
Abstract
Beta-amyloid (Aβ) is considered to be responsible for the pathogenesis of Alzheimer’s disease (AD), and accumulation and aggregation of Aβ peptide in the brains of AD patients result in activation of glial cells which, in turn, initiates neuroinflammatory responses that involve reactive oxygen intermediates and release of inflammatory cytokines. In the present study, the protective effects of S-propargyl-cysteine (SPRC), also named as ZYZ-802, a sulphur-containing amino acid, on cognitive impairment and neuronal ultrastructure damage induced by Aβ were examined in rats, and the possible mechanisms were explored. These data showed that SPRC administration at the doses of 40, 80 mg/kg by intraperitoneal injection (i.p.) may inhibit cognitive impairment and neuronal ultrastructure damage induced by intracerebroventricular (i.c.v.) injection of 10 μg of Aβ25–35 in rats. Subsequently, SPRC inhibited the expressions of tumor necrosis factor (TNF)-α, cyclooxygenase-2 (COX-2) mRNA, and protein in rat hippocampus. SPRC afforded a beneficial action on inhibitions of extracellular signal-regulated kinase (ERK1/2), as well as inhibitions of IκB-α degradation and activation of transcription factors of the nuclear factor κB (NF-κB) produced by Aβ. These findings suggested that SPRC might be a potential agent for treatment of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a representative neurodegenerative disorder characterized by gradual degeneration and loss of neurons in the brain, which correlates with the accumulation of neurofibrillary tangles and senile plaques, the two neuropathological hallmarks of the disease (Selkoe 1999). Beta-amyloid (Aβ), the major component of senile plaques, is derived from the proteolytic processing of the amyloid-β protein precursor (AβPP) and is considered to have a causal role in the development and progress of AD (Hardy and Higgins 1992), although Aβ is normally produced in the brain (Cirrito et al. 2005, 2008) and low levels are detectable in the cerebrospinal fluid of healthy subjects (Dubois et al. 2007). The mechanism underlying Aβ-induced neurotoxicity is complex, involving several pathways as signaling events (Verdile et al. 2004). Now it is found that dysregulation of the normal inflammatory processes has been implicated in the pathophysiology of AD. Elevated expression of immune response-related molecules and their corresponding receptors has been observed throughout the AD brain, and such brain-derived immune factors disrupt normal neurophysiology and contribute to cognitive dysfunction (Akiyama et al. 2000; Eikelenboom and van Gool 2004; Eikelenboom et al. 2008; Mrak 2009). Accordingly, epidemiological studies suggest that long-term treatment with non-steroidal anti-inflammatory drugs reduces the risk of AD (McGeer et al. 2006), suppresses inflammation in Tg2576 transgenic mouse with AD (Lim et al. 2000), and reduces Aβ, hyperphosphorylated Tau and memory deficits in Alzheimer mice (McKee et al. 2008).
However, a harmful side effect of long-term treatment with non-steroidal anti-inflammatory drugs to prevent AD is the gastrointestinal and occasional liver and kidney toxicity associated with the inhibition of cyclooxygenase (COX)-1 (Graupera et al. 2003; Shi and Klotz 2008), and the selective COX-2 inhibitors were ineffective (McGeer and McGeer 2007). Thus, these reasons have stimulated the research for alternative anti-inflammatory drugs that are safe in long-term treatments.
S-propargyl-cysteine (SPRC), we also named it as ZYZ-802, a structural analog of S-allylcysteine (SAC), a sulfur-containing amino acid, has cardioprotective roles in both in vitro and in vivo models (Wang et al. 2009, 2010), and it is interesting that SPRC has better cardioprotective effects than SAC in vitro and in vivo (Wang et al. 2010). Likewise, it is reported that garlic compounds containing SAC may attenuate Aβ-induced apoptosis (Peng et al. 2002), destabilize Alzheimer’s Aβ fibrils in vitro (Gupta and Rao 2007), reduce cerebral amyloid, cerebral inflammation, and tau phosphorylation in Alzheimer’s transgenic mouse model harboring Swedish double mutation (Chauhan 2006). Therefore, the current study was undertaken to investigate the effects of SPRC on cognitive deficits induced by Aβ in rats. Furthermore, we addressed the mechanisms that contribute to the neuroprotective effects of SPRC and whether it occurs via anti-inflammatory pathway.
Materials and methods
Chemicals
Cysteine (99.5%) was purchased from Hengbai Chemical Co. (Shanghai, China). Propyl bromide and propargyl bromide were purchased from Yancheng Kelida Chemical Co. (Zhejiang, China). PrimeScript ™1st Strand cDNA Synthesis Kit were from TaKaRa, China. SPRC was synthesized from the reaction of l-cysteine with propargyl bromide and purified by re-crystallization from an ethanol–water mixture as we previously reported (Wang et al. 2009).
