
Abstract
Identifying protein–protein interactions (PPIs) is critical for understanding the cellular function of the proteins and the machinery of a proteome. Data of PPIs derived from high-throughput technologies are often incomplete and noisy. Therefore, it is important to develop computational methods and high-quality interaction dataset for predicting PPIs. A sequence-based method is proposed by combining correlation coefficient (CC) transformation and support vector machine (SVM). CC transformation not only adequately considers the neighboring effect of protein sequence but describes the level of CC between two protein sequences. A gold standard positives (interacting) dataset MIPS Core and a gold standard negatives (non-interacting) dataset GO-NEG of yeast Saccharomyces cerevisiae were mined to objectively evaluate the above method and attenuate the bias. The SVM model combined with CC transformation yielded the best performance with a high accuracy of 87.94% using gold standard positives and gold standard negatives datasets. The source code of MATLAB and the datasets are available on request under smgsmg@mail.ustc.edu.cn.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Studies of protein–protein interactions (PPIs) provide critical insight into the cellular processes of DNA transcription and replication, metabolic cycles, signaling cascades, cell proliferation and apoptosis, etc. high-throughput experimental technologies, including yeast two-hybrid screen (Y2H) (Ito et al. 2000; Uetz et al. 2000), mass spectrometry protein complex identification (MS-PCI) (Ho et al. 2002) and coimmunoprecipitated protein complex (Co-IP) (von Mering et al. 2002; Ho et al. 2002), protein chips (Zhu et al. 2001) and tandem affinity purification (TAP) (Gavin et al. 2002), have been developed to elucidate and model protein interactions at genomic scale. Although genome-scale protein interaction networks have now been built and experimentally validated in several species such as Saccharomyces cerevisiae (Uetz et al. 2000; Krogan et al. 2006), Escherichia coli (Li et al., 2004), Drosophila melanogaster (Giot et al. 2003) and Helicobacter pylori (Rain et al. 2001), these interaction datasets are often noisy and always contain many false positive interactions (Uetz et al. 2000; Ito et al. 2001).Therefore, several ‘in silico’ (computed) interaction prediction methods have been developed, which provide an attracting perspective on predicting and understanding PPIs as complementary methods to experimental ones.
Among a number of computational methods that have been widely exploited for the prediction of PPIs, kernel methods attracted much attention. Similar kernels were designed for predicting interactions from sequence. Kernels, including spectrum kernel (Leslie et al. 2002), Pfam kernel (Gomez et al. 2003), constrained diffusion kernel (Koji and William 2004), pairwise kernel and sequence kernel (Ben-Hur and Noble 2006), signature product kernel (Martin et al. 2005; Faulon et al. 2008), were defined for describing the relationship between two pairs of sequences. By combining with support vector machine (SVM), which holds good generalization performance and performs better for small sample size, such kernels resulted in more accurate SVM performance when dealing with prediction problems of predicting PPIs. Meanwhile, with the increase of the number of protein sequences, sequence-based methods using various coding schemes have recently been proposed for PPIs (Sprinzak and Margalit 2001; Gomez et al. 2003; Martin et al. 2005; Shen et al. 2007; Guo et al. 2008b). Specifically, signature features of protein pair’s sequences were used to predict PPIs (Sprinzak and Margalit 2001). Gomez et al. (2003) put forward an attraction-repulsion model using the domain or motif content of a sequence to predict a candidate interaction. Martin et al. (2005) devised a protein descriptor called signature products to represent interactions between pairs of protein sequences by combining the full-length sequence information of both domains and their ligands. Shen et al. (2007) proposed a SVM model by combining a conjoint triad feature with S-kernel function of protein pairs to predict PPI network. Guo et al. (2008b) proposed a sequence-based method by combining auto covariance feature representation and SVM, and when performed on the PPI data of yeast S. cerevisiae, it achieved a very promising prediction result. Sequence-derived structural and physicochemical features of protein sequence, including Moran autocorrelation (Horne 1988), autocross-covariance transformation (Wold et al. 1993), normalized Moreau–Broto autocorrelation (Feng and Zhang 2000) and Geary autocorrelation (Sokal and Thomson 2006) have also been used for predicting PPIs and enhance the prediction ability.
