli et al. 2009). Accordingly, hDAAO is present in solution as a mixture of the (active) holoenzyme and the corresponding (inactive) apoprotein. Indeed, in the presence of a substrate (analog) the interaction between FAD and the hDAAO apoprotein moiety is 20-fold tighter and induces a “more packed” protein structure due to specific, induced conformational changes (Fig. 4).
From a physiological point of view, the weak interaction with FAD, together with the low affinity for the substrates, may have represented an evolutionary strategy to produce an enzyme that might be largely present in an active form only in the presence of high concentrations of d-serine. In fact, considering the physiological concentration of both FAD and d-serine in brain tissues in vivo, hDAAO should normally be present in the inactive apoprotein form. It is tempting to speculate that the low affinity of hDAAO for the cofactor has been selected to control d-serine concentration in brain so as to avoid excessive degradation. On the other hand, elevated levels of hDAAO activity (as in pathological conditions) presumably enhance d-serine catabolism, consequently affecting the release of the signaling molecule and its availability at synapse and thus potentially impairing NMDAR function (Fig. 1b). In this respect different lines of evidence indicate that hDAAO is involved in the NMDAR hypofunction observed in schizophrenia. Furthermore, loss of hDAAO activity and the consequent increase in d-serine levels has been shown to trigger motoneuron degeneration and to be relevant in ALS (Sasabe et al. 2007).
Concluding remarks
Maintaining physiological concentrations of d-serine, and thus the fine balance between synthesis and degradation, is crucial for the brain to develop and function correctly. For this reason, the structural/functional properties of hDAAO and the factors (effector molecules and regulatory interacting proteins) affecting its functionality should be investigated in greater depth. It is of uttermost importance to clarify the complex processes that are in play in d-serine metabolism, which we aimed to do in this review. This will help in designing new and more effective drugs for the treatment of the different pathological conditions in which NMDAR dysfunction is implicated.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- BSN:
-
Bassoon protein
- CP:
-
Choroid plexus
- CPZ:
-
Chlorpromazine
- CBIO:
-
6-Chlorobenzo[d]isoxazol-3-ol
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DAAO:
-
d-Amino acid oxidase
- d-KYN:
-
d-Kynurenine
- DOPA:
-
3,4-Dihydroxy-phenylalanine
- ECFP:
-
Enhanced cyan fluorescent protein
- EYFP:
-
Enhanced yellow fluorescent protein
- FRET:
-
Fluorescence resonance energy transfer
- hDAAO:
-
Human d-amino acid oxidase
- KYNA:
-
Kynurenic acid
- NMDA:
-
N-Methyl-d-aspartate
- NMDAR:
-
N-Methyl-d-aspartate type glutamate receptor
- pkDAAO:
-
Porcine kidney d-amino acid oxidase
- PTS1:
-
Peroxisomal targeting signal 1
- RMSD:
-
Root-mean-square deviation
- SAP:
-
Structurally ambivalent peptide
- SPR:
-
Surface plasmon resonance
- SR:
-
Serine racemase
- T m :
-
Melting temperature
References
Adage T, Trillat AC, Quattropani A, Perrin D, Cavarec L, Shaw J, Guerassimenko O, Giachetti C, Greco B, Chumakov I, Halazy J, Roach A, Zaratin P (2008) In vitro and in vivo pharmacological profile of AS057278, a selective D-amino acid oxidase inhibitor with potential antipsychotic properties. Eur Neuropsychopharmacol 18:200–214
