
Abstract
Alzheimer’s disease (AD) is associated with the inflammatory response in response to amyloid β-peptide (Aβ). Previous studies have suggested that paeoniflorin (PF) shows anti-inflammatory and neuroprotective effects in inflammation-related diseases. However, the impacts of PF on AD have not been investigated. In the present study, we showed that a 4-week treatment with PF could significantly inhibit Aβ burden, Aβ-induced over activation of astrocytes and microglia, downregulation of proinflammatory cytokines, and upregulation of anti-inflammatory cytokines in the brain. In addition, we demonstrated that chronic treatment with PF inhibited the activation of glycogen synthase kinase 3β (GSK-3β) and reversed neuroinflammtory-induced activation of nuclear factor-kappa B (NF-κB) signaling pathways. Moreover, PF exerted inhibitory effects on NALP3 inflammasome, caspase-1, and IL-1β. Collectively, in the present study, we demonstrated that PF exhibits neuroprotective effects in amyloid precursor protein (APP) and presenilin 1 (PS1) double-transgenic (APP/PS1) mice via inhibiting neuroinflammation mediated by the GSK-3β and NF-κB signaling pathways and nucleotide-binding domain-like receptor protein 3 inflammasome. Thus, these results suggest that PF might be useful to intervene in development or progression of neurodegeneration in AD through its anti-inflammatory and anti-amyloidogenic effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most prevalent form of dementia, characterized pathologically by the presence of amyloid plaques and neurofibrillary tangles in the brain, and resulting in cognitive dysfunction and memory impairment [1]. Amyloid β-peptide (Aβ) plays a central pathogenic role in AD. Aβ-induced mitochondrial dysfunction, inflammatory processes, and apoptosis may interact and amplify each other in a cycle of toxicity, leading to gradual neuronal cell death in the brain. Although many studies have focused on understanding the pathological mechanisms of AD and finding treatments, to date, no effective therapies exist to prevent, halt, or reverse AD. Thus, the development of novel, alternative, or complementary therapeutic approaches is needed.
AD is associated with increased neuroinflammation [2]. Inflammatory reactions are a crucial process of the secondary damage cascade, including inflammatory cell infiltration and neuronal and glial cell destruction. Proinflammatory cytokines, such as TNF-α and IL-1β, are primarily produced by activated microglia and astrocytes, and exhibit neuronal toxicity causing cognitive deficits [3]. The glycogen synthase kinase 3β (GSK-3β) signaling pathway plays an important role in the inflammatory response driving Aβ production in AD [4]. In addition, proinflammatory cytokines including the interleukins and TNF induce the upregulation of GSK-3β, and inhibiting GSK-3β activity results in anti-inflammatory responses [4–6]. Moreover, activation of the nuclear factor-kappa B (NF-κB) pathway and NALP3 inflammasome may be involved in modulating the release of proinflammatory cytokines in AD [7, 8]. NF-κB activity is associated with BACE1 expression and AD pathogenesis [9]. The activation of NF-κB pathway is necessary for activation of the NALP3 inflammasome [10], which enhances AD progression by mediating a harmful chronic inflammatory response, synaptic dysfunction, and cognitive impairment [8, 11]. Because of the complex AD pathogenesis, a therapy with multiple targets could be effective.
Recently, the potential use of natural herbs as neuroprotective agents has attracted attention. Paeoniflorin (PF), extracted from the root of Paeonia lactiflora Pall, has been widely used in traditional Chinese medicine for the treatment of pain, inflammation, cramps, giddiness, congestion, and degenerative disorders [12, 13]. The neuroprotective effects of PF are associated with its modulation of multiple anti-inflammatory [14, 15], anti-apoptotic [14, 16], and anti-oxidative properties [17]. The safety of PF treatment has been well established after widespread application for the treatment of Parkinson’s disease [18], acute ischemic stroke [14, 19], and AD [17] in experimental models. However, the protective effects and mechanisms of PF in AD remain unclear. Therefore, in the present study we determined the protective effects of PF in the amyloid precursor protein (APP) and presenilin 1 (PS1) double-transgenic (APP/PS1) mouse model of AD, and identified the signaling pathways involved in the actions of PF.