Despite their achievement, the existing methods for PPIs are limited by the fact that protein interaction datasets are usually incomplete and potentially unreliable (Uetz et al. 2000; Ito et al. 2001). Even reliable techniques can generate many false positive data. The absolute number of false positives may be larger than that of true positives because the expected number of negatives is several orders of magnitude higher than the number of positives. (Manly et al. 2004; Jansen and Gerstein 2004).Therefore, computational methods of assessing the reliability of each candidate protein interaction are very urgently needed. Saito et al. (2003) developed interaction generality measure to assess the credibility of PPIs using the topological properties of the interaction network structure. Meanwhile, the gene ontology (GO) is extensively exploited to analyze all kinds of high-throughput experiments (Resnik 1999; Wu et al. 2006; Guo et al. 2006, 2008a; Wang et al. 2007). Resnik’s method was proposed to determine the similarity of two GO terms based on their distances to the closest common ancestor term and/or the annotation statistics of their common ancestor terms (Resnik 1999). Although Resnik’s method is better than other methods (Guo et al. 2006), it ignores the information contained in the structure of the ontology. An improved method has been provided to encode a GO term’s semantics into a numeric value by aggregating the semantic contributions of their ancestor terms in the GO graph and the outcomes were shown to be more consistent with human perspectives (Wang et al. 2007). Thus, high-quality protein interaction data was achieved by exploring the information buried in the GO and GO annotations. Based on this idea, a new functional predictor was constructed to systematically predict the map of potential physical interactions between yeast proteins by fully exploring the knowledge buried in two GO annotations, namely, the Biological Processes and Cellular Components annotations (Wu et al. 2006; Guo et al. 2008a).
In this paper, a sequence-based method with correlation coefficient (CC) transformation was proposed to take into account the longer range relationship of amino acid residues of protein sequences and the level of CC between sequences of a protein pair. Gold standard positive and negative datasets were also constructed to objectively evaluate this method. Results suggested the competitive advantage of the method.
Materials and methods
Constructing positive and negative datasets
We chose S. cerevisiae physically interacting protein pairs that are derived from the following three popular databases as positive examples when training our classifiers. (1) DIP core dataset (Xenarios et al. 2002; Deane et al. 2002) was derived from the DIP database of DIP_20071007 (http://dip.doe-mbi.ucla.edu/dip). The original DIP core database contains 2,808 proteins and 6,459 interactions. After the protein pairs with ≤50 amino acids have been removed, the remaining dataset includes 2,800 proteins and 6,436 interactions. (2) MIPS Core dataset was gathered from the MIPS dataset (Mewes et al. 2006; Guldener et al. 2006) (http://mips.gsf.de/). MIPS dataset contains 4,556 proteins and 15,037 interactions. The MIPS Core dataset was extracted from the MIPS dataset following the principle that the selected PPIs have been validated by at least two large-scale or small-scale methods. The final MIPS Core dataset contains 829 proteins and 1,025 interactions after removing protein pairs with ≤50 amino acids. (3) BIND yeast database contains 10,517 interactions of 4,233 yeast proteins. Subset dataset with 736 proteins and 750 trusted interactions has been chosen from the BIND yeast database by multiple experimental assays (Bader et al. 2001; Ben-Hur and Noble 2006).