Almond SL, Fradley RL, Armstrong EJ, Heavens RB, Rutter AR, Newman RJ, Chiu CS, Konno R, Huston PH, Brandon NJ (2006) Behavioral and biochemical characterization of a mutant mouse strain lacking D-amino acid oxidase activity and its implications for schizophrenia. Mol Cell Neurosci 32:324–334
Arnold G, Liscum L, Holtzman E (1979) Ultrastructural localization of D-amino acid oxidase in microperoxisomes of the rat nervous system. J Histochem Cytochem 27:735–745
Banks WA, Kastin AJ (1991) Leucine modulates peptide transport system-1 across the blood-brain barrier at the stereospecific site within the central nervous system. J Pharm Pharmacol 43:252–254
Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, Wolosker H, Agam G (2007) A CSF and postmortem brain study of d-serine metabolic parameters in schizophrenia. Schizophr Res 90:41–51
Benzel I, Kew JN, Viknaraja R, Kelly F, de Belleroche J, Hirsch S, Sanderson TH, Maycox PR (2008) Investigation of G72 (DAOA) expression in the human brain. BMC Psychiatry 11:8–94
Billard JM (2008) d-serine signalling as a prominent determinant of neuronal-glial dialogue in the healthy and diseased brain. J Cell Mol Med 12:1872–1884
Burnet PW, Eastwood SL, Bristow GC, Godlewska BR, Sikka P, Walker M, Harrison PJ (2008) D-amino acid oxidase activity and expression are increased in schizophrenia. Mol Psychiatr 13:658–660
Caldinelli L, Molla G, Sacchi S, Pilone MS, Pollegioni L (2009) Relevance of weak flavin binding in human D-amino acid oxidase. Protein Sci 18:801–810
Caldinelli L, Molla G, Bracci L, Lelli B, Pileri S, Cappelletti P, Sacchi S, Pollegioni L (2010) Effect of ligand binding on human D-amino acid oxidase: implications for the development of new drugs for schizophrenia treatment. Protein Sci 19:1500–1512
Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cui J, Tu S, Sevarino KA, Nakanishi N, Tong G, Lipton SA, Zhang D (2002) Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415:793–798
Chen FC, Li WH (2001) Genomic divergences between humans and other hominoids and the effective population size on the common ancestor of humans and chimpanzee. Am J Hum Genet 68:444–456
Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, Abderrahim H, Bougueleret L, Barry C, Tanaka H, La Rosa P, Puech A, Tahri N, Cohen-Akenine A, Delabrosse S, Lissarrague S, Picard FP, Maurice K, Essioux L, Millasseau P, Grel P, Debailleul V, Simon AM, Caterina D, Dufaure I, Malekzadeh K, Belova M, Luan JJ, Bouillot M, Sambucy JL, Primas G, Saumier M, Boubkiri N, Martin-Saumier S, Nasroune M, Peixoto H, Delaye A, Pinchot V, Bastucci M, Guillou S, Chevillon M, Sainz-Fuertes R, Meguenni S, Aurich-Costa J, Cherif D, Gimalac A, Van Duijn C, Gauvreau D, Ouellette G, Fortier I, Raelson J, Sherbatich T, Riazanskaia N, Rogaev E, Raeymaekers P, Aerssens J, Konings F, Luyten W, Macciardi F, Sham PC, Straub RE, Weinberger DR, Cohen N, Cohen D (2002) Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci USA 99:13675–13680
Coyle JT (2006) Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol 26:365–384
Conti P, Tamborini L, Pinto A, Blondel A, Minoprio P, Mozzarelli A, De Micheli C (2011) Drug discovery targeting amino acid racemase. Chem Rev 111:6919–6946
Curti B, Ronchi S, Simonetta PM (1992) In: Muller F (ed) Chemistry and Biochemistry of Flavoenzyme. CRC, Boca Raton, pp 69–94
Ding X, Ma N, Nagahama M, Yamada K, Semba R (2011) Localization of d-serine and serine racemase in neurons and neuroglias in mouse brain. Neurol Sci 32:263–267