Preparation of aggregated Aβ25−35
Aβ25−35 was dissolved at the concentration of 2 μg/μl in normal saline (NS) water, which favors aggregation (Monji et al. 2002) and incubated at 37°C for 4 days before use. This procedure is known to produce insoluble precipitates and to markedly facilitate the appearance of cognitive defects in several tasks (Maurice et al. 1996; Delobette et al. 1997).
Animals
Male Sprague-Dawley (SD) rats (8–10 weeks old, weighing 300–350 g) were used in the present study. The rats were obtained from the Animal Center of the Third Military Medical University (Chongqing, China) (Certificate No. SCXK 20020003). The animals were acclimatized for 3 days at 22 ± 1°C with a 12-h light–dark cycle, and were allowed free access to food and tap water throughout the experiment. All efforts were made to minimize the number of animals used, and all animal studies were performed in accordance with the Regulations of Experimental Animal Administration issued by the State Committee of Science and Technology of the People’s Republic of China (November 14, 1988) and the Institutional Review Committee for the use of Animal.
Drug administration and surgery
Seventy-two rats were randomly assigned to six groups: sham, Aβ, Aβ+ Donepezil (DON, 1.0 mg/kg as a positive control), and Aβ+ SPRC 20, 40, 80 mg/kg groups (n = 12, respectively). Rats in sham and Aβ groups were given NS by intraperitoneal injection (i.p.) for 17 days, rats in Aβ + DON groups were administrated with DON by gavage at the dose of 1.0 mg/kg, and rats in Aβ+ SPRC 20, 40, 80 mg/kg groups were administered with SPRC at the dose of 20, 40, and 80 mg/kg once per day 3 days before surgery and thereafter continuously for 14 days. The sham and Aβ groups received the volume-matched NS as that of the SPRC.
Three days after treatment with SPRC or DON, rats were anesthetized with 50 mg/kg sodium pentobarbital (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) by i.p., and secured in a stereotaxic apparatus (SR-6N, Narishige, Japan) with rectal temperature maintained at 37°C using a heating pad. An area of skin on top of the skull was shaved and sterilized conventionally. One right small hole for needle insertion was drilled in the parietal bone posterior to bregma on either sides of the midline (coordinates: posterior −0.8 mm, medial/lateral ±1.5 mm relative to bregma, dorsal/ventral −3.8 mm below dura) (Gong et al. 2010). Aβ (10 μg in 5 μl sterile NS) was injected into right intracerebroventricular (i.c.v.) in the Aβ, Aβ+ DON and SPRC groups via a stainless steel needle using a microinjector. Aβ solution was injected into the lateral cerebral ventricle within 5 min. The sham group underwent all surgical procedures, with the exception that sterile NS was administered rather than Aβ. Following wound suturing, all rats received an intramuscular injection of 40,000 U/0.25 ml of the antibiotic penicillin (Harbin Pharmaceutical Group Co., Ltd., General Pharmaceutical Factory, Harbin, China). Rats were closely monitored during recovery and kept in a room at 22°C. Animal body weights were recorded daily, and their general behaviors were monitored. The rectal temperature was monitored by a clinical thermometer after surgery daily to ensure rats in healthy condition.
Morris water maze test
Spatial learning and memory were evaluated by the Morris water maze (Morris 1984). The apparatus and test procedure were described elsewhere (Gong et al. 2005). The Morris water maze test began on the tenth day after surgery. The procedure included two steps. The first step was the place navigation test twice per day (one time in a.m. and one time in p.m., respectively) from Day 1 to 4, in which the escape latency (the time required to escape onto the hidden platform) was recorded, and the mean escape latency from each rat in a.m. and p.m. was used to evaluate learning and memory function. Rats that found the platform were allowed to remain on the platform for 20 s and were then returned to the home cage. If a rat did not reach the platform within 120 s, it was gently guided into the platform by the experimenter, where it remained for 20 s. The second step was the spatial probe test on Day 5 after removal of the platform and after the space navigation test, which was performed to test the ability of rats to find the removed platform by memory.
Neuronal ultrastructure observation in hippocampus
For neuronal ultrastructure observation in CA1 region of hippocampus, after the behavioral tasks three rats randomly from each group were anesthetized by an overdose of 80 mg/kg sodium pentobarbital by i.p. and were perfused transcardially with 0.1 M phosphate buffer with 0.4% heparin, immediately followed by 1% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.38). Brains were removed and cut into 50 mm sections with a Vibratome. Vibratome sections were routinely embedded in glycide ether for ultrastructural examination. After light microscopic examination of the embedded Vibratome sections, selected CA1 regions of the right dorsal hippocampus were collected. After that, these tissue masses were retained in glutaraldehyde (2.5%) for 24 h. Then semi-thin sections were made, which would be stained with the Toluidine blue so as to locate pyramidal layer of hippocampal CA1 region, after a series of process of dehydration, infiltration, embedment, and polymerization. Finally, ultra-thin sections (50 nm) were cut and taken at the interval of six sections. Two copper screens were observed in compatible times, and three photos were taken from each rat at least for transmission electron microscopy (TEM).