Negative training examples play an important role for the reliability of the prediction model and influence its performances of the test examples. Four strategies as following for constructing negative training examples were employed. (1) R-NEG: the non-interacting protein pairs are assembled by randomly pairing proteins that appeared in the positive datasets. (2) BS-NEG: this method is based on an assumption that proteins occupying different subcellular localizations do not interact. The non-interacting pairs were generated from proteins in separate subcellular compartments and the negative training examples were selected from these different subsets according to the proportional law. The non-pairing proteins are derived from database Organelle DB (seen in Fig. 1). (3) IS-NEG: considering a biased estimate of the accuracy of a PPI predictor, it is necessary to generate a dataset of the non-interacting pairs with the same localization to attenuate this bias. The non-interacting protein pairs with the same localization were generated from database Organelle DB and none of them has existed in the whole DIP Core, MIPS Core and BIND interacting pairs. (4) GO-NEG: Wu et al. (2006) designed a metric called relative specificity similarity (RSS) for semantic similarity, to score the degree of functional association or the localization proximity between two proteins. It fully explored the knowledge buried in the cellular component and biological process annotations of GO for the yeast genome. Wu et al. (2006) had two conclusions: (1) interacting proteins often function in the same Biological Processes (2) interacting proteins exist in close proximity. Here, the protein pairs of GO-NEG (0 <=RSS Cellular Components <=0.4 and 0 <=RSS Biological Processes <=0.4) with lower confidence were selected as negative samples, because protein pairs with low confidence level values of RSS Biological Processes and RSS Cellular Components involve in weakly related or unrelated biological processes and localize in different cellular components. Two rational requirements should be made during the construction of the negative datasets: (1) in each case the number of negative examples is equal to the number of positive examples in the dataset. (2) Non-interacting protein pairs can not be listed in the DIP, MIPS and BIND datasets. The details of PPIs data are summarized in Table 1.

Number of proteins of S. cerevisiae distributed in different subcellular compartments from Organelle DB (http://organelledb.lsi.umich.edu/) (Wiwatwattana et al. 2007). Organelle DB presents a catalog of localized proteins and major protein complexes of eukaryote S. cerevisiae
Molecular descriptors
Twelve physicochemical properties of amino acids were chosen to reflect the amino acids characteristics. These properties include hydrophobicity (Sweet and Eisenberg 1983), hydrophilicity (Hopp and Woods 1981), polarity (Grantham 1974), polarizability (Charton and Charton 1982), solvation free energy (Eisenberg and McLachlan 1986), graph shape index (Fauchere 1988), transfer free energy (Janin 1979), amino acid composition (Grantham 1974), CC in regression analysis (Prabhakaran and Ponnuswamy 1982), residue accessible surface area in tripeptide (Chothia 1976), partition coefficient (Garel 1973) and entropy of formation (Hutchens 1970), respectively. These sequence-based physiochemical properties are employed as basis for classification. Supplementary Table S1 showed the values of the 12 physicochemical properties for each amino acid. Min–max normalization reprocessing method was used to normalize these physicochemical properties according to Eqs. 1 and 2:
where s pr is the rth descriptor value for Pth amino acid and the norm \( \left\| {\left. \cdot \right\|} \right. \) is the 2-norm for vectors. Thus each protein sequence was coded by the normalized values of 12 descriptors.
Correlation coefficient transformation was employed to transform the physicochemical descriptions into a uniform length. CC variables describe the level of correlation between two protein sequences in terms of their specific physicochemical properties, which are defined based on the distribution of amino acid properties along the sequence. Furthermore, CC variables consider the long range correlation in the protein sequences which is very important to represent the PPI information. Also, CC variables represent the co-evolution of 12 physiochemical properties between the 2 proteins at different sequence distances. Co-evolution of physiochemical properties and co-evolution in general have been successfully used to predict interacting proteins (Brenner et al. 1998; Madaoui and Guerois 2008; Yeang and Haussler 2007).
Correlation coefficient of protein sequence could be defined as:
where A and B represent two protein pairs, respectively. Ai,j and Bk,j are given by
where i and k are the position of the amino acid sequences X and Y, j is 1 of 12 physicochemical properties of amino acids, m and n are the length of amino acid sequences X and Y, respectively, d represents the distance between two different residues of protein sequence. In Eq. 3, d is the lag of CC and d = 1, 2,…, lg, where lg is the maximum d. The CC feature is calculated with 12 descriptors and 12 × lg descriptor values. Obviously, the dimension of vector space of protein sequence with CC transformation(12 × lg) is dramatically reduced compared with that of auto cross covariance (2 × 12 × 12 × lg) (Wold et al. 1993) and auto covariance (AC) transformation (2 × 12 × lg) (Guo et al. 2008b).