Materials and Methods
Animal Treatment
APPswe/PSEN1dE9 (APP/PS1, male, 4-month-old) transgenic mice and their wild-type (WT) littermates were purchased from the Model Animal Research Center of Nanjing University. The animals were kept in a temperature-controlled house under light-controlled conditions, and fed a standard laboratory diet and water ad libitum. At the age of 7 months, the mice were randomly separated into three groups (n = 20 for each group): Group 1 (PF group): APP/PS1 mice treated with PF (5 mg/kg, i.p., twice per day) for 4 weeks (PF was purchased from Nanjing ZeLang Medical Technology Co., Ltd; purity >98.5 %); Group 2, TG group: APP/PS1 mice treated with saline (2 mL/kg, i.p., twice per day) for 4 weeks; Group 3, WT group, untreated mice. All of the animal procedures and use were in accordance with the guidelines of National Institutes of Health and Southern Medical University.
Morris Water Maze
The Morris water maze (MWM) test was performed as previously described [20, 21]. The mice were gently released into the pool and allowed to find and climb onto an escape platform (12 cm in diameter) that was submerged 2 cm below the water surface for four trials per day. A different starting position was used for each trial. The duration of a trial was 90 s. Escape latencies (time spent swimming from the start point to the target) and path length (distance from start to the platform), before reaching the platform were recorded for six consecutive days and analyzed. For probe trials, the platform was removed after the last trial of the acquisition period. The mice were tested 24 h later to assess memory consolidation. The time spent in the target quadrant within 60 s was recorded. The number of times the mouse crossed the former platform location was recorded.
Immunofluorescence Staining and Immunohistochemistry
Immunohistochemistry and immunofluorescence staining were performed as previously described [21]. The antibodies used were as follows: anti-Aβ (Covance, Princeton, NJ, USA), anti-CD11b (Abcam, Cambridge, UK), and anti-GFAP (Millipore, Billerica, MA, USA). Images were acquired using a confocal microscope (Leica, Wetzlar, Germany).
Enzyme-Linked Immunosorbent Assay
To determine the levels of inflammatory cytokines (n = 4 for each group) in the mouse brain treated with or without PF, The cortex and hippocampus were homogenized by sonication in cold phosphate-buffered solution containing a protease and phosphatase inhibitor cocktail (Roche), the concentrations of TNF-α, IL-1β, IL4, and IL10 in the brain homogenates were measured using sandwich enzyme-linked immunosorbent assay (ELISA; CUSABIO, Wuhan, China) according to the manufacturer’s instructions.
The level of soluble and insoluble Aβ42 in brain samples were detected by an ELISA kit (Invitrogen, Camarillo, CA, USA) according to the manufacturer’s instructions. Briefly, The cortex and hippocampus were homogenized on ice in 200 mg/mL TPER (Pierce) with protease and phosphatase inhibitor cocktails (Roche). The homogenates were centrifuged at 20,000g for 10 min at 4 °C. The supernatants (soluble fraction) were analyzed for soluble Aβ. The pellet was resuspended in 70 % for mic acid and centrifuged at 44,000g for 10 min at 4 °C. The resulting supernatants were neutralized with 1 M Tris and then diluted in ELISA buffer for the measurement of insoluble Aβ.