Real-time RT-PCR analysis
The remaining rats from each group after the behavioral experiments were euthanized by rapid decapitation, and four right hippocampi were removed and placed into the tube containing Trizol (Huashun Bioengineering Co, Shanghai, China). Total RNA was isolated and purified with RNeasy Mini Kit (Qiagen, Valencia, CA, USA). The forward and reverse primer sequences for selected genes were designed with the ABI Primer Express software (Foster City, CA, USA) and listed in Table 1. The Power SYBR Green Master Mix (Applied Biosystems, Cheshire, UK) was used for real-time PCR analysis. The relative differences in expression between groups were expressed using cycle time (Ct) values as follows: the Ct values of the interested genes were first normalized with GAPDH of the same sample, and then the relative differences between control and treatment groups were calculated and expressed as relative increases, setting control as 100%.
Western blot analysis
The remaining rats from each group after the behavioral experiments were euthanized by rapid decapitation, and three right hippocampi were removed and rapidly frozen at −80°C. The frozen tissues were cut into small pieces, homogenized in 0.5 ml of RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris-hydrochloric acid, 2 mM phenylmethylsulfonyl fluoride pH 7.4), and incubated at 4°C overnight. The dissolved proteins were collected after centrifugation at 10,000 g for 30 min, and the supernatant was then collected. Protein concentrations were determined using the enhanced BCA protein assay kit (Biocolor Biotechnology, Shanghai, China). The proteins were then separated by SDS-polyacrylamide gel electrophoresis and transferred to a PVDF membrane; then the membrane was incubated with a primary antibody against tumor necrosis factor (TNF)-α (1:1,500, Cell Signaling Technology, Inc., Boston, MA, USA), Phospho-transcription factors of the nuclear factor κB p65 (p-NF-κB p65, 1:1,500, Cell Signaling Technology, Inc., Boston, MA, USA), IκB-α (1:1,500, Cell Signaling Technology, Inc., Boston, MA, USA), COX-2 (1:1,000, Cell Signaling Technology, Inc., Boston, MA, USA), p-ERK1/2 (1:1,500, Cell Signaling Technology, Inc., Boston, MA, USA), GAPDH (1:50,000, Kangcheng Inc., Shanghai, China), β-tubulin (1:2,000, Cell Signaling Technology, Inc., Boston, MA, USA), and then it was incubated with the appropriate secondary horseradish peroxidase-conjugated anti-rabbit IgG antibodies (1:40,000, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The membranes were visualized using an ECL system and then developed on Hyperfilm (Amersham). Immunoreactive proteins were visualized using the ECL Western blotting detection kit (Thermo Fisher Scientific Inc., Boston, MA, USA) (Liu et al. 2009).
Statistical analysis
All values were presented as mean and standard error. One-way analysis of variance (ANOVA) was used to examine statistical comparisons between groups. The statistical significance of difference between two groups was determined using two-tailed Student’s t test. All analyses were performed using SPSS 12.0. A probability value of <0.05 was taken to indicate statistical significance.
Results
Effect of SPRC on Aβ-induced cognitive impairment
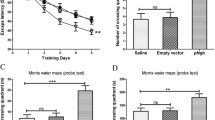
Aβ injection by i.c.v. produced less than 5% mortality. Initially, all the rats lost a few grams of body weight within 2 days after surgery. However, all of the rats had regained weight and continued to grow normally for the duration of the study. From the tenth day after surgery, all the rats received 5-day Morris water maze test. The Morris water maze test showed that the learning and memory abilities in Aβ-treated rats were significantly impaired compared with those of sham group. In the navigation test, the mean escape latency was tripled in the third and fourth days (P < 0.05; Fig. 1a), while in the spatial exploring test, the adjusted escape latency was reduced by 34% (P < 0.01; Fig. 1b), indicative of cognitive impairment. However, Aβ-induced learning and memory deficits were significantly alleviated by treatment with SPRC at the doses of 40, 80 mg/kg as well as DON compared with Aβ group on the fourth day (P < 0.05; Fig. 1a). Meanwhile, the adjusted escape latency was significantly increased by treatment with SPRC at the doses of 40, 80 mg/kg as well as DON compared with Aβ alone (P < 0.05, 0.01; Fig. 1b). These data showed that SPRC may attenuate Aβ-induced cognitive deficits in rats.