Duplantier AJ, Becker SL, Bohanon MJ, Borzilleri KA, Chrunyk BA, Downs JT, Hu LY, El-Kattan A, James LC, Liu S, Lu J, Maklad N, Mansour MN, Mente S, Piotrowski MA, Sakya SM, Sheehan S, Steyn SJ, Strick CA, Williams VA, Zhang L (2009) Discovery, SAR, and pharmacokinetics of a novel 3-hydroxyquinolin-2(1H)-one series of potent D-amino acid oxidase (DAAO) inhibitors. J Med Chem 52:3576–3585
Fernandez HH, Odin P (2011) Levodopa-carbidopa intestinal gel for treatment of advanced Parkinson’s disease. Curr Med Res Opin 27:907–919
Ferraris D, Duvall B, Ko YS, Thomas AG, Rojas C, Majer P, Hashimoto K, Tsukamoto T (2008) Synthesis and biological evaluation of D-amino acid oxidase inhibitors. J Med Chem 51:3357–3359
Foltyn VN, Bendikov I, De Miranda J, Panizzutti R, Dumin E, Shleper M, Li P, Toney MD, Kartvelishvily E, Wolosker H (2005) Serine racemase modulates intracellular d-serine levels through an alpha, beta-elimination activity. J Biol Chem 280:1754–1763
Fonda ML, Anderson BM (1968) D-Amino acid oxidase-II Studies of substrate-competitive inhibitors. J Biol Chem 243:1931–1935
Frattini LF, Piubelli L, Sacchi S, Molla G, Pollegioni L (2011) Is rat an appropriate animal model to study the involvement of d-serine catabolism in schizophrenia? Insights from characterization of D-amino acid oxidase. FEBS J 278:4362–4373
Fuchs SA, Berger R, de Koning TJ (2011) d-serine: the right or wrong isoform? Brain Res 1401:104–117
Fukushima T, Sone Y, Mitsuhashi S, Tomiya M, Toyo’oka T (2009) Alteration of kynurenic acid concentration in rat plasma following optically pure kynurenine administration: a comparative study between enantiomers. Chirality 21:468–472
Ganong AH, Lanthorn TH, Cotman CW (1983) Kynurenic acid inhibits synaptic and acidic amino acid-induced responses in the rat hippocampus and spinal cord. Brain Res 273:170–174
Gao J, Kelly JW (2008) Toward quantification of protein backbone–backbone hydrogen bonding energies: an energetic analysis of an amide-to-ester mutation in an alpha-helix within a protein. Protein Sci 17:1096–1101
Garner CC, Kindler S, Gundelfinger ED (2000) Molecular determinants of presynaptic active zones. Curr Opin Neurobiol 10:321–327
Gong XQ, Zabek RL, Bai D (2007) d-Serine inhibits AMPA receptor-mediated current in rat hippocampal neurons. Can J Physiol Pharmacol 85:546–555
Hamase K, Homma H, Takigawa Y, Fukushima T, Santa T, Imai K (1997) Regional distribution and postnatal changes of D-amino acids in rat brain. Biochim Biophys Acta 1334:214–222
Hamasu K, Shigemi K, Tsuneyoshi Y, Yamane H, Sato H, Denbow DM, Furuse M (2008) Intracerebroventricular injection of l-proline and d-proline induces sedative and hypnotic effects by different mechanisms under an acute stressful condition in chicks. Amino Acids 38:57–64
Harris CM, Molla G, Pilone MS, Pollegioni L (1999) Studies on the reaction mechanism of Rhodotorula gracilis D-amino-acid oxidase. Role of the highly conserved Tyr-223 on substrate binding and catalysis. J Biol Chem 274:36233–36240
Harris CM, Pollegioni L, Ghisla S (2001) pH and kinetic isotope effects in D-amino acid oxidase catalysis. Eur J Biochem 268:5504–5520
Hansen KB, Naur P, Kurtkaya NL, Kristensen AS, Gajhede M, Kastrup JS, Gajhede M, Kastrup JS, Traynelis SF (2009) Modulation of the dimer interface at ionotropic glutamate-like receptor δ2 by d-serine and extracellular calcium. J Neurosci 29:907–917