Western Blot
The isolated cortex and hippocampus were lysed in a radioimmunoprecipitation (RIPA) lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 % nonidet P-40, 0.5 % sodium deoxycholate) containing a cocktail of protease inhibitors (Complete; Roche, Basel, Switzerland). The lysates were electrophoresed on SDS-PAGE gels. The membrane was blocked in 5 % non-fat dry milk and 0.1 % Tween-20/Tris buffered saline (TBS) for 1 h, and was subsequently incubated overnight at 4 °C with the following primary antibodies: anti-phospho-NF-κB p65 (1:1500), anti-NF-κB p65 (1:1500), anti-phospho-IkBα (1:1000), ati- IkBα (1:1500), anti-phospho-AKT-pSer473 (1:1000), anti-AKT (1:1000), anti-phospho-GSK-3β-pSer9 (1:1000), anti- GSK3β (1:1000) and Anti-cleaved caspase-1 (Asp297) antibody (1:1000; all from Cell Signaling Technology, Beverly, MA, USA). Anti-NALP3 antibody (1:800; Santa Cruz). Secondary horseradish peroxidase (HRP)-conjugated antibodies were incubated for 2 h at room temperature (RT) and the signals were detected using SuperSignal ECL (Pierce, Rockford, IL, USA).
Statistical Analyses
All of the statistical analyses were performed using the SPSS software (version 18.0). Data are presented as mean ± standard error of the mean (SEM). Data between multiple groups were analyzed by either a one-way or a two-way analysis of variance (ANOVA), followed by Fischer’s LSD post hoc tests. Comparisons between groups were performed using Student’s two-tailed unpaired t test, and P values <0.05 were considered statistically significant in all of the analyses (*P < 0.05; **P < 0.01 and ***P < 0.001).
Results
PF Treatment Significantly Improved Memory Deficits in APP/PS1 Mice
To investigate whether PF treatment could alleviate memory impairments, the MWM test was performed to assess the effects after APP/PS1 mice received 1 month of PF treatment starting at the age of 7 months. In the hidden platform test, the mice in the TG group showed significant memory deficits compared to age-matched mice in the WT group, as indicated by the increased escape latencies and swimming distances in the consecutive trials (Fig. 1a, b). In contrast, the mice in the PF group showed significantly shorter latencies than in the TG group (Fig. 2a) and swam significantly shorter distances to reach the platform compared to TG mice on the 4th, 5th, and 6th day of the hidden platform test (Fig. 2b). The mice in the three groups displayed identical swimming speeds (Fig. 2c). In the probe trial experiment, PF treatment significantly improved the spatial memory of AD model mice as indicated by the increased number of times they swam over the target site compared to mice in the TG group (Fig. 2d). These data indicate that PF treatment significantly improved the memory deficits observed in APP/PS1 mice.

PF treatment significantly improved memory deficits in AD model mice. a MWM training showed that mice in all groups learned the task. However, PF-treated APP/PS1 mice exhibited significantly shorter escape latencies on days 4–6 of training versus vehicle-treated transgenic mice (TG group). b The PF-treated APP/PS1 mice had a shorter swimming length before escaping onto the hidden platform on days 4–6 of training. c Each group exhibited a similar latency to escape onto the visible platform. d In probe trial testing, PF-treated APP/PS1 mice crossed the former platform location more often than TG group. Data presented as mean ± SE. n = 12. *P < 0.05; **P < 0.01 using one-way ANOVA

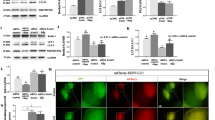
Effect of PF treatment on Aβ plaque deposition in APP/PS1 mice. The coronal sections of the cortex and hippocampus (a) in PF-treated or control APP/PS1 mice were stained with an antibody against Aβ. Scale bars: 200 μm. b Quantitative analysis of Aβ plaques in the cortex and hipppocampus of APP/PS1 mice treated with PF or vehicle. The levels of brain-soluble Aβ42 (c) and brain-insoluble Aβ42 (d) in the brains of mice from each group were measured using ELISA. n = 6 in each group. Data presented as mean ± SE, *P < 0.05; **P < 0.01, PF treated group versus vehicle-treated group
PF Decreased Neuritic Plaque Burden in APP/PS1 Transgenic Mice
To investigate whether PF could inhibit amyloid plaque deposition in APP/PS1 mice, the coronal sections of the cortex and hippocampus of PF-treated and control APP/PS1 mice were stained with an antibody against Aβ. Neuritic plaque deposition was significantly decreased in APP/PS1 mice treated with PF compared to the controls (Fig. 2a, b). Quantification showed that the number (Fig. 2c, d) of Aβ plaques was reduced in the cortex (Fig. 2c) and hippocampus (Fig. 2d) of mice in the PF group compared to the TG group. To confirm the above results, both soluble and insoluble levels of Aβ42 were detected using ELISA to investigate the effects of PF. As expected, the concentrations of soluble and insoluble of Aβ42 were significantly decreased in APP/PS1 mice treated with PF compared to the controls (Fig. 2e). These data demonstrate that PF inhibited Aβ neuritic plaque deposition in vivo.