Effect of SPRC on Aβ-induced cognitive impairment in rats. Rats were treated by slowly i.c.v. of aggregated Aβ25−35 (2 μg/μl) or NS and subjected to the Morris water maze test 10 days later. a The mean escape latency in the navigation test, b the adjusted escape latency in the spatial exploring test. Data presented as mean ± SEM, n = 11–12. *P < 0.05, **P < 0.01 versus sham; # P < 0.05, ## P < 0.01 versus Aβ alone
Effect of SPRC on Aβ-induced neuronal ultrastructure changes
The neuronal ultrastructure in hippocampal CA1 regions was observed by TEM. TEM evidence showed that the nucleus of hippocampal neurons in CA1 regions were round or oval, and the euchromatin distributed homogeneously, the structures of intracytoplasmic mitochondria and rough endoplasmic reticulum were clear, and the ribosomes abounded in sham-operated rats (Fig. 2a). On the contrary, the nucleus of hippocampal neurons subjected to Aβ were irregular, a chromatin mass formed, the perinuclear space obviously became thicker, mitochondria swelled and vacuolized, cristae arrangement became disordered, mitochondria membranes were ruptured, and the endocytoplasmic reticula were expanded. Some neurons were even obviously broken to pieces and the membrane dissolved (Fig. 2b). However, treatment with DON could obviously relieve the neuronal damage compared with those of Aβ alone: most of the nuclear chromatins distributed more homogeneously, the ultramicrostructure was similar to those of neurons in sham group with swollen mitochondria mitigated, lamellar cristae in mitochondria became clear, and the ribosomes became more abundant (Fig. 2c). Similarly, treatment with SPRC at the doses of 40, 80 mg/kg also relieved neuronal morphological damage compared with those of Aβ alone, and the ultramicrostructure was similar to those of the DON group (Fig. 2d, e), especially treatment with SPRC at the doses of 80 mg/kg, the ultramicrostructure was approximate to sham-operated group, except that mitochondria were slightly swollen (Fig. 2a, e). These evidences indicated that SPRC may attenuate Aβ-induced neuronal ultrastructure in rat hippocampi.

Effect of SPRC on Aβ-induced neuronal ultrastructure changes in hippocampus. The neuronal ultrastructure in hippocampal CA1 regions was examined by TEM after the behavioral experiments, and all the ultrastructure changes were indicated by arrows. a Sham (×11,000), and b Aβ (×11,000) injection showed remarkable ultrastructure damages of hippocampal neurons in CA1 regions. Treatment with c DON (×11,000), as well as d, e SPRC 40, 80 mg/kg (×11,000) significantly attenuated the damages subjected to Aβ
Effect of SPRC on the expressions of TNF-α and COX-2 mRNA
The expressions of TNF-α and COX-2 mRNA in hippocampus were detected by real-time RT-PCR. It was found that i.c.v. Aβ significantly increased the expressions of TNF-α and COX-2mRNA, which were 1.7- and 1.8-fold compared with sham group, respectively (P < 0.05; Fig. 3a–b). However, compared with Aβ group, treatments with SPRC at the doses of 20–80 mg/kg significantly decreased the expression of TNF-α mRNA (P < 0.05; Fig. 3a). Similarly, treatments with SPRC at the doses of 40, 80 mg/kg as well as DON also inhibited the expression of COX-2 mRNA (P < 0.01, 0.05; Fig. 3b). These results showed that SPRC may inhibit the overexpressions of TNF-α and COX-2 mRNA induced by Aβ in rat hippocampi.

Effect of SPRC on the expressions of TNF-α and COX-2 mRNA. The expressions of TNF-α and COX-2 mRNA in hippocampus were detected by real-time RT-PCR. a The expressions of TNF-α mRNA, b the expressions of COX-2 mRNA. Data presented as mean ± SEM, n = 4. *P < 0.05 versus sham; # P < 0.05, ## P < 0.01 versus Aβ
Effect of SPRC on the expressions of TNF-α and COX-2 protein
To explore the mechanism of action of SPRC on amelioration of Aβ-induced cognitive impairment, the expressions of TNF-α and COX-2 protein were determined by Western blot. Consistent with the mRNA expressions, i.c.v. Aβ significantly increased the expressions of TNF-α and COX-2 protein (P < 0.01; Figs. 4a, b, 5a, b). However, after treatment with SPRC at the doses of 40, 80 mg/kg, the TNF-α and COX-2 protein expressions significantly decreased compared with Aβ group (P < 0.01; Figs. 4a, b, 5a, b). It was interesting that DON, a typical cholinesterase inhibitor, also inhibited the COX-2 protein expression compared with Aβ group (P < 0.05; Fig. 5a, b).