Hashimoto A, Kumashiro S, Nishikawa T, Oka T, Takahashi K, Mito T, Takashima S, Doi N, Mizutani Y, Yamazaki T, Kaneko T, Ootomo E (1993) Embryonic development and postnatal changes in free D-aspartate and d-serine in the human prefrontal cortex. J Neurochem 61:348–351
Hashimoto A, Oka T, Nishikawa T (1995) Anatomical distribution and postnatal changes in endogenous free D-aspartate and d-serine in rat brain and periphery. Eur J Neurosci 7:1657–1663
Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, Shinoda N, Nakazato M, Kumakiri C, Okada S, Hasegawa H, Imai K, Iyo M (2003) Decreased serum levels of d-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry 60:572–576
Hashimoto A, Yoshikawa M, Niwa A, Konno R (2005) Mice lacking D-amino acid oxidase activity display marked attenuation of stereotypy and ataxia induced by MK-801. Brain Res 1033:210–215
Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, Catinari S, Ermilov M (2005) d-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry 57:577–585
Holtze M, Asp L, Schwieler L, Engberg G, Karlsson H (2008) Induction of the kynurenine pathway by neurotropic influenza A virus infection. J Neurosci Res 86:3674–3683
Horiike K, Tojo H, Arai R, Nozaki M, Maeda T (1994) D-amino-acid oxidase is confined to the lower brain stem and cerebellum in rat brain: regional differentiation of astrocytes. Brain Res 652:297–303
Inoue T, Hamase K, Morikawa A, Zaitsu K (2000) Determination of minute amounts of d-leucine in various brain regions of rat and mouse using column-switching high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 744:213–219
Ishii K, Ogaya T, Song Z, Iizuka H, Fukushima T (2010) Changes in the plasma concentrations of D-kynurenine and kynurenic acid in rats after intraperitoneal administration of tryptophan enantiomers. Chirality 22:901–906
Iwana S, Kawazoe T, Park HK, Tsuchiya K, Ono K, Yorita K, Sakai T, Kusumi T, Fukui K (2008) Chlorpromazine oligomer is a potentially active substance that inhibits human D-amino acid oxidase, product of a susceptibility gene for schizophrenia. J Enzyme Inhib Med Chem 23:901–911
Kapoor R, Lim KS, Cheng A, Garrick T, Kapoor V (2006) Preliminary evidence for a link between schizophrenia and NMDA-glycine site receptor ligand metabolic enzymes, d-amino acid oxidase (DAAO) and kynurenine aminotransferase-1 (KAT-1). Brain Res 1106:205–210
Kartvelishvily E, Shleper M, Balan L, Dumin E, Wolosker H (2006) Neuron-derived d-serine release provides a novel means to activate N-methyl-D-aspartate receptors. J Biol Chem 281:14151–14162
Kawazoe T, Tsuge H, Pilone MS, Fukui K (2006) Crystal structure of human D-amino acid oxidase: context-dependent variability of the backbone conformation of the VAAGL hydrophobic stretch located at the si-face of the flavin ring. Protein Sci 15:2708–2717
Kawazoe T, Tsuge H, Imagawa T, Aki K, Kuramitsu S, Fukui K (2007) Structural basis of D-DOPA oxidation by D-amino acid oxidase: alternative pathway for dopamine biosynthesis. Biochem Biophys Res Commun 355:385–391
Kvajo M, Dhilla A, Swor DE, Karayiorgou M, Gogos JA (2008) Evidence implicating the candidate schizophrenia/bipolar disorder susceptibility gene G72 in mitochondrial function. Mol Psychiatry 13:685–696
Kessler M, Terramani T, Lynch G, Baudry M (1989) A glycine site associated with N-methyl-d-aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem 52:1319–1328
Konno R, Yasumura Y (1983) Mouse mutant deficient in D-amino acid oxidase activity. Genetics 103:277–285
Konno R, Sasaki M, Asakura S, Fukui K, Enami J, Niwa A (1997) D-amino-acid oxidase is not present in the mouse liver. Biochim Biophys Acta 1335:173–181