PF Inhibited Activation of Microglia and Astrocytes in APP/PS1 Mice
Activated microglia and astrocytes are associated with amyloid plaques, which contribute to an inflammatory process causing brain injury and further neurodegeneration [2, 21–24]. Therefore, anti-inflammatory therapy has been considered a strategy for slowing the progression of AD [9, 24, 25]. To investigate whether PF could prevent glial activation in APP/PS1 mice, we compared astrocytic and microglial reactivity in the different groups. Astrocytes displayed reactive changes in the cortex and hippocampus of mice in the TG group, as characterized by increased expression of GFAP (an astrocyte marker, Fig. 3a) and the number of activated GFAP-positive cells compared to the WT group (Fig. 3b). However, PF treatment significantly inhibited the astrocytic activation in the brain of APP/PS1 mice (Fig. 3a, b).

PF inhibits activation of microglia and astrocytes in APP/PS1 mice. Immunofluorescence staining for astrocytes with anti-GFAP (a) and microglia with anti-CD11b (b) in the cortex and hippocampus. Quantitative image analysis of % area of GFAP+ cells (c) and CD11b+ cells (d) in the cortex and hippocampus of each group of mice. Data presented as mean ± SE. n = 6. *P < 0.05; **P < 0.01 using one-way ANOVA
The CD11b (a microglial marker) staining showed that the microglia in the TG group displayed an activated microglial morphology characterized by large cell bodies and thick processes (Fig. 3c). The number of activated microglia and the soma volume of microglia were markedly inhibited by the PF treatment (Fig. 3c, d). These results indicate that chronic PF treatment attenuates glial activation induced by amyloid plaques.
PF Modulated Neuroinflammation Via Downregulation of Proinflammatory Cytokines and Upregulation of Anti-inflammatory Cytokines
Reducing proinflammatory cytokines prevents neuronal dysfunction in AD [26]. Thus, we examined whether the beneficial effects of PF treatment were associated with a reduction of proinflammatory factors. APP/PS1 mice had significantly higher levels of proinflammatory cytokines, including TNF-α and IL-1β, than the mice in the WT group (Fig. 4a, b). However, PF treatment significantly decreased the TNF-α and IL-1β levels (Fig. 4a, b), and significantly upregulated the anti-inflammatory cytokines IL-10 and IL-4 levels in the cortex and hippocampus of APP/PS1 mice (Fig. 4c, d). These findings suggest that the beneficial effects of PF treatment were associated with a modulation of inflammatory responses in APP/PS1 mice.