Effect of SPRC on the expression of TNF-α protein. The expression of TNF-α protein was determined by Western blot analysis. a Western blot of TNF-α protein contents, b TNF-α protein contents were plotted for sham, Aβ, Aβ+ DON, and Aβ+ various doses of SPRC, respectively. The relative optical density was normalized to GAPDH. Data presented as mean ± SEM, n = 3. **P < 0.01 versus sham; ## P < 0.01 versus Aβ

Effect of SPRC on the expression of COX-2 protein. The expression of COX-2 protein was determined by Western blot analysis. a Western blot of COX-2 protein contents, b COX-2 protein contents were plotted for sham, Aβ, Aβ+ DON, and Aβ+ various doses of SPRC, respectively. The relative optical density was normalized to β-tubulin. Data presented as mean ± SEM, n = 3. **P < 0.01 versus sham; # P < 0.05, ## P < 0.01 versus Aβ
Effect of SPRC on ERK1/2 activation, IκB-α degradation, and NF-κB p65 phosphorylation
To further explore the mechanisms underlying the protective effect of SPRC against Aβ neurotoxicity, ERK1/2 activation, IκB-α degradation and NF-κB p65 phosphorylation were also analyzed by Western blot.
It has been shown that Aβ treatment led to an activation of ERK1/2 and this is related to cell death (Florent et al. 2006). The present study found that i.c.v. Aβ significantly resulted in an elevation of phosphorylated ERK1/2 (P < 0.01; Fig. 6a, b). However, after treatments with SPRC at the doses of 40, 80 mg/kg notably repressed an elevation of phosphorylated ERK1/2 (P < 0.05, 0.01; Fig. 6a, b). It was interesting that DON also decreased an elevation of phosphorylated ERK1/2 (P < 0.01; Fig. 6a, b).

Effects of SPRC on ERK1/2 activation, IκB-α degradation and NF-κB phosphorylation. ERK1/2 activation, IκB-α degradation, and NF-κB p65 phosphorylation were detected by Western blot analysis. a Western blot of various protein contents. Protein contents were plotted for sham, Aβ, Aβ+ DON, and Aβ+ various doses of SPRC, respectively. b ERK1/2 and c IκB-α degradation, d NF-κB p65 phosphorylation. The relative optical density was normalized to GAPDH. Data presented as mean ± SEM, n = 3. **P < 0.01 versus sham; # P < 0.05, ## P < 0.01 versus Aβ
NF-κB is inactive in the cytosol because it is bound to IκB, and becomes active after IκB has been phosphorylated and subsequently degraded. It is found that i.c.v. Aβ significantly decreased the IκB-α protein levels (P < 0.01; Fig. 6a, c). However, treatment with DON or SPRC was found to significantly repress the Aβ-induced IκB-α degradation (P < 0.01; Fig. 6a, c). RelA/p65 is a subunit of the NF-κB transcription complex, which plays a crucial role in inflammatory and immune responses. After the Aβ-induced phosphorylation and degradation of IκB-α, p65, a part of p65/p50 heterodimer of NF-κB, translocates to the nucleus and then phosphorylates. Accordingly, i.c.v. Aβ significantly enhanced the NF-κB p65 phosphorylation (P < 0.01; Fig. 6a, d). However, treatment with SPRC or DON was found to significantly repress the Aβ-induced NF-κB p65 phosphorylation (P < 0.01, 0.05; Fig. 6a, d). Taken together, these findings indicate that the inhibition of Aβ-induced production of TNF-α and COX-2 by SPRC or DON is, at least partly, mediated by suppressing Aβ-induced IκB-α degradation and thereafter activation of NF-κB.
Discussion
The accumulation of Aβ plaques in the brains of patients could be a critical event and plays an important role in initiating the inflammatory and neurotoxic cascade that results in synaptic dysfunction and neuronal damage in AD. Significant neuronal damage in the CA1 region of the hippocampus and memory deficits were observed following Aβ25–35 injection by i.c.v. in rats (Stepanichev et al. 2004; Cheng et al. 2006; Ji et al. 2008). In the present study, it was found that cognitive deficits and neuronal ultrastructure in the CA1 region of the hippocampus after Aβ25–35 injection by i.c.v. in rats were consistent with the reports (Stepanichev et al. 2004; Tohda et al. 2004; Cheng et al. 2006; Ji et al. 2008). Aβ25–35 is the core fragment of full-length Aβ1–42 (Yankner et al. 1990). A number of studies have demonstrated that the acute injection of this peptide into rat cerebral ventricle resulted in neurotoxic effects similar to those produced by the Aβ1–40 (Kowall et al. 1991; Yamada and Nabeshima 2000; Stepanichev et al. 2004; Tohda et al. 2004), although it is still not clear whether the Aβ25–35 fragment occurs in the AD brains.