Korostishevsky M, Kaganovich M, Cholostoy S, Ashkenazy M, Ratner Y, Dahary D, Bernstein J, Bening-Abu-Shach U, Ben-Asher E, Lancet D, Ritsner M, Navon R (2004) Is the G72/G30 locus associated with schizophrenia? Single nucleotide polymorphism, haplotypes, and gene expression analysis. Biol Psychiatry 56:169–176
Krebs HA (1935) Metabolism of amino-acids: deamination of amino acids. Biochem J 29:1620–1644
Labrie V, Roder JC (2010) The involvement of the NMDA receptor d-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci Biobehav Rev 34:351–372
Lange JH, Venhorst J, van Dongen MJ, Frankena J, Bassissi F, de Bruin NM, den Besten C, de Beer SB, Oostenbrink C, Markova N, Kruse CG (2011) Biophysical and physicochemical methods differentiate highly ligand-efficient human D-amino acid oxidase inhibitors. Eur J Med Chem 46:4808–4819
Lu M, Fan Y, Tang M, Qian X, Ding J, Hu G (2011) Potentiation of d-serine involves degeneration of dopaminergic neurons in MPTP/p mouse model of Parkinson’s disease. CNS Neurosci Ther 17:796–798
Lugo-Huitrón R, Blanco-Ayala T, Ugalde-Muñiz P, Carrillo-Mora P, Pedraza-Chaverrí J, Silva-Adaya D, Maldonado PD, Torres I, Pinzón E, Ortiz-Islas E, López T, García E, Pineda B, Torres-Ramos M, Santamaría A, La Cruz VP (2011) On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol Teratol 33:538–547
Madeira C, Freitas ME, Vargas-Lopes C, Wolosker H, Panizzutti R (2008) Increased brain D-amino acid oxidase (DAAO) activity in schizophrenia. Schizophr Res 101:76–83
Maekawa M, Watanabe M, Yamaguchi S, Konno R, Hori Y (2005) Spatial learning and long-term potentiation of mutant mice lacking D-amino-acid oxidase. Neurosci Res 53:34–38
Martineau M, Baux G, Mothet JP (2006) d-serine signalling in the brain: friend and foe. Trends Neurosci 29:481–491
Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B (1996) Crystal structure of D-amino acid oxidase: a case of active site mirror-image convergent evolution with flavocytochrome b2. Proc Natl Acad Sci USA 93:7496–7501
Mitchell J, Paul P, Chen HJ, Morris A, Payling M, Falchi M, Habgood J, Panoutsou S, Winkler S, Tisato V, Hajitou A, Smith B, Vance C, Shaw C, Mazarakis ND, de Belleroche J (2010) Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc Natl Acad Sci USA 107:7556–7561
Molla G, Porrini D, Job V, Motteran L, Vegezzi C, Campaner S, Pilone MS, Pollegioni L (2000) Role of arginine 285 in the active site of Rhodotorula gracilis D-amino acid oxidase. A site-directed mutagenesis study. J Biol Chem 275:24715–24721
Molla G, Sacchi S, Bernasconi M, Pilone MS, Fukui K, Pollegioni L (2006a) Characterization of human D-amino acid oxidase. FEBS Lett 580:2358–2364
Molla G, Bernasconi M, Sacchi S, Pilone MS, Pollegioni L (2006b) Expression in Escherichia coli and in vitro refolding of the human protein pLG72. Protein Expr Purif 46:150–155
Moreno S, Nardacci R, Cimini A, Cerù MP (1999) Immunocytochemical localization of D-amino acid oxidase in rat brain. J Neurocytol 28:169–185
Morikawa A, Hamase K, Zaitsu K (2003) Determination of d-alanine in the rat central nervous system and periphery using column-switching high-performance liquid chromatography. Anal Biochem 312:66–72
Mothet JP, Parent AT, Wolosker H, Brady RO Jr, Linden DJ, Ferris CD, Rogawsky MA, Snyder SH (2000) d-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci USA 97:4926–4931