PF modulated neuroinflammation via downregulation of proinflammatory cytokines and upregulation of anti-inflammatory cytokines. a–d The levels of TNF-α, IL-1β, IL-10, and IL-4 in the brains of mice from each group were analyzed using ELISA; PF treatment significantly decreased the TNF-α and IL-1β levels (a, b), while upregulating the anti-inflammatory cytokines IL-10 and IL-4 levels (c, d) in the cortex and hippocampus of APP/PS1 mice. Data presented as mean ± SE. n = 6. *P < 0.05 using one-way ANOVA
PF Treatment Inhibited GSK-3β and NF-κB Activation and Increased the Activation of AKT in APP/PS1 Mice
We showed that PF decreased Aβ production and modulated inflammatory responses in AD mice. To further elucidate the potential mechanism of the effects of PF, we determined the activity of AKT, GSK-3β, and NF-κB, which play a pivotal role in the inflammatory response and regulation of Aβ production in AD [4, 14, 27]. The protein levels of Akt phosphorylated at serine 473 (p-AKT), total Akt, GSK-3β phosphorylated at serine 9 (p-GSK3β-pSer9), total GSK-3β, NF-κB p65 phosphorylated at serine 536 (p-NF-κB p65), total NF-κB p65, total I-κBa and I-κBa phosphorylated at serine 32 (p-I-κBa) were detected by Western blot analysis (Fig. 5a). PF increased the p-AKT (Fig. 5a, b), and p-GSK3β-pSer9 (pSer9, the inhibitory phosphorylation site, Fig. 5a, c) levels in the cortex and hippocampus of APP/PS1 mice. However, the protein levels of p-NF-κB p65 (Fig. 5a, d) and p-I-κBa (Fig. 5a, e) were significantly decreased in the PF-treated group. Collectively, these results indicate that PF increased the activity of AKT, and inhibited the activation of GSK-3β and NF-κB p65.

PF treatment inhibited GSK-3β and NF-κB activation and increased the activation of AKT in APP/PS1 mice. The levels of p-AKT, AKT, p-GSK3β(pSer9), GSK3β, p-NF-κB p65, NF-κB p65, IκBa and p-IκBa in each group of mice were analyzed by Western blot analysis (a). The relative levels of p-AKT (b), p-GSK-3β (c), p-NF-κB p65 (d), p-IκBa (e) were quantified. n = 6 in each group. Data presented as mean ± SE, *P < 0.05; **P < 0.01, PF treated group versus the vehicle-treated group
PF Inhibits NALP3 Inflammasome Activation in APP/PS1 Mice
We showed that PF inhibited the activity of NF-κB p65 in APP/PS1 mice. Previous studies have shown that the activity of NF-κB is necessary for NALP3 inflammasome activation [8, 10, 11], which is important for inflammation and brain injury in AD [8, 11]. Hence, we examined whether PF could modulate the activation of NALP3 inflammasome by Western blot analysis. As shown in Fig. 6, PF treatment significantly reduced the NALP3 levels in APP/PS1 mice (Fig. 6a, b). In addition, PF treatment decreased the caspase-1 p20 subunit levels in AD mice (Fig. 6a, b). Collectively, these results suggest that PF inhibited NALP3 inflammasome activation in APP/PS1 mice.

PF inhibited NALP3 inflammasome activation in APP/PS1 mice. a The levels of NALP3 and caspase-1 p20 subunit in mice from each group were analyzed using Western blot with β-actin as a loading control. b Quantification of the band intensity of NALP3 and caspase-1 p20 subunit levels. PF significantly decreased the NALP3 and caspase-1 p20 subunit levels. Data presented as mean ± SE. n = 6. *P < 0.05 using one-way ANOVA
Discussion
The Aβ peptide is considered to be the root cause of AD; however, the neuroinflammatory process mediated by Aβ plaque-induced microglial cells and astrocytes also contributes to AD pathogenesis [2, 28]. It was also reported that several anti-inflammatory compounds 4-O-methylhono-kiol, thiacremonone and obovatol improved memory functions in AD animal models [29–31]. Therefore, therapeutic strategies targeting the inflammatory response can inhibit the progression of tissue damage, providing an extended therapeutic window for AD. Recent studies suggest that PF has potent neuroprotective effects by inhibiting inflammatory responses [13, 14, 16, 18], and increases the possibility of using PF in AD therapy. Although PF has shown protective effects in models of Parkinson’s disease [18], acute ischemic stroke [14, 19], and experimental autoimmune encephalomyelitis [19], its effects and underlying mechanisms on AD remain unclear. In this study, we used the APP/PS1 mouse model in which Aβ deposition and neuroinflammation were present in the early stages of life [32] to assess the efficacy of PF on AD-related cognitive and neuropathological outcomes. We found that PF inhibited memory impairment, suppressed amyloidogenesis and neuroinflammatory, inhibited NF-κB activity and NALP3 inflammasome activation in APP/PS1 mice. These results indicate that anti-neuroinflammatory effects of PF may be associated with suppressed amyloidogenesis, which targeting NF-κB activity and NALP3 inflammasome activation, and thus improve memory dysfunction.