We examined the effects of SPRC on Aβ-induced cognitive deficits and neuronal ultrastructure damage in the CA1 region of the hippocampus using DON as a positive control in rats. To our knowledge, it is first found that SPRC, especially at the doses of 40 and 80 mg/kg, could significantly ameliorate Aβ-induced cognitive deficits and neuronal ultrastructure damage. SPRC, a sulphur-containing amino acid, is a novel structural analog of SAC containing cysteine structure. Although garlic compounds containing SAC may attenuate Aβ-induced apoptosis (Peng et al. 2002), destabilizes Alzheimer’s Aβ fibrils in vitro (Gupta and Rao 2007), reduces cerebral amyloid, cerebral inflammation, and Tau phosphorylation in Alzheimer’s transgenic mouse model harboring Swedish double mutation (Chauhan 2006), effects of SAC on cognitive deficits and neuronal ultrastructure are still unclear. Taken together, the present study first found that SPRC, a sulfur-containing amino acid, may attenuate Aβ-induced cognitive deficits and neuronal ultrastructure, and thus SPRC might be a potential agent for treatment of AD.
To investigate the mechanisms underlying the effects of SPRC, the expressions of TNF-α and COX-2 mRNA were detected by real-time RT-PCR, and the protein expressions were determined by Western blot, respectively. TNF signaling has been strongly implicated in AD pathology and overwhelming evidence now suggests that TNF-driven processes may contribute to cognitive dysfunction and accelerated progression of AD (Flick and Gifford 1986; Kitazawa et al. 2005). It has been proposed that elevated levels of pro-inflammatory cytokines, including TNF-α, may inhibit phagocytosis of Aβ in AD brains, thereby hindering efficient plaque removal by resident microglia (Koenigsknecht-Talboo and Landreth 2005). In the present study, we found that Aβ by i.c.v. significantly enhanced the production of TNF-α mRNA and protein, which is consistent with the report (Kuang et al. 2009). COX-2 is an important mediator that is involved in the inflammatory cascade in AD (Cheng et al. 2006). The production of COX-2 mRNA and protein were seen in hippocampus in Aβ25–35 treated rats similar to the report (Cheng et al. 2006), suggesting that COX-2 plays a role in the inflammatory cascade in the present study. COX-2 is a rate-limiting enzyme that converts arachidonic acid to prostaglandins including PGE2, a potent inflammatory mediator. The overexpression of COX-2 and the subsequent increase of PGE2 production may, in turn, further support the inflammatory cycle (Cheng et al. 2006). Our results first revealed that SPRC may inhibit the mRNA and protein expressions of TNF-α and COX-2, suggesting that inhibitions of TNF-α and COX-2 productions are involved in protective effects of SPRC on Aβ-induced cognitive deficits and neuronal ultrastructure. It was interesting that DON, a typical cholinesterase inhibitor, also inhibited Aβ-induced COX-2 production in the present study. DON has an anti-inflammatory activity via direct or indirect activation of cholinergic receptors, and may affect signaling pathways associated with inflammation in vivo and in vitro (D’Intino et al. 2005; Lee et al. 2007; Tyagi et al. 2007; Hwang et al. 2010).
The signal transduction pathways involved in Aβ-dependent neurotoxicity have become a major focus in AD research. There is a general consensus that Aβ induces ERK1/2 abnormalities as shown in the brain of AD patients (Webster et al. 2006) and animal models (Arancio et al. 2004; Dineley et al. 2001). The present study found that Aβ injection by i.c.v. significantly activated ERK1/2. An activation of ERK1/2 was reported in hippocampal slices after treatment with soluble Aβ1–42 (Dineley et al. 2001), and its inhibition could attenuate cellular injury and neuronal death, indicating a pro-death signaling role of ERK1/2 (Murray et al. 1998; Lee et al. 2005). However, treatments with SPRC or DON notably decreased an elevation of phosphorylated ERK1/2. Thus, it is concluded that inhibition of ERK1/2 activation is implicated in protective effects of SPRC on Aβ-induced cognitive deficits and neuronal ultrastructure.
To further explore the mechanisms underlying the inhibitory effect of SPRC on the expressions of TNF-α and COX-2, the current study analyzed the IκB-α and NF-κB p65 phosphorylation by Western blot. It has been well established that neuroinflammation is involved in AD pathogenesis (Schwab and McGeer 2008). The current study found that Aβ injection by i.c.v. significantly triggered the degradation of IκB-α and the subsequent phosphorylation of the p65 subunit of NF-κB. Among the several transcriptional factors activated by inflammatory responses during viral and bacterial infections, NF-κB is known to up-regulate the expressions of cytokines, chemokines, adhesion molecules, acute phase proteins, and inducible effector enzymes that are involved in innate immune response (Ghosh and Karin 2002). In unstimulated cells, NF-κB dimers are bound to inhibitory κB (IκBs), and as a result, are retained in the cytoplasm. However, when the cells are stimulated with pro-inflammatory stimuli, IκBs are rapidly phosphorylated and degraded via IκB kinase complex, and the free NF-κB is translocated to the nucleus, where it binds to target sites and induces the transcriptions of pro-inflammatory mediators (Li and Verma 2002). So it can be concluded that the free NF-κB is translocated to the nucleus, where it binds to target sites and induces the transcriptions of pro-inflammatory mediators, including TNF-α and COX-2. However, treatment with SPRC or DON inhibits the degradation of IκB and the subsequent phosphorylation of the p65 subunit of NF-κB in the nucleus, by which the generations of TNF-α and COX-2 are repressed.