SVM optimization and evaluation of performance
Kernel method address the classification problem by mapping the data into a high dimensional feature space, where each coordinate corresponds to one feature of the data items. The advantage of the kernel method is in the feature space no need computing the coordinates of the data, but rather simply computing the inner products between all pairs of data. This operation is often computationally cheaper than the explicit computation of the coordinates. The representative kernel-based methods include SVM (Vapnik 1998), kernel Fisher’s linear discriminant analysis (Baudat and Anouar 2000) and kernel principal components analysis (Scholkopf et al. 1998), which have effectively solved many practical problems.
Inverse problems of matrix are often ill-posed and always encountered in many machine learning methods such as LDA, CCA and SVM. To solve these problems numerically one must introduce some additional assumption on the smoothness or a bound on the norm. The popular method is often known as regularization and regularization constant C is usually applied to improve the generalization performance and diminish the complexity of model.
Cross validation is a popular model evaluation method which validates how well a model generalizes to new data. K-fold cross validation divides the dataset into k subsets and repeats k times. Each time, one of the k subsets is used as the test set and the rest k − 1 subsets are put together to form a training set. Then the average error across all k trials is computed. The advantage of this validation method is that it matters less how the data gets divided. Every data point gets to be in a test set exactly once and in a training set k − 1 times.
LIBSVM (http://www.csie.ntu.edu.te/~cjlin/libsvm) was used to do classification. Radial Basis Function \( k(x,x^{\prime}) = { \exp }( - \gamma \left\| {x - x^{\prime}} \right\|^{2} ) \)was selected as the kernel function and the optimized parameters (C, γ) were obtained with a grid search approach. The prediction performances are evaluated as follows:
where TP is the true positive, FN is the false negative, FP is the false positive, TN is the true negative and MCC denotes Mathews CC.
The P value was calculated to evaluate the credibility of the prediction performance. P value is defined as
where z and z observed refer to the value of sensitivity, precision, MCC and accuracy with the above validated and randomly generated observation datasets, respectively. Randomly generalized 1,000 observation datasets were constructed from the protein pairs of the validated datasets and the size of each randomly generalized dataset is equal to that of the validated datasets.
Results and discussions
Assessment of prediction ability with different positive and negative datasets
Support vector machine prediction models were constructed to evaluate the performance of the three positive datasets of S. cerevisiae and the four negative datasets respectively. Twelve combinations were built with the above datasets. 50% of the protein pairs were randomly selected from the positive and negative dataset as the training set and the remaining 50% were chosen as the test set. For instance, the final data set consisted of 2,050 protein pairs when datasets of MIPS Core plus GO-NEG are trained with the SVM method and the training set and test set include 1,025 and 1,025 protein pairs respectively. The regularization parameter C and parameter γ were selected by applying fivefold cross-validation. This process has been repeated five times.