Activated astrocytes and microglia are closely associated with amyloid plaques in AD, and may play a role in the neurotoxicity observed in AD because of the inflammatory reaction they generate [2]. In response to amyloid plaques, actived glial cells are accompanied by functional changes, such as increasing expression of proinflammatory factors TNF-α and IL-1β, which further induce brain tissue damage [2, 33]. Our data showed that GFAP and CD11b-positive cells were increased in the brain of APP/PS1 mice, and PF administration significantly reduced the number of these cells. Simultaneously, the levels of TNF-α and IL-1β in the brain were also increased, and PF treatment significantly downregulated the levels of these two proinflammatory factors and upregulated the levels of anti-inflammatory factors in the brain of APP/PS1 mice. Because inhibition of the glial inflammatory response is considered a promising target for the treatment of AD [9, 29, 30], our data firstly demonstrated the antiinflammatory effects of PF in AD animal model.
GSK-3β plays an important role in the inflammatory response driving Aβ production in AD [4, 5]. Several proinflammatory cytokines including the interleukins and TNF activate GSK-3β signaling activity, and inhibition causes anti-inflammatory effects [4, 5]. Furthermore, the activation of NF-κB and subsequent degradation of I-kBa are key events in the inflammatory response of AD [4, 10, 27, 34–36]. Numerous studies have suggested that GSK-3β regulates gene transcription in an NF-κB-dependent manner, and GSK-3β and NF-κB signaling pathways are involved in regulating inflammatory responses and Aβ production [4, 34, 35]. The activation of NF-κB signaling is necessary for activation of the NALP3 inflammasome [36]. In the present study, APP/PS1 mice was characterized by a decreased phosphorylation state of Akt and GSK-3β, which was associated with augmented neuroinflammation as evidenced by increased NF-κB phosphorylation and TNF-α as well as IL-1β. These changes were normalized with PF administration. PF enhanced Akt and GSK3β-pSer9(the inhibitory phosphorylation site) phosphorylation (inhibited the activity of GSK-3β), and significantly inhibited the activation of NF-κB and NALP3 inflammasome, thus leading to decreased production of downstream cytokines (TNF-α and IL-1β) and Aβ deposits in APP/PS1 mice. The results suggest that interfering with the GSK-3β and NF-κB pathways and NALP3 inflammasome can result in PF-mediated neuroprotective effects in AD mice. However, the links about GSK-3β and NF-κB pathways which contributed to Aβ reduction and inflammatory response is still unknown in APP/PS1 mice, further study are needed.
A previous study suggested the activation of NALP3–caspase-1 pathway by Aβ may represent a critical component of the inflammatory response in AD [8, 11]. The NALP3 inflammasome is required for the Aβ-induced activation of caspase-1, the release of mature IL-1β and the secretion of proinflammatory and potentially neurotoxic cytokines and chemokines in AD [8, 11]. NALP3 or caspase-1 knockout largely protected AD transgenic model by mediating a harmful chronic inflammatory tissue response, mediating synaptic dysfunction and enhancing Aβ clearance [11]. NALP3 inflammasome inhibition represents a novel therapeutic target for AD [8, 11, 27]. The present study showed that PF exerted inhibitory effects on NALP3 inflammasome, caspase-1, and IL-1β, which may explain the memory improvement and protective effects on Aβ pathology.