In summary, the present study showed that SPRC, a sulfur-containing amino acid, may inhibit cognitive impairment and neuronal ultrastructure damage induced by Aβ for the first time. SPRC afforded a beneficial action on inhibitions of ERK1/2, as well as IκB-α degradation and NF-κB activation produced by Aβ. These findings suggested that SPRC might be a potential agent for treatment of AD.
References
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM et al (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421
Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D et al (2004) RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J 23:4096–4105
Chauhan NB (2006) Effect of aged garlic extract on APP processing and tau phosphorylation in Alzheimer’s transgenic model Tg2576. J Ethnopharmacol 108:385–394
Cheng G, Whitehead SN, Hachinski V, Cechetto DF (2006) Effects of pyrrolidine dithiocarbamate on beta-amyloid (25–35)-induced inflammatory responses and memory deficits in the rat. Neurobiol Dis 23:140–151
Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC et al (2005) Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48:913–922
Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM et al (2008) Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58:42–51
D’Intino G, Paradisi M, Fernandez M, Giuliani A, Aloe L, Giardino L et al (2005) Cognitive deficit associated with cholinergic and nerve growth factor down-regulation in experimental allergic encephalomyelitis in rats. Proc Natl Acad Sci USA 102:3070–3075
Delobette S, Privat A, Maurice T (1997) In vitro aggregation facilities beta-amyloid peptide-(25–35)-induced amnesia in the rat. Eur J Pharmacol 319:1–4
Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD (2001) Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci 21:4125–4133
Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J et al (2007) Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 6:734–746
Eikelenboom P, van Gool WA (2004) Neuroinflammatory perspectives on the two faces of Alzheimer’s disease. J Neural Transm 111:281–294
Eikelenboom P, Veerhuis R, Familian A, Hoozemans JJ, van Gool WA, Rozemuller AJ (2008) Neuroinflammation in plaque and vascular β-amyloid disorders: clinical and therapeutic implications. Neurodegener Dis 5:190–193
Flick DA, Gifford GE (1986) Production of tumor necrosis factor in unprimed mice: mechanism of endotoxin-mediated tumor necrosis. Immunobiology 171:320–328
Florent S, Malaplate-Armand C, Youssef I, Kriem B, Koziel V, Escanye MC et al (2006) Docosahexaenoic acid prevents neuronal apoptosis induced by soluble amyloid-beta oligomers. J Neurochem 96:385–395
Ghosh S, Karin M (2002) Missing pieces in the NF-kappaB puzzle. Cell 109:S81–S96
Gong QH, Wu Q, Huang XN, Sun AS, Shi JS (2005) Protective effects of Ginkgo biloba leaf extract on aluminum-induced brain dysfunction in rats. Life Sci 77:140–148
Gong QH, Wang Q, Pan LL, Liu XH, Huang H, Zhu YZ (2010) Hydrogen sulfide attenuates lipopolysaccharide-induced cognitive impairment: a pro-inflammatory pathway in rats. Pharmacol Biochem Behav 96:52–58
Graupera M, García-Pagán JC, Abraldes JG, Peralta C, Bragulat M, Corominola H et al (2003) Cyclooxygenase-derived products modulate the increased intrahepatic resistance of cirrhotic rat livers. Hepatology 37:172–181
Gupta VB, Rao KS (2007) Anti-amyloidogenic activity of S-allyl-l-cysteine and its activity to destabilize Alzheimer’s beta-amyloid fibrils in vitro. Neurosci Lett 429:75–80
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185
Hwang J, Hwang H, Lee HW, Suk K (2010) Microglia signaling as a target of donepezil. Neuropharmacology 58:1122–1129
Ji C, Aisa HA, Yang N, Li Q, Wang T, Zhang L et al (2008) Gossypium herbaceam extracts inhibited NF-κB activation to attenuate spatial memory impairment and hippocampal neurodegeneration induced by amyloid-β in rats. J Alzheimers Dis 14:271–283
Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM (2005) Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci 25:8843–8853
Koenigsknecht-Talboo J, Landreth GE (2005) Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci 25:8240–8249
Kowall NW, Beal MF, Busciglio J, Duffy LK, Yankner BA (1991) An in vivo model for the neurodegenerative effects of beta amyloid and protection by substance P. Proc Natl Acad Sci USA 88:7247–7251
Kuang X, Du JR, Chen YS, Wang J, Wang YN (2009) Protective effect of Z-ligustilide against amyloid beta-induced neurotoxicity is associated with decreased pro-inflammatory markers in rat brains. Pharmacol Biochem Behav 92:635–641