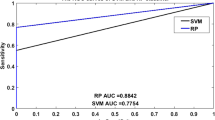
Tables 2 and 3 illustrate the average prediction performance of the SVM models. The models with positive datasets DIP Core, MIPS Core and BIND and negative dataset R-NEG yield the Accuracy of 80.7, 83.6 and 76.9%, respectively. These accuracies are higher than those with negative dataset IS-NEG and lower than those with negative dataset BS-NEG. The simple uniform random choice of non-interacting protein pairs (R-NEG) yields an unbiased estimate of the true distribution when predicting PPIs (Ben-Hur and Noble 2006). However, imposing the constraint of non co-localization induces a different distribution on the features and the resulting biased distribution of negative examples leads to over-optimistic estimates of classifier accuracy. So selecting the non-interacting protein pairs from the same co-localization is an useful method to attenuate this prediction bias. The SVM models based on three positive datasets and the negative dataset BS-NEG yield the Accuracy of 82.7, 85.8 and 77.8%, respectively, whereas, the Accuracy with negative dataset IS-NEG are 79.5, 81.8 and 75.8%, respectively, and these results are apparently lower than those with negative dataset BS-NEG. The SVM models with three positive datasets and GO-NEG yield the Accuracy of 84.91, 87.94 and 82.21%, respectively, which achieves the best results compared with other three negative datasets. The average prediction performance, i.e., Sensitivity, Precision, MCC and Accuracy based on MIPS Core and GO-NEG are 86.86, 89.52, 75.98 and 87.94%, respectively. Therefore, the model based on the positive dataset MIPS Core and negative dataset GO-NEG achieves the best performance. GO-NEG generally outperforms R-NEG, BS-NEG and IS-NEG based on positive dataset MIPS Core in terms of prediction performance. The average Accuracy based on positive dataset MIPS Core and negative dataset GO-NEG is more than 5.2, 2.5, 7.5% higher than that on negative dataset R-NEG, BS-NEG and IS-NEG, respectively. Moreover, the average Accuracy based on GO-NEG and MIPS Core is more than 3.6% higher than that on GO-NEG and DIP Core. The average Accuracy of three SVM models based on three positive datasets and negative dataset GO-NEG is 85%, which yields more than 5.7, 3.5, 7.6% higher than that on negative dataset R-NEG, BS-NEG and IS-NEG, respectively. It illustrates that the SVM model with CC transformation has enough generalization ability.
Gold standard positives and gold standard negatives dataset
As shown in Tables 2 and 3, the performances of DIP Core are inferior to that of MIPS Core. A possible explanation is summarized: the protein interaction pairs of DIP Core may lack biological significance or be noisy. Consequently, there may be a number of random protein pairs in the simulation dataset of DIP Core. We found that negative dataset GO-NEG yielded better prediction performances than other three negative datasets. The dataset GO-NEG has two advantages over conventional three datasets. Protein pairs of GO-NEG both involved in weakly related or unrelated biological processes are localized in different cellular components. Thus, the resulting GO-NEG dataset is less biased compared with other three negative datasets. Moreover, the assignment of protein pairs into categories with different RSS values is statistically significant (Wu et al. 2006). Thus, the GO-NEG dataset is less biased compared with those constructed using other three strategies.
The null hypothesis statistical test are also used to evaluate the prediction performance with P value that could be computed with Eq. 9. The P value is compared with the significance level (i.e., 0.05) of the null hypothesis test: if the P value is less than 0.05, the null hypothesis is rejected and the difference between two prediction models is considered to be statistically significant. The results of P value could be summarized as follows: (1) for the dataset MIPS Core, its prediction performance are statistically significant with all four negative datasets. (2) For the dataset BIND and DIP Core, the prediction performance are not statistically significant with four negative datasets. Some interacting protein pairs in positive dataset BIND and DIP Core may be noisy or lack of biological significance compared with other randomly generalized observation datasets.
We evaluated the 12 combinations of the three positive datasets and the four negative datasets. The prediction performance of the other eleven dataset combinations were much lower than that obtained from the combination of GSPs and GSNs, indicating that the datasets of the other eleven combinations contain high rate false positives. And these results demonstrate that high-quality datasets have a strong effect on the performance of computational methods for the prediction of PPIs.
Performance comparison of CC and AC
The prediction results of SVM prediction models with CC and AC transformation of protein sequence for five test sets are shown in Table 4. The results showed that SVM prediction model with CC transformation outperforms that with AC transformation in terms of performances. Moreover, the average values of Sensitivity, Precision, MCC and Accuracy with CC transformation are more than 2, 6.6, 9.5 and 3.8% higher than those with AC transformation. The standard deviation of Accuracy is as low as 0.92, indicating that data points are close to the mean. Furthermore, the Accuracy with CC and AC transformation based on 7 physicochemical properties of 20 amino acids (Guo et al. 2008b) are 81.26 and 81.79%, respectively. It shows that the value of Accuracy with CC transformation increase dramatically than that of AC transformation when the number of physicochemical properties is increasing. CC variables consider the co-evolution of physiochemical properties between the two proteins and the long range correlation, which leads to a significant increase in Accuracy. Above all, the performance can be substantially improved by selecting the appropriate coding scheme.