Collectively, chronic PF treatment alleviates memory deficits and reduces amyloid plaque burden in APP/PS1 transgenic mice. PF affected the pathogenesis of AD via interference of GSK-3β and NF-κB pathways, and suppression of the NLRP3 inflammasome activity or inflammasome-derived cytokines (TNF-α and IL-1β). Although the mechanisms of PF involved in anti-amyloidogenesis and anti-neuroinflammation in AD requires further elucidation, our data suggest that the application of PF might be an effective treatment for AD.
References
Cummings JL, Cole G (2002) Alzheimer disease. JAMA 287:2335–2338
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421
Fan R, Xu F, Previti ML, Davis J, Grande AM, Robinson JK, Van Nostrand WE (2007) Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J Neurosci 27:3057–3063
Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W (2013) Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Investig 123:224–235
Yuskaitis CJ, Jope RS (2009) Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell Signal 21:264–273
Beurel E, Jope RS (2009) Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J Neuroinflammation 6:9
Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A (2008) Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. J Cell Mol Med 12:2255–2262
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55:697–711
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E (2009) Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183:787–791
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678
Wu SH, Wu DG, Chen YW (2010) Chemical constituents and bioactivities of plants from the genus Paeonia. Chem Biodivers 7:90–104
Nizamutdinova IT, Jin YC, Kim JS, Yean MH, Kang SS, Kim YS, Lee JH, Seo HG, Kim HJ, Chang KC (2008) Paeonol and paeoniflorin, the main active principles of Paeonia albiflora, protect the heart from myocardial ischemia/reperfusion injury in rats. Planta Med 74:14–18
Guo RB, Wang GF, Zhao AP, Gu J, Sun XL, Hu G (2012) Paeoniflorin protects against ischemia-induced brain damages in rats via inhibiting MAPKs/NF-kappaB-mediated inflammatory responses. PLoS ONE 7:e49701
Jiang D, Chen Y, Hou X, Xu J, Mu X, Chen W (2011) Influence of Paeonia lactiflora roots extract on cAMP-phosphodiesterase activity and related anti-inflammatory action. J Ethnopharmacol 137:914–920
Wang D, Wong HK, Feng YB, Zhang ZJ (2013) Paeoniflorin, a natural neuroprotective agent, modulates multiple anti-apoptotic and pro-apoptotic pathways in differentiated PC12 cells. Cell Mol Neurobiol 33:521–529
Zhong SZ, Ge QH, Li Q, Qu R, Ma SP (2009) Peoniflorin attentuates Abeta(1–42)-mediated neurotoxicity by regulating calcium homeostasis and ameliorating oxidative stress in hippocampus of rats. J Neurol Sci 280:71–78
Liu HQ, Zhang WY, Luo XT, Ye Y, Zhu XZ (2006) Paeoniflorin attenuates neuroinflammation and dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease by activation of adenosine A1 receptor. Br J Pharmacol 148:314–325
Liu DZ, Xie KQ, Ji XQ, Ye Y, Jiang CL, Zhu XZ (2005) Neuroprotective effect of paeoniflorin on cerebral ischemic rat by activating adenosine A1 receptor in a manner different from its classical agonists. Br J Pharmacol 146:604–611
Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858
Zhang M-Y, Zheng C-Y, Zou M-M, Zhu J-W, Zhang Y, Wang J, Liu C-F, Li Q-F, Xiao Z-C, Li S (2014) Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol Aging 35:2713–2725
Mrak RE, Griffin WS (2005) Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 26:349–354