Lee SY, Lee JW, Lee H, Yoo HS, Yun YP, Oh KW et al (2005) Inhibitory effect of green tea extract on beta-amyloid-induced PC12 cell death by inhibition of the activation of NF-kappaB and ERK/p38 MAP kinase pathway through antioxidant mechanisms. Brain Res Mol Brain Res 140:45–54
Lee JH, Park SY, Shin YW, Kim CD, Lee WS, Hong KW (2007) Concurrent administration of cilostazol with donepezil effectively improves cognitive dysfunction with increased neuroprotection after chronic cerebral hypoperfusion in rats. Brain Res 1185:246–255
Li Q, Verma IM (2002) NF-kappaB regulation in the immune system. Nat Rev Immunol 2:725–734
Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B et al (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci 20:5709–5714
Liu XH, Chen PF, Pan LL, Zhu YZ (2009) 4-Guanidino-n-butyl syringate (leonurine) protects H9c2 rat ventricular cells from hypoxia-induced apoptosis. J Cardiovasc Pharmacol 54:437–444
Maurice T, Lockhart BP, Privat A (1996) Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res 706:181–193
McGeer PL, McGeer EG (2007) NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging 28:639–647
McGeer PL, Rogers J, McGeer EG (2006) Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis 9:271–276
McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S et al (2008) Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res 1207:225–236
Monji A, Utsumi H, Ueda T, Imoto T, Yoshida I, Hashioka SK et al (2002) Amyloid-beta-protein (Abeta) (25–35)-associated free radical generation is strongly influenced by the aggregational state of the peptides. Life Sci 70:833–841
Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11:47–60
Mrak RE (2009) Neuropathology and the neuroinflammation idea. J Alzheimers Dis 18:473–481
Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ (1998) Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc Natl Acad Sci USA 95:11975–11980
Peng Q, Buz’Zard AR, Lau BH (2002) Neuroprotective effect of garlic compounds in amyloid-beta peptide-induced apoptosis in vitro. Med Sci Monit 8:BR328–BR337
Schwab C, McGeer PL (2008) Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J Alzheimers Dis 13:359–369
Selkoe DJ (1999) Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 399:A23–A31
Shi S, Klotz U (2008) Clinical use and pharmacological properties of selective COX-2 inhibitors. Eur J Clin Pharmacol 64:233–252
Stepanichev MY, Zdobnova IM, Zarubenko II, Moiseeva YV, Lazareva NA, Onufriev MV (2004) Amyloid beta(25–35)-induced memory impairments correlate with cell loss in rat hippocampus. Physiol Behav 80:647–655
Tohda C, Matsumoto N, Zou K, Meselhy MR, Komatsu K (2004) Abeta(25–35)-induced memory impairment, axonal atrophy, and synaptic loss are ameliorated by M1, A metabolite of protopanaxadiol-type saponins. Neuropsychopharmacology 29:860–868
Tyagi E, Agrawal R, Nath C, Shukla R (2007) Effect of anti-dementia drugs on LPS induced neuroinflammation in mice. Life Sci 80:1977–1983
Verdile G, Fuller S, Atwood CS, Laws SM, Gandy SE, Martins RN (2004) The role of beta amyloid in Alzheimer’s disease: still a cause of everything or the only one who got caught? Pharmacol Res 50:397–409
Wang Q, Liu HR, Mu Q, Rose P, Zhu YZ (2009) S-propargyl-cysteine protects both adult rat hearts and neonatal cardiomyocytes from ischemia/hypoxia injury: the contribution of the hydrogen sulfide-mediated pathway. J Cardiovasc Pharmacol 54:139–146
Wang Q, Wang XL, Liu HR, Rose P, Zhu YZ (2010) Protective effects of cysteine analogues on acute myocardial ischemia: novel modulators of endogenous H(2)S production. Antioxid Redox Signal 12:1155–1165
Webster B, Hansen L, Adame A, Crews L, Torrance M, Thal L et al (2006) Astroglial activation of extracellular-regulated kinase in early stages of Alzheimer disease. J Neuropathol Exp Neurol 65:142–151
Yamada K, Nabeshima T (2000) Animal models of Alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacol Ther 88:93–113
Yankner BA, Duffy LK, Kirschner DA (1990) Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250:279–282
Acknowledgments
This work was supported by the National Basic Research Program of China (973 Program, Grant No. 2010CB912600), National natural Science foundation of China (Grant No. 30888002) and National Drug Innovative Program (2009ZX09301-011).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gong, QH., Pan, LL., Liu, XH. et al. S-propargyl-cysteine (ZYZ-802), a sulphur-containing amino acid, attenuates beta-amyloid-induced cognitive deficits and pro-inflammatory response: involvement of ERK1/2 and NF-κB pathway in rats. Amino Acids 40, 601–610 (2011). https://doi.org/10.1007/s00726-010-0685-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-010-0685-1