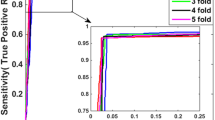
The prediction results for SVM classifier with CC transformation of different d values are shown in Fig. 2. SVM classifier with CC based on MIPS Core and GO-NEG is constructed by optimal parameters C = 75, γ = 0.1125. SVM classifier with CC based on DIP Core and GO-NEG is built with optimal parameters C = 120, γ = 0.22. Figure 2 shows that the maximum value, 87.94%, of an average Accuracy of MIPS Core is achieved and the corresponding d value is 24, and those of DIP Core are 84.7 and 31%, respectively. The results also illustrate that CC transformation would lose useful classification information of protein sequence with d value less than 24 amino acids and introduce noise to diminish average Accuracy with d value larger than 24 amino acid. Moreover, SVM prediction model with MIPS Core is computationally more efficient than that with DIP Core.

The average accuracy of SVM classifier with CC transformation of different d values, respectively
There are two possible reasons that the SVM prediction model with CC transformation outperforms that with AC transformation. First, CC transformation adequately considers the neighboring effect of protein sequence and describes the level of CC between two protein sequences. Second, the dimension of vector space of protein sequence with CC transformation is dramatically reduced compared with that of AC transformation. It also demonstrates that eigenvectors with CC transformation include less noisy data and it efficiently attenuates the biased selection of positive and negative examples.
Performance of dataset H. pylori
In order to evaluate the practical prediction ability of the SVM prediction model with CC transformation towards cross-species analysis, dataset H. pylori was constructed. The H. pylori dataset contains 1,458 interacting protein pairs and 1,458 non-interacting protein pairs (Rain et al. 2001). 50% of the protein pairs were randomly selected from the interacting and non-interacting protein pairs as the training set and the remaining 50% were chosen as the test set. The regularization parameter C and parameter γ were selected by applying fivefold cross-validation. The performance of this method in predicting such samples is summarized in Table 5. The average prediction performance, i.e., Sensitivity, Precision, MCC and Accuracy achieved by SVM with CC transformation are 81.25, 80.53, 72.26 and 83.68%, respectively. It is shown that SVM method achieves better performance than Boosting (Friedman 2001) and Linear Regression with L1 regularization (the Lasso) (Efron et al. 2004). All these results demonstrate that this SVM classifier is also able to achieve better performance towards cross-species dataset.
Conclusions
We have developed a simple and elegant sequenced-based approach to predict PPIs. The prediction model was constructed by using CC transformation and SVM. CC transformation of protein sequences was calculated with different lags along a protein chain in terms of their specific physicochemical properties and account for longer range relationship. It also describes the level of CC between two protein sequences. Moreover, we evaluated high-throughput experimental interaction datasets using the three positive datasets and the four negative datasets including GSPs and GSNs. And the results demonstrate that a high-quality positive and negative dataset has a strong effect on the performance of any of the computational methods for prediction of PPIs and plays an important role in the inference of interacting protein pairs with high confidence. Protein pairs both involved in weakly related or unrelated biological processes and localized in different cellular components are chosen and assembled into GSNs. Thus, the resulting GSNs dataset is less biased compared with other three negative datasets. This analysis is expected to also provide a new approach for predicting PPIs from protein sequences with high-quality GO-based annotations.
In conclusion, the proposed sequence-based method using SVM and CC transformation will be a powerful tool to predict PPIs and expedite the study of protein networks.