Zhang R, Miller RG, Madison C, Jin X, Honrada R, Harris W, Katz J, Forshew DA, McGrath MS (2013) Systemic immune system alterations in early stages of Alzheimer’s disease. J Neuroimmunol 256:38–42
Nunes AF, Amaral JD, Lo AC, Fonseca MB, Viana RJ, Callaerts-Vegh Z, D’Hooge R, Rodrigues CM (2012) TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-beta deposition in APP/PS1 mice. Mol Neurobiol 45:440–454
Ding Y, Qiao A, Wang Z, Goodwin JS, Lee ES, Block ML, Allsbrook M, McDonald MP, Fan GH (2008) Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J Neurosci 28:11622–11634
He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y (2007) Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol 178:829–841
Shi JQ, Zhang CC, Sun XL, Cheng XX, Wang JB, Zhang YD, Xu J, Zou HQ (2013) Antimalarial drug artemisinin extenuates amyloidogenesis and neuroinflammation in APPswe/PS1dE9 transgenic mice via inhibition of nuclear factor-kappaB and NLRP3 inflammasome activation. CNS Neurosci Ther 19:262–268
Wyss-Coray T, Rogers J (2012) Inflammation in Alzheimer disease—a brief review of the basic science and clinical literature. Cold Spring Harbor Perspect Med 2:a006346
Lin GH, Lee YJ, Choi DY, Han SB, Jung JK, Hwang BY, Moon DC, Kim Y, Lee MK, Oh KW, Jeong HS, Leem JY, Shin HK, Lee JH, Hong JT (2012) Anti-amyloidogenic effect of thiacremonone through anti-inflamation in vitro and in vivo models. JAD 29:659–676
Lee YJ, Choi DY, Choi IS, Kim KH, Kim YH, Kim HM, Lee K, Cho WG, Jung JK, Han SB, Han JY, Nam SY, Yun YW, Jeong JH, Oh KW, Hong JT (2012) Inhibitory effect of 4-O-methylhonokiol on lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment via inhibition of nuclear factor-kappaB in vitro and in vivo models. J Neuroinflam. 9:35
Choi DY, Lee JW, Peng J, Lee YJ, Han JY, Lee YH, Choi IS, Han SB, Jung JK, Lee WS, Lee SH, Kwon BM, Oh KW, Hong JT (2012) Obovatol improves cognitive functions in animal models for Alzheimer’s disease. J Neurochem 120:1048–1059
Puli L, Pomeshchik Y, Olas K, Malm T, Koistinaho J, Tanila H (2012) Effects of human intravenous immunoglobulin on amyloid pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J Neuroinflam. 9:105
Ruan L, Kang Z, Pei G, Le Y (2009) Amyloid deposition and inflammation in APPswe/PS1dE9 mouse model of Alzheimer’s disease. Curr Alzheimer Res 6:531–540
Takada Y, Fang X, Jamaluddin MS, Boyd DD, Aggarwal BB (2004) Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J Biol Chem 279:39541–39554
Steinbrecher KA, Wilson W 3rd, Cogswell PC, Baldwin AS (2005) Glycogen synthase kinase 3beta functions to specify gene-specific NF-kappaB-dependent transcription. Mole Cell Biol 25:8444–8455
Paris D, Patel N, Quadros A, Linan M, Bakshi P, Ait-Ghezala G, Mullan M (2007) Inhibition of Abeta production by NF-kappaB inhibitors. Neurosci Lett 415:11–16
Acknowledgments
This study was supported by funding from the “National Natural Science Foundation of guangdong’’ (Grant No. 32215050) and “Scientific research and innovation project of Jinan University” (Grant No. 21615336). This work was also supported by the “Scientific research and innovation project of Luoyang”(Grant No. 150415), the “Natural Scientific Research funds of China” (No. 81301116) and “China Postdoctoral Science Foundation”(Grant No. 2012M521922).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhang, HR., Peng, JH., Cheng, XB. et al. Paeoniflorin Atttenuates Amyloidogenesis and the Inflammatory Responses in a Transgenic Mouse Model of Alzheimer’s Disease. Neurochem Res 40, 1583–1592 (2015). https://doi.org/10.1007/s11064-015-1632-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1632-z