Mustafa AK, Ahmad AS, Zeynalov E, Gazi SK, Sikka G, Ehmsen JT, Barrow RK, Coyle JT, Snyder SH, Doré S (2010) Serine racemase deletion protects against cerebral ischemia and excitotoxicity. J Neurosci 30:1413–1416
Nagata Y, Yamamoto K, Shimojo T, Konno R, Yasumura Y, Akino T (1992) The presence of free d-alanine, d-proline and d-serine in mice. Biochim Biophys Acta 1115:208–211
Nagata Y, Horiike K, Maeda T (1994) Distribution of free d-serine in vertebrate brains. Brain Res 634:291–295
Neims AH, Zieverink WD, Smilack JD (1966) Distribution of D-amino acid oxidase in bovine and human nervous tissues. J Neurochem 13:163–168
Ono K, Shishido Y, Park HK, Kawazoe T, Iwana S, Chung SP, Abou El-Magd RM, Yorita K, Okano M, Watanabe T, Sano N, Bando Y, Arima K, Sakai T, Fukui K (2009) Potential pathophysiological role of D-amino acid oxidase in schizophrenia: immunohistochemical and in situ hybridization study of the expression in human and rat brain. J Neural Transm 116:1335–1347
Otte DM, Bilkei-Gorzo A, Filiou MD, Turck CW, Ylmaz O, Holst MI, Schilling K, Abou-Jamra R, Schumacher J, Benzel I, Kunz WS, Beck H, Zimmer A (2009) Behavioral changes in G72/G30 transgenic mice. Eur Neuropharmacol 19:339–348
Owald D, Sigrist SJ (2009) Assembling the presynaptic active zone. Curr Opin Neurobiol 19:311–318
Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH (2006) Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell 125:775–784
Pérez-de la Cruz V, Amori L, Sathyasaikumar KV, Wang XD, Notarangelo FM, Wu HQ, Schwarcz R (2012) Enzymatic transamination of D-kynurenine generates kynurenic acid in rat and human brain. J Neurochem (ahead of print)
Perkins MN, Stone TW (1982) An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res 247:184–187
Pilone MS, Pollegioni L, Casalin P, Curti B, Ronchi S (1989) Properties of D-amino-acid oxidase from Rhodotorula gracilis. Eur J Biochem 180:199–204
Pilone MS (2000) D-Amino acid oxidase: new findings. Cell Mol Life Sci 57:1732–1747
Pollegioni L, Butò S, Tischer W, Ghisla S, Pilone MS (1993) Characterization of D-amino acid oxidase from Trigonopsis variabilis. Biochem Mol Biol Int 31:709–717
Pollegioni L, Diederichs K, Molla G, Umhau S, Welte W, Ghisla S, Pilone MS (2002) Yeast D-amino acid oxidase: structural basis of its catalytic properties. J Mol Biol 324:535–546
Pollegioni L, Piubelli L, Sacchi S, Pilone MS, Molla G (2007a) Physiological functions of D-amino acid oxidases: from yeast to humans. Cell Mol Life Sci 64:1373–1394
Pollegioni L, Sacchi S, Caldinelli L, Boselli A, Pilone MS, Piubelli L, Molla G (2007b) Engineering the properties of D-amino acid oxidases by a rational and a directed evolution approach. Curr Protein Pept Sci 8:600–618
Pollegioni L, Sacchi S (2010) Metabolism of the neuromodulator d-serine. Cell Mol Life Sci 67:2387–2404
Popiolek M, Ross JF, Charych E, Chanda P, Gundelfinger ED, Moss SJ, Brandon NJ, Pausch MH (2011) D-amino acid oxidase activity is inhibited by an interaction with bassoon protein at the presynaptic active zone. J Biol Chem 286:28867–28875
Porter DJ, Voet JG, Bright HJ (1977) Mechanistic features of the D-amino acid oxidase reaction studied by double stopped flow spectrophotometry. J Biol Chem 252:4464–4473
Puyal J, Martineau M, Mothet JP, Nicolas MT, Raymond J (2006) Changes in d-serine levels and localization during postnatal development of the rat vestibular nuclei. J Comp Neurol 497:610–621
Raibekas AA, Fukui K, Massey V (2000) Design and properties of human D-amino acid oxidase with covalently attached flavin. Proc Natl Acad Sci USA 97:3089–3093