References
Bader GD, Donaldson I, Wolting C, Ouellette BF, Pawson T, Hogue CW (2001) BIND-the biomolecular interaction network database. Nucleic Acids Res 29:242–245
Baudat G, Anouar F (2000) Generalized discriminant analysis using a kernel approach. Neural Comput 12:2385–2404
Ben-Hur A, Noble WS (2006) Choosing negative examples for the prediction of protein–protein interactions. BMC Bioinformatics 7:S2
Brenner SE, Chothia C, Hubbard TJ (1998) Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc Natl Acad Sci USA 95:6073–6078
Charton M, Charton BI (1982) The structural dependence of amino acid hydrophobicity parameters. J Theor Biol 99:629–644
Chothia C (1976) The nature of the accessible and buried surfaces in proteins. J Mol Biol 105:1–12
Deane CM, Salwinski L, Xenarios I, Eisenberg D (2002) Protein interactions: Two methods for assessment of the reliability of high throughput observations. Mol Cell Proteomics 1:349–356
Efron B, Hastie T, Johnstone I, Tibshirani R (2004) Least angle regression. Ann Stat 32:407–499
Eisenberg D, McLachlan AD (1986) Solvation energy in protein folding and binding. Nature 319:199–203
Fauchere JL (1988) Amino acid side chain parameters for correlation studies in biology and pharmacology. Int J Pept Protein Res 32:269–278
Faulon JL, Misra M, Martin S, Sale K, Sapra R (2008) Genome scale enzyme-metabolites and drug-target interaction predictions using the signature molecular descriptor. Bioinformatics 24:225–233
Feng ZP, Zhang CT (2000) Prediction of membrane protein types based on the hydrophobic index of amino acids. J Protein Chem 19:269–275
Friedman JH (2001) Greedy function approximation: a gradient boosting machine. Ann Stat 29:1189–1232
Garel JP (1973) Coefficients de partage d’aminoacides, nucleobases, nucleosides et nucleotides dans un systeme solvant salin. J Chromatogr 78:381–391
Gavin AC et al (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415:141–147
Giot L et al (2003) A protein interaction map of Drosophila melanogaster. Science 302:1727–1736
Gomez SM, Noble WS, Rzhetsky A (2003) Learning to predict protein–protein interactions. Bioinformatics 19:1875–1881
Grantham R (1974) Amino acid difference formula to help explain protein evolution. Science 185:862–864
Guldener U, Munsterkotter M, Oesterheld M, Pagel P, Ruepp A, Mewes HW, Stumpflen V (2006) MPact: the MIPS protein interaction resource on yeast. Nucleic Acids Res 34:D436–D441
Guo X et al (2006) Assessing semantic similarity measures for the characterization of human regulatory pathways. Bioinformatics 22:967–973
Guo J, Wu XM, Zhang DY, Lin K (2008a) Genome-wide inference of protein interaction sites: lessons from the yeast high-quality negative protein–protein interaction dataset. Nucleic Acids Res 36:2002–2011
Guo YZ, Yu LZ, Wen ZN, Li ML (2008b) Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res 36:3025–3030
Ho Y et al (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415:180–183
Hopp TP, Woods KR (1981) Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci USA 78:3824–3828
Horne DS (1988) Prediction of protein helix content from an autocorrelation analysis of sequence hydrophobicities. Biopolymers 27:451–477
Hutchens JO (1970) Heat capacities, absolute entropies, and entropies of formation of amino acids and related compounds. In: Sober HA (ed) Handbook of biochemistry, 2nd edn. Chemical Rubber Co., Cleveland, pp B60–B61
Ito T et al (2000) Toward a protein–protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc Natl Acad Sci USA 97:1143–1147
Ito T et al (2001) A comprehensive two-hybrid analysis to explore the yeast protein ineractome. Proc Natl Acad Sci USA 98:4569–4574
Janin J (1979) Surface and inside volumes in globular proteins. Nature 277:491–492
Jansen R, Gerstein M (2004) Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Curr Opin Microbiol 7:535–545
Koji T, William SN (2004) Learning kernels from biological networks by maximizing entropy. Bioinformatics 20:i326–i333
Krogan NJ et al (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440:637–643
Leslie C, Eskin E, Noble WS (2002) The spectrum kernel: a string kernel for SVM protein classification. In: Proceedings of the Pacific symposium on biocomputing, New Jersey. World Scientific, Singapore, pp 564–575
Li S et al (2004) A map of the interactome network of the metazoan c elegans. Science 303:540–543
Madaoui H, Guerois R (2008) Coevolution at protein complex interfaces can be detected by the complementarity trace with important impact for predictive docking. Proc Natl Acad Sci USA 105:7708–7713
Manly KF, Nettleton D, Hwang JT (2004) Genomics, prior probability, and statistical tests of multiple hypotheses. Genome Res 14:997–1001
Martin S, Roe D, Faulon JL (2005) Predicting protein–protein interactions using signature products. Bioinformatics 21:218–226
Mewes HW et al (2006) MIPS: analysis and annotation of proteins from whole genomes in 2005. Nucleic Acids Res 34:D169–D172
Prabhakaran M, Ponnuswamy PK (1982) Shape and surface features of globular proteins. Macromolecules 15:314–320
Rain JC et al (2001) The protein–protein interaction map of Helicobacter pylori. Nature 409:211–215
Resnik P (1999) Semantic similarity in a taxonomy: an information-based measure and its application to problems of ambiguity in natural language. J Artif Intell Res 11:95–130
Saito R et al (2003) Construction of reliable protein–protein interaction networks with a new interaction generality measure. Bioinformatics 19:756–763
Scholkopf B, Smola A, Muller KR (1998) Nonlinear component analysis as a kernel eigenvalue problem. Neural Comput 10:1299–1319
Shen JW et al (2007) Predicting protein–protein interactions based only on sequences information. Proc Natl Acad Sci USA 104:4337–4341
Sokal RR, Thomson BA (2006) Population structure inferred by local spatial autocorrelation: an example from an Amerindian tribal population. Am J Phys Anthropol 129:121–131
Sprinzak E, Margalit H (2001) Correlated sequence-signatures as markers of protein–protein interaction. J Mol Biol 311:681–692
Sweet RM, Eisenberg D (1983) Correlation of sequence hydrophobicities measures similarity in three-dimensional protein structure. J Mol Biol 171:479–488
Uetz P et al (2000) A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 403:623–627
Vapnik V (1998) Statistical learning theory. Wiley, New York
Von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P (2002) Comparative assessment of large scale data sets of protein–protein interactions. Nature 417:399–403
Wang JZ, Du ZD, Payattakool R, Yu PS, Chen CF (2007) A new method to measure the semantic similarity of GO terms. Bioinformatics 23:1274–1281
Wiwatwattana N, Landau CM, Cope GJ, Harp GA, Kumar A (2007) Organelle DB: an updated resource of eukaryotic protein localization and function. Nucleic Acids Res 35:D810–D814
Wold S et al (1993) DNA and peptide sequences and chemical processes mutlivariately modelled by principal component analysis and partial least-squares projections to latent structures. Anal Chim Acta 277:239–253
Wu X, Zhu L, Guo J, Zhang DY, Lin K (2006) Prediction of yeast protein–protein interaction network: insights from the gene ontology and annotations. Nucleic Acids Res 34:2137–2150
Xenarios I et al (2002) Dip, the database of interacting proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res 30:303–305
Yeang CH, Haussler D (2007) Detecting coevolution in and among protein domains. PLoS Comput Biol 3:e211
Zhu H et al (2001) Global analysis of protein activities using proteome chips. Science 293:2101–2105
Acknowledgments
This work was supported by the grants of the National Science Foundation of China, Nos. 60472111 and 30570368, the grant from the National Basic Research Program of China (973 Program), No. 2007CB311002, the grants from the National High Technology Research and Development Program of China (863 Program), Nos. 2007AA01Z167 and 2006AA02Z309, the grant of Oversea Outstanding Scholars Fund of CAS, No. 2005-1-18, HFUT, No. 070403F and the Knowledge Innovation Program of the Chinese Academy of Sciences (0823A16121).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Shi, MG., Xia, JF., Li, XL. et al. Predicting protein–protein interactions from sequence using correlation coefficient and high-quality interaction dataset. Amino Acids 38, 891–899 (2010). https://doi.org/10.1007/s00726-009-0295-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-009-0295-y