Redzic ZB, Segal MB (2004) The structure of the choroid plexus and the physiology of the choroid plexus epithelium. Adv Drug Deliv Rev 56:1695–1716
Romano D, Molla G, Pollegioni L, Marinelli F (2009) Optimization of human D-amino acid oxidase expression in Escherichia coli. Protein Expr Purif 68:72–78
Rosenberg D, Kartvelishvily E, Shleper M, Klinker CM, Bowser MT, Wolosker H (2010) Neuronal release of d-serine: a physiological pathway controlling extracellular d-serine concentration. FASEB J 24:2951–2961
Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT (2006) Neurobiology of schizophrenia. Neuron 52:139–153
Rossi DJ, Brady JD, Mohr C (2007) Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci 10:1377–1386
Sacchi S, Lorenzi S, Molla G, Pilone MS, Rossetti C, Pollegioni L (2002) Engineering the substrate specificity of D-amino-acid oxidase. J Biol Chem 277:27510–27516
Sacchi S, Bernasconi M, Martineau M, Mothet JP, Ruzzene M, Pilone MS, Pollegioni L, Molla G (2008) pLG72 modulates intracellular d-serine levels through its interaction with D-amino acid oxidase: effect on schizophrenia susceptibility. J Biol Chem 283:22244–22256
Sacchi S, Cappelletti P, Giovannardi S, Pollegioni L (2011) Evidence for the interaction of D-amino acid oxidase with pLG72 in a glial cell line. Mol Cell Neurosci 48:20–28
Sacchi S, Rosini E, Pollegioni L, Molla G (2012) D-amino acid oxidase as a novel class of drug for schizophrenia therapy. Curr Pharm Design (ahead of print)
Saitoh Y, Katane M, Kawata T, Maeda K, Sekine M, Furuchi T, Kobuna H, Sakamoto T, Inoue T, Arai H, Nakagawa Y, Homma H (2012) Spatiotemporal localization of D-amino acid oxidase and D-aspartate oxidases during development in Caenorhabditis elegans. Mol Cell Biol 32:1967–1983
Sasabe J, Chiba T, Yamada M, Okamoto K, Nishimoto I, Matsuoka M, Aiso S (2007) d-serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J 26:4149–4159
Sasabe J, Miyoshi Y, Suzuki M, Mita M, Konno R, Matsuoka M, Hamase K, Aiso S (2012) D-amino acid oxidase controls motoneuron degeneration through d-serine. Proc Natl Acad Sci USA 109:627–632
Sasaki M, Konno R, Nishio M, Niwa A, Yasumura Y, Enami J (1992) A single-base-pair substitution abolishes D-amino-acid oxidase activity in the mouse. Biochim Biophys Acta 1139:315–318
Schell MJ, Brady RO Jr, Molliver ME, Snyder SH (1997) d-serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci 17:1604–1615
Sinnokrot MO, Valeev EF, Sherrill CD (2002) Estimates of the ab initio limit for pi–pi interactions: the benzene dimer. J Am Chem Soc 124:10887–10893
Smith SM, Uslaner JM, Yao L, Mullins CM, Surles NO, Huszar SL, McNaughton CH, Pascarella DM, Kandebo M, Hinchliffe RM, Sparey T, Brandon NJ, Jones B, Venkatraman S, Young MB, Sachs N, Jacobson MA, Hutson PH (2009) The behavioral and neurochemical effects of a novel D-amino acid oxidase inhibitor compound 8 [4H-thieno [3,2-b]pyrrole-5-carboxylic acid] and d-serine. J Pharmacol Exp Ther 328:921–930
Snyder SH, Kim PM (2000) D-amino acids as putative neurotransmitters: focus on d-serine. Neurochem Res 25:553–560
Song Y, Feng Y, Lu X, Zhao S, Liu CW, Liu YM (2008) D-Amino acids in rat brain measured by liquid chromatography/tandem mass spectrometry. Neurosci Lett 445:53–57
Sparey T, Abeywickrema P, Almond S, Brandon N, Byrne N, Campbell A, Hutson PH, Jacobson M, Jones B, Munshi S, Pascarella D, Pike A, Prasad GS, Sachs N, Sakatis M, Sardana V, Venkatraman S, Young MB (2008) The discovery of fused pyrrole carboxylic acids as novel, potent D-amino acid oxidase (DAO) inhibitors. Bioorg Med Chem Lett 18:3386–3391
Stevens ER, Esguerra M, Kim PM, Newman EA, Snyder SH, Zahs KR, Miller RF (2003) d-serine and serine racemase are present in the vertebrate retina and contribute to the physiological activation of NMDA receptors. Proc Natl Acad Sci USA 100:6789–6794
Strohmaier J, Georgi A, Schirmbeck F, Schmael C, Jamra RA, Schumacher J, Becker T, Hofels S, Klopp N, Illig T, Propping P, Cichon S, Nothen MM, Rietschell M, Shulze TG (2007) No association between the serine racemase gene (SRR) and schizophrenia in a German case-control sample. Psychiatr Genet 17:125
Takarada T, Takahata Y, Iemata M, Hinoi E, Uno K, Hirai T, Yamamoto T, Yoneda Y (2009) Interference with cellular differentiation by d-serine through antagonism at N-methyl-D-aspartate receptors composed of NR1and NR3A subunits in chondrocytes. J Cell Physiol 22:756–764
Tsai G, Yang P, Chung LC, Lange N, Coyle JT (1998) d-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 44:1081–1089
Umhau S, Pollegioni L, Molla G, Diederichs K, Welte W, Pilone MS, Ghisla S (2000) The x-ray structure of D-amino acid oxidase at very high resolution identifies the chemical mechanism of flavin-dependent substrate dehydrogenation. Proc Natl Acad Sci USA 97:12463–12468
Verrall L, Walker M, Rawlings N, Benzel I, Kew JN, Harrison PJ, Burnet PW (2007) D-Amino acid oxidase and serine racemase in human brain: normal distribution and altered expression in schizophrenia. Eur J Neurosci 26:1657–1669
Verrall L, Burnet PW, Betts JF, Harrison PJ (2010) The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. Mol Psychiatry 15:122–137
Weimar WR, Neims AH (1977) The development of D-amino acid oxidase in rat cerebellum. J Neurochem 29:649–656
Williams SM, Diaz CM, Macnab LT, Sullivan RK, Pow DV (2006) Immunocytochemical analysis of d-serine distribution in the mammalian brain reveals novel anatomical compartmentalizations in glia and neurons. Glia 53:401–411
Wolosker H, Sheth KN, Takahashi M, Mothet JP, Brady RO Jr, Ferris CD, Snyder SH (1999a) Purification of serine racemase: biosynthesis of the neuromodulator d-serine. Proc Natl Acad Sci USA 96:721–725
Wolosker H, Blackshaw S, Snyder SH (1999b) Serine racemase: a glial enzyme synthesizing d-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc Natl Acad Sci USA 96:13409–13414
Wolosker H, Dumin E, Balan L, Foltyn VN (2008) D-amino acids in the brain: d-serine in neurotransmission and neurodegeneration. FEBS J 275:3514–3526
Yamada K, Ohnishi T, Hashimoto K, Ohba A, Iwayama-Shigeno Y, Toyoshima M, Okuno A, Takao H, Toyota T, Minabe Y, Nakamura K, Shimizu E, Itokawa M, Mori N, Iyo M, Yoshikawa T (2005) Identification of multiple serine racemase (SRR) mRNA isoforms and genetic analyses of SRR and DAO in schizophrenia and d-serine levels. Biol Psychiatry 57:1493–1503
Acknowledgments
This work was supported by grants from Fondo di Ateneo per la Ricerca to L. Pollegioni, S. Sacchi and G. Molla. We are grateful for the support from Consorzio Interuniversitario per le Biotecnologie and Centro Grandi Attrezzature, Università degli studi dell’Insubria.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sacchi, S., Caldinelli, L., Cappelletti, P. et al. Structure–function relationships in human d-amino acid oxidase. Amino Acids 43, 1833–1850 (2012). https://doi.org/10.1007/s00726-012-1345-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-012-1345-4